
Abstract
Sideromycins are antibiotics covalently linked to siderophores. They are actively transported into gram-positive and gram-negative bacteria. Energy-coupled transport across the outer membrane and the cytoplasmic membrane strongly increases their antibiotic efficiency; their minimal inhibitory concentration is at least 100-fold lower than that of antibiotics that enter cells by diffusion. This is particularly relevant for gram-negative bacteria because the outer membrane, which usually forms a permeability barrier, in this case actively contributes to the uptake of sideromycins. Sideromycin-resistant mutants can be used to identify siderophore transport systems since the mutations are usually in transport genes. Two sideromycins, albomycin and salmycin, are discussed here. Albomycin, a derivative of ferrichrome with a bound thioribosyl-pyrimidine moiety, inhibts seryl-t-RNA synthetase. Salmycin, a ferrioxamine derivative with a bound aminodisaccharide, presumably inhibts protein synthesis. Crystal structures of albomycin bound to the outer membrane transporter FhuA and the periplasmic binding protein FhuD have been determined. Albomycin and salmycin have been used to characterize the transport systems of Escherichia coli and Streptococcus pneumoniae and of Staphylococcus aureus, respectively. The in vivo efficacy of albomycin and salmycin has been examined in a mouse model using Yersinia enterocolitica, S. pneumoniae, and S. aureus infections. Albomycin is effective in clearing infections, whereas salmycin is too unstable to lead to a large reduction in bacterial numbers. The recovery rate of albomycin-resistant mutants is lower than that of the wild-type, which suggests a reduced fitness of the mutants. Albomycin could be a useful antibiotic provided sufficient quantities can be isolated from streptomycetes or synthesized chemically.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron, an essential element, is contained in active centers of many redox enzymes. Under oxic conditions, it occurs in nature as an insoluble Fe3+ hydroxide polymer and in eukaryotic hosts tightly bound to transferrin and lactoferrin. However, bacterial and fungal siderophores, with their high Fe3+ specificity and affinity, make Fe3+ available. The microorganisms synthesize and secrete the siderophores, which are then taken up as Fe3+-complexes via specific active transport systems. Gram-negative bacteria concentrate Fe3+-siderophores at the cell surface by binding them to very specific high-affinity transport proteins. The Fe3+-siderophores are then transported across the outer membrane into the periplasm and from there across the cytoplasmic membrane into the cytoplasm, where iron is released from the siderophores and incorporated into heme and non-heme iron proteins.
The term sideromycin was coined for naturally occurring Fe3+-siderophores that are covalently linked to an antibiotic moiety. Sideromycins were actually discovered prior to siderophores in a sceening for new antibiotics and were originally called sideramines or siderochromes (Nüesch and Knüsel 1967). Siderophores were later found to antagonize the activity of sideromycins, and it was initially assumed that the siderophores competed with the sideromycins at the sideromycin target sites. It was then shown that siderophores and sideromycins compete for common uptake systems.
Sideromycins are taken up by Fe3+-siderophore transport systems
Crystal structures of the proteins responsible for the transport of Fe3+-siderophores across the outer membrane have been determined. The various proteins are similar in structure and consist of 22 β strands that form β barrels with an internal pore completely closed by a globular domain, called the plug (summarized by Wiener 2005; Krewulak and Vogel 2008; Schalk 2008). Up to ten amino acid residues at the surface of the transporters bind the Fe3+-siderophores very tightly with K D values in the range of 1 nM.
Structural transitions must occur to release the Fe3+-siderophores and to move the plugs to open the pores. Both processes require energy, which is not available in the outer membrane. Energy is provided by the proton-motive force of the cytoplasmic membrane and is transduced from there to the outer membrane by the Ton protein complex, consisting of TonB, ExbB, and ExbD. These three proteins are embedded in the cytoplasmic membrane (Fig. 1). The stoichiometry of the TonB:ExbB:ExbD proteins in cells, but not necessarily in the complex, is 1:7:2. TonB interacts with the outer membrane transport proteins, as has been revealed by genetic, biochemical, and protein crystallographic methods. The C-proximal end of TonB forms a three-stranded β-sheet, to which the β-strand of the N-terminal end of the transporters binds (Fig. 1). Prior to the crystallographic proof of binding, the N-terminal end was presumed to bind to TonB based on various pieces of evidence: sequence similarities among transporters, mutations that inactivate the transporters, and suppressor mutations in the C-proximal end of TonB that restore the activity of the TonB box mutants. This region was therefore designated the TonB box (Schramm et al. 1987). The Ton complex responds to the proton-motive force by altering the confirmation of TonB to its energized form (Larsen et al. 1999). It is assumed that interaction of the energized form with the outer membrane transporters then alters the conformation of the transporters, resulting in the release of the bound Fe3+-siderophores, the opening of a pore in the transporters, and the diffusion of the Fe3+-siderophores into the periplasm.

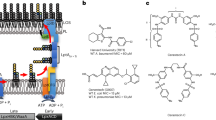
Albomycin and ferrichrome transport system of E. coli. Both compounds are transported by the same proteins: FhuA in the outer membrane, FhuD in the periplasm, and FhuB in the cytoplasmic membrane. FhuC is the ATPase that provides the energy for transport across the cytoplasmic membrane. The proteins TonB, ExbB, and ExbD form a complex that provides energy for transport across the outer membrane. The transmembrane topology of the cytoplasmic membrane proteins but not the stoichiometry of TonB, ExbB, and ExbD is shown. The complete crystal structures of FhuA (red) and FhuD (green) loaded with albomycin and the crystal structures of periplasmic portions of TonB and ExbD are shown. The 22 anti-parallel β-strands of FhuA form a β-barrel (blue-green) that is closed by a globular domain (yellow). The TonB box of FhuA forms with TonB a four-stranded β-sheet. The structure of the linkage between the membrane segment and the crystallized domain of TonB is unknown
Only a few naturally occurring sideromycins have been found, among them albomycin and salmycin (Fig. 2). A wide spectrum of siderophore–antibiotic conjugates have been chemically synthesized. Although the antibiotic moieties account for a substantial part of the sideromycins and drastically change the shape of the siderophore from which they are derived, the sideromycins are nevertheless recognized and transported by the Fe3+-siderophore transport proteins. The transport proteins identify the Fe3+-complexes and tolerate additions to an astounding degree. It seems that whatever is bound to the active site of the transport proteins is also transported. The most extreme case is the transport of a rifamycin derivative, designated CGP 4832, by the FhuA outer membrane transporter—CGP 4832 in the crystal structure with FhuA shows no structural similarity with ferrichrome, the substrate of FhuA (Ferguson et al. 2001). Of the ten amino acid residues of FhuA that bind ferricrocin, a derivative of ferrichrome, nine also bind CGP 4832. The rifamycin derivative, however, is not transported across the cytoplasmic membrane, probably because it is not recognized by the FhuBD proteins that transport ferrichrome into the cytoplasm (Fig. 1).

Chemical structures of ferrichrome, albomycin, ferrioxamine B, and salmycin. The co-crystals of ferrichrome and albomycin with FhuA reveal their structures in the protein-bound form (Fig. 1)
Transport of albomycin into E. coli
Among the sideromycins, albomycin has been studied in most detail. Albomycin consists of a siderophore structurally similar to ferrichrome with a bound thioribosyl pyrimidine antibiotic (Fig. 2). Inside E. coli cells, the antibiotic is cleaved off the Fe3+ carrier by peptidase N, encoded by pepN. The antibiotic stays inside the cells, whereas the iron-free carrier is excreted. pepN mutants are albomycin resistant; therefore, release of the antibiotic from the carrier is required for activity. Without cleavage, albomycin functions only as a siderophore and carries iron into cells (Hartmann et al. 1979; Braun et al. 1983).
Albomycin is taken up via the ferrichrome transport system across the outer membrane and the cytoplasmic membrane of E. coli and other gram-negative bacteria. It binds to FhuA well above the surface of cells. It assumes two different conformations in the FhuA–albomycin crystal structure: an extended form and a compact form (Ferguson et al. 2000). The extended form has the same ten amino acid binding sites as ferrichrome. Four additional residues bind the antibiotic.
After the FhuA- and Ton-dependent uptake across the outer membrane, albomycin binds to FhuD in the periplasm (Fig. 1). Albomycin and ferrichrome bound to FhuD have dissociation constants of 5.4 and 1 μM, respectively (Rohrbach et al. 1995). FhuD then delivers albomycin to FhuB. FhuD binds to FhuB in this process, as indicated by chemical cross-linking of substrate-loaded as well as unloaded FhuD to FhuB and the prevention of FhuB proteolysis when FhuB is incubated with FhuD (Rohrbach et al. 1995). The two independently folded domains of FhuD are connected by an α-helix that confers rigidity to the molecule. The large movement of the two domains upon substrate binding and release observed in most other periplasmic binding proteins (Venus flytrap-like fashion) does not occur in FhuD. A molecular dynamics simulation has revealed a closure of 6° of a C-terminal domain upon release of the siderophore; this movement seems to be sufficient for the distinction of unloaded and loaded FhuD by FhuB (Krewulak et al. 2005).
In contrast to most dimeric ABC transport proteins in the cytoplasmic membrane, FhuB consists of a single polypeptide with 12 transmembrane segments (Groeger and Köster 1998). The N- and C-terminal halves are homologous. FhuB point mutations in regions conserved among ABC transporters confer albomycin resistance and lack of ferrichrome transport (Köster and Böhm 1992). It is not known whether there is specific albomycin binding to FhuB in the cytoplasmic membrane. In this and other ABC import systems, substrate specificity is primarily determined by binding of the substrate to the periplasmic binding protein (Davidson et al. 2008). Binding of substrate-loaded FhuD to FhuB presumably opens a channel in FhuB and triggers ATP hydrolysis by FhuC.
FhuC is a typical NBD protein (ATPase), as found in other ABC transporters. Mutations in predicted ATP binding sites inactivate FhuC, abolish ferrichrome transport, and confer albomycin resistance (Schultz-Hauser et al. 1992).
Comparison of the crystal structures of four ABC transporters (Hollenstein et al. 2007) and biochemical studies of ABC transporters reconstituted in liposomes (Davidson et al. 2008) reveal the following common features, which might also apply to ferrichrome and albomycin transport via FhuBCD. The channel in the cytoplasmic membrane is formed by 12 α-helices. They assume an outward-facing or inward-facing conformation. Unidirectional transport is achieved by the ATP-driven conversion of the inward-facing state to the outward-facing state. Binding of two ATP molecules at the interface between the two nucleotide-binding domains (ATPases) closes the gap between the two ATP-binding sites and also causes closure of the helices that bind to the two NBD molecules. The helices flip from the inward-facing to the outward-facing conformation. This structural transition might be transmitted to the substrate-loaded binding protein, which opens and releases the substrate to the inner membrane transporter. ATP hydrolysis and dissociation of ADP + Pi from the ABC transporter reset the inward-facing conformation, allowing the substrate to enter the cytoplasm.
Transport of albomycin and salmycin into Streptococcus pneumoniae and Staphylococcus aureus
Since salmycin inhibits almost exclusively gram-positive bacteria (see below), uptake of salmycin has been studied in the gram-positive S. pneumoniae and S. aureus. Mutants of both organisms display cross-resistance to salmycin and albomycin, and ferrichrome and ferrioxamine B inhibit the toxicity of both antibiotics. These results indicate that salmycin and albomycin are taken up by the same transport system.
The target site of albomycin is known, but that of salmycin is not. Salmycin consists of a ferrioxamine B derivative (danoxamine) and an aminodisaccharide (Vértesy et al. 1995). It inhibits an unidentified step of protein synthesis. Target-site mutants common to albomycin and salmycin cannot be isolated because the completely different antibiotics exclude the same toxicity mechanism. Since albomycin is more readily available, mostly it has been used to isolate transport mutants of Streptococcus peumoniae and to characterize the mutated genes and their products. A few S. aureus mutants have been selected with salmycin (Gwinner and Braun, unpublished results).
Several iron transport systems have been predicted in S. pneumoniae based on comparison of its genome sequence with other genome sequences (Brown et al. 2002). These systems have been shown experimentally to be related to iron transport, but siderophores have not been identified. The identification of siderophores in S. pneumoniae is difficult because the bacterium only grows on complex media, such as blood agar. The use of sideromycins circumvents this problem since resistant mutants can be isolated on complex media and these mutants usually have modified transport proteins (Pramanik and Braun 2006). Using this approach, the albomycin transport genes were identified (Fig. 3). Additional mutants were isolated by transposon mutagenesis and insertion duplication mutagenesis. Chromosomal fhuB, fhuD, and fhuG mutants were isolated. The transport defects were verified by comparing growth promotion of the wild-type and the mutants by the Fe3+-hydroxamates and by comparing the transport rates of radiolabeled [55Fe3+]ferrichrome and radiolabeled [55Fe3+]ferrioxamine B of the wild-type with that of mutants. In addition, the Fe3+-hydroxamates cross-streaked on nutrient agar with the antibiotics interfered with growth inhibition of the antibiotics. The FhuD binding protein has been isolated and shown to bind ferrichrome, ferrioxamine B, albomycin, and salmycin (Pramanik and Braun 2006). This study shows that the S. pneumoniae genome encodes a single Fe3+-hydroxamate transport system, through which ferrichrome, ferrioxamine B, and the structurally related antibiotics albomycin and salmycin are taken up into the cells.

Arrangement of the fhu genes on the genome of S. pneumoniae and S. aureus. The number of nucleotides (nt, nucleotides; mnt, million nucleotides) between the genes is indicated; a negative number indicates that the genes overlap
The transport system of S. aureus for Fe3+-hydroxamates has been characterized without the use of sideromycins (Sebulsky et al. 2003; Speziali et al. 2006). S. aureus grows on simple defined media, which allow addition of Fe3+-siderophores as the sole iron source. The transport system exhibits broad substrate specificity and takes up ferrichrome and the Fe3+-complexes of aerobactin, coprogen, Desferal®, and rhodotorulic acid. Five involved genes have been identified, four of which are required to promote growth by ferrichrome on iron-limited medium. The genome of S. aureus contains two fhuD genes, which are located distant from the fhuBCG genes (Fig. 3). Both fhuD genes encode substrate-binding proteins that are anchored by an N-terminal lipid to the cytoplasmic membrane. Mutant H430 fhuD1 is sensitive to salmycin and albomycin and transports ferrioxamine and ferrichrome at rates similar to the wild-type. Mutant H364 fhuD2 transports ferrioxamine B with only 10% of the wild-type rate and is nearly salmycin resistant; in contrast, the ferrichrome transport rate is 20% of the wild-type rate, which is sufficient to confer full albomycin sensitivity (Gwinner and Braun, unpublished results). Mutant H431 fhuD1 fhuD2 does not transport either siderophore and is resistant to both antibiotics. The fhuD2 mutant has a lower growth response to ferrichrome and ferrioxamine B than the fhuD1 mutant and a narrower specificity, i.e., aerobactin and coprogen do not support growth, in contrast to the fhuD1 mutant, which grows on all four Fe3+-hydroxamates (Sebulsky and Heinrichs 2001). FhuD2 is the preferred binding protein for the antibiotics and the Fe3+-hydroxamates. A spontaneous salmycin-resistant fhuD2 mutant with a mutation that truncates the protein from 302 to 279 residues still confers albomycin sensitivity. Either the C-proximal end is important for salmycin binding or the expression level of the protein is insufficient for salmycin uptake but sufficient for albomycin uptake. Additional spontaneous salmycin-resistant mutants with deletions in the fhuB gene or insertions in the fhuG gene that encode the transport proteins in the cytoplasmic membrane are cross-resistant to albomycin (Gwinner and Braun, unpublished results). These studies demonstrate a single Fe3+-hydroxamate transport system in S. aureus through which ferrichrome, ferrioxamine B, and the structurally related antibiotics albomycin and salmycin are taken up into cells.
Active transport strongly reduces the minimal inhibitory concentration
For many antibiotics, the diffusion rate across the cell membranes into the cytoplasm is poor. This is frequently a serious problem in antibiotic therapy against especially gram-negative bacteria since the outer membrane reduces permeation to such an extent that the minimal inhibitory concentration required reaches toxic levels. In contrast, for sideromycins, the outer membranes and the cytoplasmic membranes do not serve as permeability barriers, but rather actively contribute to the entry of the antibiotics to their targets in the cytoplasm. Active transport reduces the minimal inhibitory concentrations more than a 100-fold (Braun 1999). This is illustrated by the following examples.
In a screening for new antibiotics in Streptomyces species, the seryl thioribosyl pyrimidine moiety of albomycin free of the siderophore (designated SB-217452) was isolated and shown to inhibit in vitro the seryl-tRNA synthetases of S. aureus and rats (Stefanska et al. 2000). SB-217452 structurally resembles seryl adenylate, which explains the inhibition of seryl-tRNA synthetases. The high in vitro activity against isolated seryl-t-RNA synthetase (Table 1) conflicts with the poor in vivo activity against gram-positive and gram-negative bacteria. Without a siderophore carrier, the antibiotic moiety is 30,0000-fold less effective against S. aureus and E. coli because it barely enters these cells. Other bacteria are somewhat more sensitive to SB-217452, but still 1,000-fold less sensitive than to albomycin.
Another example is the semi-synthetic rifamycin derivative CGP 4832, which is >200-fold more active against E. coli and Salmonella than rifamycin. The enhanced activity of CGP 4832 rests on transport by the FhuA transporter across the outer membrane into the periplasm (Pugsley et al. 1987). Energy-coupled transport results in an accumulation in the periplasm, which accelerates diffusion across the cytoplasmic membrane, giving rise to an increased cytoplasmic concentration. The higher efficacy of CGP 4832 is not caused by a stronger inhibition of the DNA-dependent RNA polymerase than rifamycin. Ferrimycin A1 and danomycin are other sideromycins derived from ferrioxamine B that are taken up by the ferrioxamine B transport system. Ferrimycin A1 specifically inhibits protein synthesis, as has been shown by incorporation of radiolabeled phenylalanine into whole cells of S. aureus (Nüesch and Knüsel 1967; Urban et al. 2007).
Besides sideromycins of the Fe3+-hydroxamate type, natural sideromyins of the Fe3+-catecholate type occur. Microcins, for example, are antibacterial peptides, some of which are modified covalently by Fe3+-catecholates (Braun et al. 2002; Miethke and Marahiel 2007). In microcin MccE492m (m = modified), a linear trimer of N-(2,3-dihydroxybenzoyl)-l-serine (DHBS) is bound to the C-terminal serine 84 via a β-d-glucose linker (Thomas et al. 2004). DHBS is the precursor of the cyclic trimer enterobactin, which is a potent siderophore of E. coli and related bacteria. The DHBS monomer, dimer, and trimer also bind iron and serve as Fe3+-siderophores. They are taken up into E. coli via the FepA, Fiu, and Cir outer membrane transporters. These transporters are also required to confer high sensitivity to MccE492m. The minimal inhibitory concentration of MccE492m required to kill E. coli B is 0.04 μM, whereas 0.3 μM is required for the unmodified MccE492. The fepA fiu double mutant exhibits an eight-fold and the fepA fiu cir triple mutant a 200-fold reduction in sensitivity to MccE492m. tonB mutants show a strong reduction in sensitivity, whereas, unexpectedly, an exbBD deletion mutant is unaffected. This is a surprising result because TonB requires ExbBD to function. Since ExbBD can be partially replaced by TolQR (Braun and Herrmann 1993), it would be worthwile to study the TolQR dependence of MccE492m uptake.
A number of catechol-derived antibiotic conjugates that confer 100–10,000-fold higher antibiotic activity than the antibiotic without iron carrier have been chemically synthesized (compiled in Braun 1999). Cephalosporins linked to Fe3+-catecholates are taken up by the Fe3+-catechol transporters across the outer membranes of E. coli and Pseudomonas aeruginosa, and their activity exceeds that of the unsubstituted cephalosporins by several orders of magnitude. Since cephalosporins inhibit cross-linking of murein (peptidoglycan) in the periplasm, they must be transported only across the outer membrane. Susceptibility of E. coli to Fe3+-catechol cephalosporins is decreased more than 1,000-fold by a mutation in tonB, which is required for active transport, and 100–1,000-fold in mutants that lack two or three of the catecholate outer membrane transporters FepA, Fiu, and Cir (Watanabe et al. 1987; Curtis et al. 1988; Silley et al. 1990). For high efficacy of the conjugates, iron must be present to form the Fe3+-complexes, but the iron concentration must be low to de-repress transcription of the transport genes in the strains tested. Such conditions occur at sites of infections in mammals.
When the Fe3+-siderophore conjugates are transported by a single outer membrane protein, resistant bacteria frequently develop. Such mutants show a reduced growth rate if the particular Fe3+-siderophore transport system is needed at the site of infection or at the location in the environment. Slower growth of pathogenic bacteria would increase the chance of the native and the acquired immune systems to cope with the infection. This also holds true for tonB mutants because many bacteria encode only one tonB gene, and this gene is essential for the uptake of all Fe3+-siderophores and heme. When more than one tonB gene is encoded, the TonB proteins display specificity for certain iron transport systems (Cescau et al. 2007; Wyckoff et al. 2007).
To overcome the resistance problem, a cephalosporin was chemically conjugated to hydroxamates and catecholates that use different transporters (Ghosh et al. 1996). The mixed-ligand siderophore–β-lactam conjugates are less active than the cephalosporin, except for one compound, which inhibits Acinetobacter at a much lower concentration (0.03 μg/ml) than cephalosporin (64 μg/ml). Despite the mixed ligand and the lower frequency of double mutants than single mutants, resistant E. coli and P. aeruginosa cells developed on agar plates. Such multiple mutants may be subject to iron starvation, which limits proliferation and pathogenicity in the hosts.
Activity spectrum of albomycin and salmycin
Defined bacterial laboratory strains and clinical isolates have been used to determine growth inhibition by albomycin and salmycin. Albomycin is highly active against gram-negative Enterobacteriaceae except Proteus and related Morganella strains (Table 1). Proteus and Morganella are transport deficient (Pramanik et al. 2007) because they lack the fhu transport genes, as has been shown for several Proteus mirabilis strains (Pramanik et al. 2007; Pearson et al. 2008). This result is unexpected since the genomes of all other tested Enterobacteriaceae encode the fhu transport system. Albomycin kills only a few gram-positive bacteria, among them pathogens of great clinical importance, such as S. aureus and S. pneumoniae. Salmycin displays the same activity spectrum with gram-positive bacteria as albomycin, except that salmycin but not albomycin is active against Streptococcus agalactiae. In contrast to albomycin, salmycin is inactive against most gram-negative bacteria (Table 2). Exceptions are Hafnia alvei, Citrobacter freundii, and some strains of Yersinia enterocolitica, which are sensitive to 10 ng/ml salmycin, the concentration that inhibits gram-positive bacteria. Resistance of the gram-negative bacteria to salmycin is most likely caused by the specificity of the outer membrane transporters, which exclude salmycin. After transformation of an E. coli K-12 strain with the foxA gene of Y. enterocolitica, E. coli becomes sensitive to salmycin. The foxA gene encodes the outer membrane transport protein that takes up ferrioxamine B, the iron carrier of salmycin. This also shows that the E. coli FhuBCD proteins transport salmycin.
In general, outer membrane transporters are more specific than cytoplasmic membrane transporters. For example, E. coli transports ferrichrome, aerobactin, and ferrioxamine B by a common transport system across the cytoplasmic membrane but uses specific transporters across the outer membrane for each of the Fe3+-hydroxamates.
In vivo activities of albomycin and salmycin in mouse infection models
To test the protection by albomycin of mice against infection by pathogenic bacteria, the gram-positive S. pneumoniae strain D39 and the gram-negative Y. enterocolitica strain 8081 serotype O:8 were used; bacterial infection models have been established in these strains (Pramanik et al. 2007). A single dose of 1 mg/kg body weight of intraperitoneally injected albomycin given 12 h post-infection cleared S. pneumoniae administered into the peritoneum of BALBc mice with a lethal dose of 400 colony forming units (CFU). At 7 days post-infection, all albomycin-treated mice had survived and contained no detectable CFU in the blood, liver, and spleen. In an experiment with 10 mg/kg body weight of albomycin administered 8 and 14 h post-infection, 1,000 CFU/ml were found in the blood after 24 h; untreated mice contained 3 × 1010 CFU/ml. The values obtained with albomycin were very similar to the values obtained with amoxicillin.
Under laboratory conditions, spontaneous albomycin-resistant bacteria occur at a frequency of 10−4 to 10−5. To examine whether the emergence of resistant bacteria precludes the use of albomycin, growth of an albomycin-resistant strain was compared with growth of a sensitive strain. Mice were infected with a mixture of wild-type (98.6%) and albomycin-resistant (1.4%) S. pneumoniae cells. After 12, 20, 28, and 36 h post-infection, blood was collected from the tail vein, and the number of resistant and sensitive bacteria were determined. At 20 h post-infection, the percentage of mutant cells in untreated mice decreased to 0–0.46% of the total bacterial cells, which indicates that the mutant has a reduced fitness in the mice. At 12 h post-infection, the mutant cells amounted to 0–2% of the total cells in the albomycin-treated mice (5 mg/kg body weight). At 36 h post-infection, the percentage of albomycin-resistant bacteria increased to 86–97%, which indicates eradication of most of the sensitive bacteria and selection for the resistant bacteria.
Mice are also cleared from Y. enterocolitica by albomycin. An inoculum of 5 × 104 CFU per C57BL/6 mouse yields a strong increase in the number of bacteria in the spleen (Table 3). Administration of albomycin 24 h post-infection strongly reduces the CFU in the spleen. The number of albomycin-resistant bacteria increases in mice treated with albomycin. However, the CFU is nearly 3–4 orders of magnitude lower than in untreated mice, which gives the immune system a chance to cope with the infection. To examine proliferation of resistant bacteria, mice were inoculated with a surplus of sensitive over resistant bacteria. Results obtained with S. pneumoniae are shown in Fig. 4. In untreated mice, more wild-type than mutant bacteria were found in the spleen. In the albomycin-treated mice, the mutant bacteria overrode the wild-type bacteria, which are killed by albomycin. In a similar experiment done with Y. enterocolitica, wild-type bacteria proliferate faster than albomycin-resistant bacteria (Pramanik et al. 2007).

Competitiveness of the albomycin-resistant S. pneumoniae mutant D39T1 with the wild-type parent strain D39 in mice. Mice (n = 12) were infected with a mixture of 1,000 CFU of strain D39 and 14 CFU of strain D39T1. After 12 h post-infection, six mice received placebo (PBS) and six mice received albomycin (5 mg/kg body weight, thrice a day). In 8 h intervals, blood samples were collected from the tail vein of each mouse and the bacterial cells were counted. All PBS-treated mice died within 26 h post-infection. Open squares, CImutant from PBS-treated mice; filled triangles, CImutant of albomycin-treated mice. CImutant = percentage of mutant CFU in the total CFU/percentage of mutant CFU in the inoculum CFU. A CImutant of 1 indicates that the mutant proliferates as well as the wild-type, and a CFUmutant <1 indicates that the mutant multiplies less than the wild-type
To examine the in vivo efficacy of salmycin, protection of mice by treatment with salmycin against S. aureus infections (infection dosis: 108 bacterial cells) was tested. Salmycin A (10 mg/kg body weight) administered 6 and 24 h post-infection reduced the CFU of the S. aureus MRSA strain COL in kidneys of CD1 mice 30-fold 48 h post-infection and 80-fold 72 h post-infection (Fig. 5). For comparison, in a control experiment with a mixture of vancomycin and rifamycin (vancomycin alone was ineffective), each at 10 μg/kg body weight, the CFU was reduced 5 × 103- and 2 × 104-fold, respectively. Salmycin A is effective, but should be administered at shorter intervals since it rapidly loses activity. The optimal treatment was not established.

Salmycin treatment of infected mice. Mice were infected with S. aureus on day 0, and antibiotics were administered 6 and 24 h post-infection. The bacterial numbers (CFU) were determined in the kidney 72 h post-infection. For comparison, infected mice were treated with a mixture of rifamycin and vancomycin. n, number of mice. Dotted line indicates detection limit
References
Braun V (1999) Active transport of siderophore-mimicking antibacterials across the outer membrane. Drug Resist Updates 2:363–369. doi:10.1054/drup.1999.0107
Braun V, Herrmann C (1993) Evolutionary relationship of uptake systems for biopolymers in Escherichia coli: cross-complementation between the TonB–ExbB–ExbD and the TolA–TolQ–TolR proteins. Mol Microbiol 8:261–268. doi:10.1111/j.1365-2958.1993.tb01570.x
Braun V, Günthner K, Hantke K, Zimmermann L (1983) Intracellular activation of albomycin in Escherichia coli and Salmonella typhimurium. J Bacteriol 156:308–315
Braun V, Patzer SI, Hantke K (2002) Ton-dependent colicins and microcins: modular design and evolution. Biochimie 84:365–380. doi:10.1016/S0300-9084(02)01427-X
Brown JS, Gilliland SM, Ruiz-Albert J, Holden DW (2002) Characterization of Pit, a Streptococcus pneumoniae iron uptake ABC transporter. Infect Immun 70:4389–4398. doi:10.1128/IAI.70.8.4389-4398.2002
Cescau S, Cwerman H, Letoffe S, Delepelaire P, Wandersman C, Biville F (2007) Heme acquisition by hemophores. Biometals 20:603–613. doi:10.1007/s10534-006-9050-y
Curtis NA, Eisenstadt RL, East SJ, Cornford RJ, Walker LA, White AJ (1988) Iron-regulated outer membrane proteins of Escherichia coli K-12 and mechanism of action of catechol-substituted cephalosporins. Antimicrob Agents Chemother 32:1879–1886
Davidson AL, Dassa E, Orelle C, Chen J (2008) Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev 72:317–364. doi:10.1128/MMBR.00031-07
Ferguson AD, Braun V, Fiedler HP, Coulton JW, Diederichs K, Welte W (2000) Crystal structure of the antibiotic albomycin in complex with the outer membrane transporter FhuA. Protein Sci 9:956–963
Ferguson AD, Ködding J, Walker G, Bös C, Coulton JW, Diederichs K, Braun V, Welte W (2001) Active transport of an antibiotic rifamycin derivative by the outer-membrane protein FhuA. Structure 9:707–716. doi:10.1016/S0969-2126(01)00631-1
Ghosh A, Ghosh M, Niu C, Malouin F, Moellmann U, Miller MJ (1996) Iron transport-mediated drug delivery using mixed-ligand siderophore-beta-lactam conjugates. Chem Biol 3:1011–1019. doi:10.1016/S1074-5521(96)90167-2
Groeger W, Köster W (1998) Transmembrane topology of the two FhuB domains representing the hydrophobic components of bacterial ABC transporters involved in the uptake of siderophores, haem and vitamin B12. Microbiology 144:2759–2769
Hartmann A, Fiedler HP, Braun V (1979) Uptake and conversion of the antibiotic albomycin by Escherichia coli K-12. Eur J Biochem 99:517–524. doi:10.1111/j.1432-1033.1979.tb13283.x
Hollenstein K, Dawson RJP, Locher KP (2007) Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol 17:412–418. doi:10.1016/j.sbi.2007.07.003
Köster W, Böhm B (1992) Point mutations in two conserved glycine residues within the integral membrane protein FhuB affect iron(III) hydroxamate transport. Mol Gen Genet 232:399–407. doi:10.1007/BF00266243
Krewulak KD, Vogel HJ (2008) Structural biology of bacterial iron uptake. Biochim Biophys Acta 1778:1781–1804. doi:10.1016/j.bbamem.2007.07.026
Krewulak KD, Shepherd CM, Vogel HJ (2005) Molecular dynamics simulations of the periplasmic ferric-hydroxamate binding protein FhuD. Biometals 18:375–386. doi:10.1007/s10534-005-3712-z
Larsen RA, Thomas MG, Postle K (1999) Protonmotive force, ExbB and ligand-bound FepA drive conformational changes in TonB. Mol Microbiol 31:1809–1824. doi:10.1046/j.1365-2958.1999.01317.x
Miethke M, Marahiel MA (2007) Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev 71:413–451. doi:10.1128/MMBR.00012-07
Nüesch J, Knüsel F (1967) Sideromycins. In: Gottlieb D, Shaw PD (eds) Antibiotics, vol 1. Springer, Heidelberg, pp 499–541
Pearson MM et al (2008) Complete genome sequence of uropathogenic Proteus mirabilis, a master of both adherence and motility. J Bacteriol 190:4027–4037. doi:10.1128/JB.01981-07
Pramanik A, Braun V (2006) Albomycin uptake via a ferric hydroxamate transport system of Streptococcus pneumoniae R6. J Bacteriol 188:3878–3886. doi:10.1128/JB.00205-06
Pramanik A et al (2007) Albomycin is an effective antibiotic, as exemplified with Yersinia enterocolitica and Streptococcus pneumoniae. Int J Med Microbiol 297:459–469. doi:10.1016/j.ijmm.2007.03.002
Pugsley AP, Zimmerman W, Wehrli W (1987) Highly efficient uptake of a rifamycin derivative via the FhuA-TonB-dependent uptake route in Escherichia coli. J Gen Microbiol 133:3505–3511
Rohrbach MR, Braun V, Köster W (1995) Ferrichrome transport in Escherichia coli K-12: altered substrate specificity of mutated periplasmic FhuD and interaction of FhuD with the integral membrane protein FhuB. J Bacteriol 177:7186–7193
Schalk IJ (2008) Metal trafficking via siderophores in Gram-negative bacteria: specificities and characteristics of the pyoverdine pathway. J Inorg Biochem 102:1159–1169. doi:10.1016/j.jinorgbio.2007.11.017
Schramm E, Mende J, Braun V, Kamp RM (1987) Nucleotide sequence of the colicin B activity gene cba: consensus pentapeptide among TonB-dependent colicins and receptors. J Bacteriol 169:3350–3357
Schultz-Hauser G, Köster W, Schwarz H, Braun V (1992) Iron(III) hydroxamate transport in Escherichia coli K-12: FhuB-mediated membrane association of the FhuC protein and negative complementation of fhuC mutants. J Bacteriol 174:2305–2311
Sebulsky MT, Heinrichs DE (2001) Identification and characterization of fhuD1 and fhuD2, two genes involved in iron-hydroxamate uptake in Staphylococcus aureus. J Bacteriol 183:4994–5000. doi:10.1128/JB.183.17.4994-5000.2001
Sebulsky MT, Shilton BH, Speziali CD, Heinrichs DE (2003) The role of FhuD2 in iron(III)-hydroxamate transport in Staphylococcus aureus: demonstration that FhuD2 binds iron(III)-hydroxamates but with minimal conformational change and implication of mutations on transport. J Biol Chem 278:49890–49900. doi:10.1074/jbc.M305073200
Silley P, Griffiths JW, Monsey D, Harris AM (1990) Mode of action of GR69153, a novel catechol-substituted cephalosporin, and its interaction with the TonB-dependent iron transport system. Antimicrob Agents Chemother 34:1806–1808
Speziali CD, Dale SE, Henderson JA, Vines ED, Heinrichs DE (2006) Requirement of Staphylococcus aureus ATP-binding cassette-ATPase FhuC for iron-restricted growth and evidence that it functions with more than one iron transporter. J Bacteriol 188:2048–2055. doi:10.1128/JB.188.6.2048-2055.2006
Stefanska AL, Fulston M, Houge-Frydrych CS, Jones JJ, Warr SR (2000) A potent seryl tRNA synthetase inhibitor SB-217452 isolated from a Streptomyces species. J Antibiot (Tokyo) 53:1346–1353
Thomas X, Destoumieux-Garzón D, Peduzzi J, Afonso C, Blond A, Birlirakis N, Goulard C, Dubost L, Thai R, Tabet JC, Rebuffat S (2004) Siderophore peptide, a new type of post-translationally modified antibacterial peptide with potent activity. J Biol Chem 279:28233–28242. doi:10.1074/jbc.M400228200
Urban A, Eckermann S, Fast B, Metzger S, Gehling M, Ziegelbauer K, Rübsamen-Waigmann H, Freiberg C (2007) Novel whole-cell antibiotic biosensors for compound discovery. Appl Environ Microbiol 73:6436–6443. doi:10.1128/AEM.00586-07
Vértesy L, Aretz W, Fehlhaber H-W, Kogler H (1995) Salmycin A–D, Antibiotika aus Streptomyces violaceus, DSM 8286, mit Siderophor-Aminoglycosid-Struktur. Helv Chim Acta 78:46–60. doi:10.1002/hlca.19950780105
Watanabe NA, Nagasu T, Katsu K, Kitoh K (1987) E-0702, a new cephalosporin, is incorporated into Escherichia coli cells via the TonB-dependent iron transport system. Antimicrob Agents Chemother 31:497–504
Wiener MC (2005) TonB-dependent outer membrane transport: going for Baroque? Curr Opin Struct Biol 15:394–400. doi:10.1016/j.sbi.2005.07.001
Wyckoff EE, Mey AR, Payne SM (2007) Iron acquisition in Vibrio cholerae. Biometals 20:405–416. doi:10.1007/s10534-006-9073-4
Acknowledgments
We thank D. E. Heinrichs for providing the S. aureus fhuD mutants; K. A. Brune for critical reading of the manuscript; and the Landesstiftung Baden-Würtemberg, the Deutsche Forschungsgemeinschaft, and the Fonds der Chemischen Industrie for financial support. Experiments with mice followed animal ethics guidelines under biosafety level containment two in specific pathogen-free facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Braun, V., Pramanik, A., Gwinner, T. et al. Sideromycins: tools and antibiotics. Biometals 22, 3–13 (2009). https://doi.org/10.1007/s10534-008-9199-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-008-9199-7