
Abstract
Trachycarpus fortunei (Arecaceae: Coryphoideae) is an Asian palm that was introduced during the nineteenth century in southern Switzerland and northern Italy as an ornamental plant. In the recent decades, the palm has become an aggressive invasive species in the region. Before this study, the genetic structure and diversity of the naturalised populations were unknown. We aimed at understanding the dynamics of invasion and at comparing the results obtained with two types of markers. This genetic approach aimed at tracing back as far as possible the source of invasive populations comparing historical information found in literature and invasive genetic patterns. The genetic diversity was analysed using eight microsatellites (five were developed for that purpose) and 31′000 SNPs identified through GBS analyses. Genetic analyses were carried out for 200 naturalised individuals sampled from 21 populations in the Canton Ticino (Switzerland) and the provinces of Lombardy and Piedmont (Italy). The observed general panmixia indicates that the expansion of T. fortunei is active in its naturalised areas. The genetic pattern found for both SNPs and microsatellites appears to be related to the colonisation process, with a lack of geographic structure and bottleneck signatures occurring at the colonisation front, distantly from historical sites. This study gives a better understanding of the expansion of T. fortunei and adds new insights to its ecology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Current climate changes favour invasion processes and modify ecosystems’ dynamics, including changes in plant communities, as demonstrated in the mountain range of the Alps (Carraro et al. 2001; Walther 2003; Pauli et al. 2007; Walther et al. 2007). Such modifications are also found in the lowlands of Southern Alps and more particularly in the Insubric region which extends from Lake Maggiore to Lake Garda. This region is considered as transitional between the warm temperate climate with evergreen broad-leaved forests and the temperate climate with deciduous and frost-resistant forests (Klötzli 1988; Walther 2001). Ongoing changes in species distributions have been partially explained by the rising winter temperatures measured in the last few decades (Klötzli et al. 1996; Carraro et al. 2001; Walther et al. 2007). For instance, the number of evergreen broad-leaved alien (i.e. exotic) tree species is increasing in Switzerland (Gianoni et al. 1988; Klötzli et al. 1996; Walther 2000, 2001; Berger and Walther 2006; Walther et al. 2007). They can negatively impact ecosystems, because they are likely to become the dominant element in an otherwise deciduous vegetation and, therefore, outcompete or suppress other plants due to their tallness and shade tolerance (Koike 2001; Richardson and Rejmánek 2011). Trachycarpus fortunei (Hook.) H. Wendl. (Arecaceae), commonly called the hemp palm, is one of the most invasive palms worldwide (Fehr et al. 2020). The species represents a spectacular example of evergreen broad-leaved forests in the Insubric region (Walther et al. 2007; Fehr and Burga 2016; Fehr et al. 2020) and the study system of the present paper.
Originally native from South-East China (Dransfield et al. 2008), T. fortunei was introduced in Europe for the first time at the beginning of the nineteenth century (Siebold 1856; Gay 1861; Jacobi 1998; see Appendix S2 in Walther et al. 2007). In the middle of the nineteenth century, Robert Fortune explored the Chusan province (China) and brought seeds of T. fortunei to western botanical gardens (Gay 1861; Gardener 1971). An increasing number of introductions (Dutch, English and French) of T. fortunei took place during the nineteenth century and the large distribution to horticulturists began in the second half of the this century (Gay 1861). In the Insubric region, currently known for its rich exotic flora (Banfi and Galasso 2010; Schoenenberger et al. 2014), T. fortunei was introduced as an ornamental plant, especially on the renowned Isola Madre, i.e. the first botanical garden with exotic plants in the Lake Maggiore region (Schröter 1936; Pfister 1977; Walther 2000). Trachycarpus fortunei was recorded for the first time in southern Switzerland in 1882 (Kny 1882; see Appendix S2 in Walther et al. 2007). Exotic plants have always been an important touristic attraction around the different lakes (Lodari 2002; Muzzarini 2007), and T. fortunei is now deeply rooted in the gardening and landscape practices of the Insubric region.
By the 1950s, T. fortunei had already escaped from private and public gardens into natural environments, as it succeeded in colonising some shady and humid sites in Ticino (Schmid 1956; see Appendix S2 in Walther et al. 2007). In the 1970s, small sterile palms were found in forest herb layers and in the 1980s, they were seen in the shrub layer (Zuber 1979; Gianoni et al. 1988; see Appendix S2 in Walther et al. 2007). Seedlings from naturalised palms persisted and started to compete with the local native flora (Walther 2000; Walther et al. 2007). Since several years, naturalised adult palms occur at reproductive stage in the tree layer (Fehr and Burga 2016). The species has become strongly invasive (Fehr et al. 2020) in forests of southern Switzerland (Info Flora 2014) and in northern Italy (Banfi and Galasso 2010; Galasso et al. 2020) and was accordingly listed on the Swiss Blacklist of invasive neophytes (Info Flora 2014). The rapid expansion of this species has prompted recent ecological studies on T. fortunei (e.g. Walther et al. 2007; Fehr and Burga 2016; Fehr et al. 2020). Walther et al. (2007) have determined that the mean temperature of + 2.2 °C during the coldest month is the lowest temperature that the palm tolerates to successfully spread. In addition, Fehr and Burga (2016) assessed some ecological factors that also explain the palm’s spread in the region and discussed the roles played by the environmental dynamics. In particular, the high propagule pressure due to cultivated palms and the increasing portion of forests in which management was abandoned fuelled the colonisation dynamic of Trachycarpus fortunei.
Both ecology and genetics are important aspects to consider in plant invasions. Therefore, genetics of invasive plants has become an increasingly explored field in the last decades (Excoffier and Ray 2008; Currat et al. 2008; Olivieri 2008; Smith et al. 2020). Several studies have demonstrated that genetics could play a more important role than environmental conditions to explain species invasiveness: high genetic variations allow responding to natural selection and adapting rapidly to new areas (Lavergne and Molofsky 2007; Keller et al. 2014), hybridisation processes and gene rearrangements allow increasing the invasive potential through new gene combinations (Hovick and Withney 2014; Keller et al. 2014), epistasis can modify genetics trade-offs and finally some genes increase invasive characters (Wolf et al. 2000). Through the increase of gene rearrangements, invasions have also been suggested to favour genetic revolutions (Hallatschek et al. 2007; Excoffier and Ray 2008; Olivieri 2008; Smith et al. 2020). For instance, Klopfstein et al. (2006) have demonstrated that low-frequency alleles can reach higher allelic frequencies during species spatial expansions, a phenomenon called allele surfing. Using microbial material, Hallatschek et al. (2007) have demonstrated that complex patterns could emerge with species expansions, such as patches with low genetic diversity and allele frequency gradients among the latter. Regarding the native range of T. fortunei, a recent study (Mao et al. 2020) has analysed its population genetics in one of the Chinese provinces, Guizhou, where the species is native. Using Sequence-related amplified polymorphism (SRAP markers), Mao et al. (2020) found a high genetic diversity and a large genetic differentiation among populations. Their results suggest that Trachycarpus fortunei has a high potential to respond to selection and breeding in its native range.
Here, we focused on the genetic variability of T. fortunei populations in the Insubric region. The present study aimed at (1) comparing the results obtained with eight microsatellites on the one hand and single-nucleotide polymorphism (SNPs) on the other; (2) understanding the dynamics of the invasion process through the analysis of the genetic variation and its distribution within and among populations, while also discussing the possible source of naturalised populations of T. fortunei in the Insubric region; and (3) comparing the information found in the literature on the first colonised sites in the region and the genetic patterns of the invasion. The colonisation direction is expected to stand from the lake shore vertically up to the mountain slope, following the feeding behaviour of birds to move from gardens with cultivated palms (historical places) to their perching and nesting sites in the surrounding, peri-urban forests and habitats.
Materials and methods
Sampling
The study area is located in the western side of the Insubric region and is characterised by mild, sunny, and dry winters (absolute temperatures are moderate and regulated by the Alps as a barrier) and by warm and sunny summers (Klötzli 1988; Walther 2001; Fehr and Burga 2016). Rainfall and thunderstorms are severe during summer and the annual precipitation mean ranges between 1300 to 2300 mm (climatic normals 1981–2010; MeteoSwiss 2021). The comparison of the climatic diagrams of Locarno in Switzerland (deciduous broad-leaved vegetation) with different locations in Asia (evergreen broad-leaved vegetation) by Walther (2001) reveals many similarities.
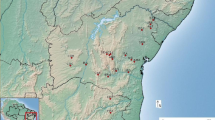
Within this region, we aimed at sampling the most important populations of T. fortunei, around the two main lakes of Ticino (Lake Maggiore and Lake Lugano), taking also into account the Italian border area. All samples (assigned to locations) consisted of leaf material instantly dried in small plastic bags filled with silica-gel and afterwards stored at room temperature at the Conservatoire et Jardin botaniques de Genève (CJBG). A total of 200 individuals were sampled in April and May 2019 from 21 populations across Ticino (Switzerland), Lombardy and Piedmont (Italy), with 10 individuals/population in most cases, but seven for Isole di Brissago and three for Isola Madre (Fig. 1 and Table 1). Populations 1 to 14 belong to the Lake Maggiore region whereas populations 15 to 21 were sampled in the Lake Lugano region. All the altitudes have been recorded for each population. Historical populations were defined based on literature and all date back from the nineteenth century. They are indicated with an asterisk in Fig. 1 and Table 1. Conversely, more recently naturalised populations were defined by their greater distance from historical localities and/or by a higher altitude (distance from lake level) and therefore named “naturalised populations”. Based on literature (Kny 1882; Schmid 1956; Zuber 1979; Agliati and Mondada 1987; Walther et al. 2000; see Appendix S2 in Walther et al. 2007), the populations of Isole di Brissago and Solduno (next to Locarno) were considered as the historical ones for the upper part of Lake Maggiore. On the middle part of the Lake Maggiore, the population of Isola Madre was considered as the first historical introduction (Schröter 1936; Pfister 1977; Walther et al. 2000). For Lake Lugano, the historical locations were most probably concentrated at the lake’s shore (such as Caslano, Melide and Morcote populations), also traditionally known as touristic sites. Conversely, Biasca, Castelrotto, Gandria, Intragna, Manno, Melano, Pianezzo, Quartino, Tegna and Verscio populations were considered as more isolated, because distantly located from the two lakes (and at higher altitudes) and/or from populations that are larger and older according to historical records.

Sampling map of Trachycarpus fortunei in Canton Ticino (southern Switzerland), Lombardy and Piedmont provinces (northern Italy) for both types of markers (microsatellites and SNPs). Historical naturalised populations were defined based on literature and are indicated with an asterisk
DNA extraction and quantification
DNA was extracted with the kit NucleoSpin Plant II of Macherey–Nagel following the manufacturer’s protocol. DNA quantification was performed using the Qbit dsDNA BR kit of LifeTechnologies and the Qbit machine of Invitrogen. DNA extracts were stored at −20 °C for further use.
Molecular analyses: microsatellite markers
Three different strategies were followed to find usable microsatellite markers for T. fortunei. First, seventeen microsatellite markers identified on the European palm Chamaerops humilis (Arecaceae, Coryphoideae; Molecular Ecology Resources Primer Development Consortium 2013) were tested (Suppl. Table 1). Second, three microsatellite markers identified on the date palm Phoenix dactylifera (Arecaceae, Coryphoideae): Pdac-011 (PdAG1-SSR1), Pdac-012 (PdCUC3-ssr1) and Pdac-015 (mPdIRD13) (Suppl. Table 2), selected from a batch of 18 microsatellites (Aberlenc-Bertossi et al. 2014; Zehdi-Azouzi et al. 2015), were also tested for amplification on T. fortunei. Finally, new microsatellite markers were directly characterized on T. fortunei using the methodology of Estoup and Martin (1996).
To identify new microsatellite markers, total DNA from a single individual was digested according to Estoup and Martin (1996) with three blunt end enzymes (HaeIII, AluI, RsaI from Promega). Fragments between 250 and 600 bp were selected using polyethylene glycol (PEG) beads and different washes. The fragments were then ligated into a pUC19 vector blunt end digested with SmaI. Transformation was conducted with Escherichia coli DH5α competent bacteria (from Thermo Fisher) following manufacturer’s protocol and grown for 16 h on plates. A total of 1′302 recombinant colonies were first transferred on gridded plates then onto nylon membranes and used for hybridisation with digoxigenin-labelled microsatellite probes. Four digoxigenin-labelled probes (AC)12, (AG)12, (TCT)8, (AAT)4 were obtained with the DIG Oligonucleotide Tailing kit of Boehringer Mannheim. The immunological detection of microsatellites was realised with the DIG Nucleic Acid Detection kit of Roche. The positive recombinant colonies containing putative microsatellites were sequenced on an automated Beckman-Coulter Sequencer (GenomeLab GeXP) using M13 forward and reverse primers to access the inserted sequence. The sequence data analyses and alignments were performed with the Sequencher software version 5.4.6 (Gene Codes Corporation, Ann Arbor, Michigan, USA) and the BiodEdit software (Hall 1999). For the ten colonies that gave true microsatellite sequences, primers were designed with the Primer Designing Tool from NCBI and named according to their respective number on the gridded plates.
For all tested microsatellite markers, PCR amplifications were conducted on eight individuals from geographically remote populations using the FastStart Taq DNA Polymerase kit of Roche with the following amounts per individual: 10 × Buffer (with MgCl2): 2.0 μl; dNTPs 10 mM: 0.4 μl; BSA 5%: 0.2 μl; Primer F (Forward) and R (reverse) 10 μM: 1.0 μl each; Taq Polymerase 5U/μl: 0.15 μl; Nuclease-Free water: 14.25 μl and extracted DNA: 1.0 μl (see Suppl. Tables 1 and 2 for PCR conditions). The resulting PCR products were checked visually on 2% agarose gels.
Out of the 30 tested microsatellites, eight microsatellites were finally selected based on their amplification success and polymorphism and were subsequently used for the study (Suppl. Table 3). Our final batch of microsatellites was composed of one of the 17 microsatellites identified on C. humilis, two of the three tested microsatellites identified on P. dactylifera and five microsatellites out of the ten that were developed for the present study. The forward primers for each were labelled with 5’ fluorescent base (Biomers). The multiplex PCR mix was prepared for one individual as: 2 × Master Mix: 10.0 μl; Q-Solution: 4.0 μl; 6 Primers (4 μM, 3 fluorescent Forward and 3 Reverse): 1.0 μl each and extracted DNA: 1.0 μl, using the MultiPlex PCR kit of Qiagen. The annealing temperatures for the three mixes as well as PCR conditions are indicated in Suppl. Table 3. Genotyping was achieved on an automatic Beckman-Coulter Sequencer (GenomeLab GeXP) and alleles, named according to their length, were recorded on an Excel spread sheet for further analyses.
Molecular analyses: genotyping by sequencing (GBS)
GBS was used to identify thousands of SNPs on the sampled individuals, following the protocol developed by Elshire et al. (2011). GBS libraries construction, sequence data generation and SNP identification were performed at the University of Minnesota (USA), which transmitted a VCF file. Briefly, the genomic DNA of T. fortunei samples was digested using restriction enzyme BamHI. Single-end 100 bp reads were produced, then demultiplexed fastq files were generated using Illumina bcl2fastq software, adapter sequences were trimmed from the 5` and 3` ends of reads using Cutadapt. The Stacks version 2.0 (Catchen et al. 2013) denovo_map.pl workflow was used to generate a raw VCF file, first to generate the loci catalogue from a random selection of 20 samples, then default parameters were used to produce the raw VCF file for all samples. This VCF file was filtered using VCFtools to remove variants with minor allele frequency < 1%, variants with genotype rate success < 80%, and samples with genotype rate success < 50%.
Bioinformatic analyses
The bioinformatics analyses aimed at analysing microsatellite markers and SNPs on the 21 sampled populations. Using indices of population genetics, the dynamic of the invasion was analysed using genetic variances and their distribution within and among populations. Bioinformatics analyses were carried out using both types of markers, when possible, for comparison.
Genetic diversity
The genetic diversity (He) across all populations and for each population (for both microsatellites and SNP markers), and for each microsatellite locus (locus-by-locus analyses) was analysed with the Hierfstat package (Goudet 2005) in R (R Core Team 2021). He was calculated for both type of markers, together with their standard deviations. For microsatellites, 95% and 99% confidence intervals were calculated when normal distribution and homoscedasticity of He values were confirmed at the 5% level using the Shapiro–Wilk and Bartlett tests, respectively. Differences in genetic diversity among loci and among populations were additionally tested using the Wilcoxon–Mann–Whitney signed rank test and the Kruskal–Wallis rank test, respectively, using a false discovery rate correction (Benjamini and Yekutieli 2001).
Inbreeding and structure indices
The Arlequin software (Excoffier and Lisher 2010) was used to compute the inbreeding coefficients (Fis) over all populations and per population (microsatellites and SNP markers) as well as per locus for microsatellite data. Analyses of molecular variances (AMOVA) were used to test for any significant deficit or excess of heterozygotes. The Arlequin software was also used to compute the Fst indices overall populations and per populations pair (Suppl. Tables 4, 5, 6, for microsatellites and SNP markers). The Fis and Fst values were considered as significantly different from 0 using a resampling procedure with 10′000 permutations. In addition, the mean Fst per population with their standard deviation and the 95% confidence intervals were calculated (when normal distribution and homoscedasticity of Fst values were confirmed among populations using Shapiro–Wilk and Bartlett tests, respectively, at the 5% level). For microsatellite markers, the genetic structure was also estimated using the Rst index (Slatkin 1995) that considers the differences in repetition numbers (length of PCR) in addition to allele frequencies. The differences between Fst and Rst (for which the 95% confidence intervals were also calculated), overall populations and per each population were tested using the Wilcoxon-Mann–Whitney signed rank test. For all these tests, a nominal level of 5% was used.
Dispersion dynamics (microsatellites)
Regarding microsatellite data, the effective number of alleles (Nae) was calculated according to Maruyama (1970). The allelic richness per population and per locus was computed with the Fstat software version 2.9.4 (Goudet 1995) with a minimum number of three individuals corresponding to the size of the Isola Madre population. Differences in allelic richness among populations were tested using a Kruskal–Wallis rank test. The number of rare alleles was calculated as the number of alleles with frequencies lower than 5% within populations. Linkage disequilibrium among loci was tested with the Arlequin software (Excoffier and Lisher 2010) and the significance level assessed using a resampling procedure of 10′000 permutations. Corrections for multiple tests were applied, using a false discovery rate correction (Benjamini and Yekutieli 2001). Bottleneck footprints were tested within populations with the Bottleneck software (Piry et al. 1999) version 1.2.02 using 1000 iterations and a significance level of 5% to detect recent effective population size reductions. Heterozygote excesses were tested using the Wilcoxon test under the SMM (stepwise mutation model) as it is the most precise model for microsatellites according to Di Rienzo et al. (1994) and Zhu et al. (1999). Although a high number of samples per population (30 individuals) is preconized, eight loci are sufficient to detect bottleneck signs according to Luikart et al. (1998). Except for populations of Isola Madre and Isole di Brissago, the number of analysed individuals (10 meaning 20 gene copies) was therefore sufficient according to Piry et al. (1999). The different indices (population with significant deficits or excess of heterozygotes, populations with higher Fst indices, populations with significant pairs of loci in linkage disequilibrium, populations that displays significant bottlenecks) were mapped (Fig. 2, displayed in the ArcGis and Photoshop software).

Synthesis and comparison of the two types of markers (microsatellites and SNPs) in Trachycarpus fortunei. White circles: Hardy–Weinberg equilibrium (both markers); Blue circles: Significant excess of heterozygotes: Manno (both markers), Intragna and Pianezzo (SNPs); Red circles: Significant excess of homozygotes: Isole di Brissago and Verscio (microsatellites); Red squares: Highest mean pairwise Fst indices: Cannobio, Intragna, Isola Madre and Manno (both markers), Tegna (microsatellites) and Gandria (SNPs); LD: More than one pair of loci in significant in linkage disequilibrium (microsatellites): Cannobio, Caslano, Gandria, Ghiffa and Manno; *: Significant signs of bottlenecks (microsatellites): Gandria, Intragna, Maccagno, Melano and Ronco s. Ascona. Historical naturalised populations were defined based on literature and are indicated with an asterisk
Diversity and structure plots
Principal components analyses (PCA, Suppl. Fig. 1) were performed with the Hierfstat package (Goudet 2005) in R (R Core Team 2021) for microsatellites and the SmartPCA software (Patterson et al. 2006) for SNPs. To represent Reynold’s distances (Reynolds et al. 1983) on graphs, non-metric multidimensional scaling analyses with a minimal stress value (MDS, Suppl. Fig. 2) were performed in R (R Core Team 2021) with the Hierfstat, Mass, Vegan and Reshape2 packages and using the Reynold’s matrices computed in Arlequin (Excoffier and Lisher 2010). The distinction between the two lakes was analysed to search for a possible genetic structure on a larger scale. Graphic representations and ellipsoids of 95% confidence intervals of the centroid for each lake region (PCA plots, Fig. 3) were performed in R using the Ggplot2 package (Wickham 2016). In addition to ellipsoids of 95% confidence intervals, analyses of variance (AMOVA) considering the two lake regions (Lake Maggiore and Lake Lugano) were performed. Two indices (Fsc, representing the proportion of variation due to differences among populations within a group, and Fct, representing the proportion of variation due to differences between groups of populations) were used to test whether the grouping criterion is significant or not at the 5% level (resampling with 10′000 permutations). A Bayesian approach was used to infer K, the optimal number of groups in the datasets (Pritchard et al. 2000). This method uses multi-locus genotypes and allows finding the best number of populations in a dataset, assuming that each inferred population follows panmixia. For microsatellite data, the Structure software version 2.3.4 (Pritchard et al. 2000) was used to compute the admixture rates per individual according to the K clusters, and the Structure Harvester software (Earl and Vonholdt 2012) was used to determine the more likely K using the Evanno’s DeltaK method (Evanno et al. 2005). Each run, from K = 2 to K = 10, was repeated 20 times and performed with 100′000 burn-in iterations and 100′000 subsequent Monte Carlo Markov chains. For SNP data, the FastStructure and ChooseK softwares (Raj et al. 2014) were used. For both markers, the results for the 21 analysed populations (Suppl. Fig. 3) were then mapped with individuals coloured according to their assignment to the K panmictic populations (Fig. 4, displayed in the ArcGis and Photoshop software). The mean number of populations assigned per individual (proportions higher than 5%) was calculated to quantify the general admixture per individual.

Genetic analyses in Trachycarpus fortunei: PCA Plots on microsatellites (A) and SNP markers (B) on 200 and 199 individuals, respectively. The individuals are named according to their population (abbreviations) and coloured according to the two regions (Lake Maggiore in blue and Lake Lugano in purple). The ellipsoids represent the 95% confidence intervals of each region centroid

Genetic analyses in Trachycarpus fortunei: structure Plots mapped on geography with microsatellites (A) and SNP markers (B) on the 21 analysed populations with individuals coloured according to Structure and FastStructure results into two or seven panmictic populations, respectively. Historical naturalised populations were defined based on literature and are indicated with an asterisk
Results
Data generation
Over the 200 sampled individuals, one individual was excluded due to a low number of reads and was not considered in the following analyses. Over the 199 remaining samples, a total of 73′878 markers were identified and 30′862 markers (that represent 19′095 loci) were selected after filtering. For the analyses with Arlequin software version 3.5.2.2 (Excoffier and Lisher 2010), the allowed level of missing data was reduced to 5%, which led to a subsample of 13′931 markers out of 30′862.
Genetic diversity, inbreeding and structure indices
Across the study range, the mean genetic diversity is 0.522 (with a standard deviation of 0.048) for microsatellite markers and 0.263 (with a standard deviation of 0.013) for SNP markers. The Fis inbreeding coefficient is 0.028 with a non-significant p value (0.138) for microsatellite markers and -0.058 with a significant p value (0.001) for SNP markers. The Fst index is equal to 0.068 for microsatellite markers and equal to 0.079 for SNP markers, both with a significant p value (0.000).
For the microsatellite locus-by-locus analyses (Table 2), the comparison between the number of allele (Na) and the effective number of alleles (Nae) shows that four loci (named 11, 12, 24 and 63) have Nae close to their corresponding Na values. The total number of alleles is 27, the total effective number of alleles is 19.92 and the total allelic richness is 19.29. The loci 11, 12, 230 and 348 display significantly higher genetic diversity than the four others (p < 0.018). Heterozygosities (Ho) are lower than expected under Hardy–Weinberg for seven loci out of eight, and the Fis coefficients indicate that three of them (24, 230 and 312) exhibit significant heterozygote deficiencies at the 5% level.
The analyses on each population are reported in Table 3. Based on microsatellite markers, genetic diversity is not significantly different among populations (p > 0.05 and overlapping 95% and 99% confidence intervals). The population from Isole di Brissago, that also has the highest allelic richness, presents the highest genetic diversity (0.622). The Isola Madre (sampling reduced at three samples) and Intragna populations exhibit the lowest genetic diversity values (0.417 and 0.427, respectively). The Fis coefficients show that Manno population displays a significant heterozygote excess (p < 0.05) whereas Isole di Brissago and Verscio populations present a significant deficit of heterozygotes. The Fst indices are significant (p < 0.05) for most pairs of populations (Suppl. Table 4) and the mean pairwise Fst per population indicates that Cannobio, Intragna, Isola Madre, Manno and Tegna populations are overall more differentiated than the others. Fst values were not significantly different from Rst values (Suppl. Table 5) across populations (p > 0.05), except in three populations (Gandria, Intragna and Manno) for which Rst were significantly higher than Fst (p < 0.05).
Based on SNP markers (Table 3), the standard deviations of genetic diversity per population are largely overlapping. The Solduno population displays the highest genetic diversity (0.278). Cannobio, Intragna, Isola Madre, Manno and Melide populations present lower genetic diversity. The Fis coefficient per population indicates that three populations (Intragna, Manno and Pianezzo) show a significant excess of heterozygotes (p < 0.05). The Fst indices are significant (p < 0.05) for all pairs of populations (Suppl. Table 6). The mean pairwise Fst per population indicates that Cannobio, Gandria, Intragna, Isola Madre and Manno populations are overall more differentiated than the others.
Dispersion dynamic indices (microsatellites)
The differences in allelic richness, calculated for three individuals, are not significantly different among populations (p > 0.05). Populations with the highest number of rare alleles are Maccagno, Manno and Ronco s. Ascona (four rare alleles), followed by Calde, Melano and Morcote (three), while the remaining populations have two or less. The Biasca, Cannobio, Caslano, Gandria, Ghiffa, Isole di Brissago, Maccagno, Manno, Morcote, Pianezzo and Quartino populations display pairs of loci in significant linkage disequilibrium (p < 0.018). Five populations present bottleneck footprints with significant excess of heterozygotes under the SMM model (Gandria, Intragna, Maccagno, Melano and Ronco s. Ascona; p < 0.05).
General indices: comparisons of marker types
The synthesis map (Fig. 2) illustrates the different indices and compares the two types of markers. Isole di Brissago and Solduno populations show the highest genetic diversity values with microsatellite and SNP data, respectively. Conversely, Intragna and Isola Madre populations display low genetic diversity with both markers. Globally, Fis values reveal that populations are at Hardy–Weinberg equilibrium with microsatellite markers, while they present a slight excess of heterozygotes with SNP data. For Fis values per population, significant excess in heterozygotes were observed in the Manno population with both markers. Two additional populations (Isole di Brissago and Verscio) present a deficit of heterozygotes with microsatellite markers and two additional populations (Intragna and Pianezzo) display an excess of heterozygotes with SNP markers. The genetic structure is low with both markers. The mean pairwise Fst per population indicate that Cannobio, Intragna, Isola Madre and Manno populations are overall more differentiated than other populations with both markers. Within the latter, two population (Intragna and Manno) show Rst indices significantly higher than Fst indices with microsatellite data.
Diversity and structure plots
Individuals are graphically represented on the PCA plots (Fig. 3, Suppl. Fig. 1) and populations on the MDS plots (Suppl. Fig. 2). Axes 1 and 2 of the PCA plots represent 8.3% and 6.1% of the variability for microsatellites and 8.8% and 5.9% for SNPs, respectively. Individuals of a given population tend to be found close to each other but they are never grouped all together. For microsatellite markers, a total of 13 individuals are found outside the 95% confidence interval: nine of them belong to five populations from the Lake Maggiore region (Cannobio, Ghiffa, Isole di Brissago, Maccagno and Tegna) and the remaining to four populations of the Lake Lugano region (Castelrotto, Gandria, Manno and Melano). For SNP markers, only six individuals are found outside the 95% confidence interval: two belong to the Lake Maggiore region (Isola Madre) and the remaining to two populations of the Lake Lugano region (Caslano and Gandria). With both markers, the two lakes’ ellipses are largely overlapping. The low differentiation between the two lake regions is confirmed by AMOVA with significantly higher Fsc values (0.075 for SNPs and 0.069 for microsatellites, respectively; both p = 0.000) than Fct values (0.002 for SNPs and 0.000 for microsatellites, respectively; p equal to 0.110 and 0.470, respectively).
No obvious structural patterns are observed among populations (Suppl. Fig. 3). For microsatellite markers, the most likely number of clusters is K = 2. Each individual is, on average, assigned to 1.9 populations. According to Evanno’s method, the DeltaK is equal to 76.74 for K = 2 (with Mean LnP(K) = −2906.27), while it ranges between 0.07 and 2.09 for higher K values. For SNP markers, the most likely number of clusters is K = 7 and the mean number of assigned populations per individual is equal to 2.9. With SNP markers, two individuals of Isola Madre population are completely attributed to one cluster. Individuals of Caslano, Gandria and Manno populations also display more similar and exclusive patterns. The results of structure analyses are also illustrated on the sampling map (Fig. 4). With microsatellite markers, two populations are exclusively attributed to one panmictic population (Intragna and Ronco s. Ascona). The Biasca, Isola Madre, Quartino and Tegna populations of the Lake Maggiore region, and Castelrotto and Manno populations of the Lake Lugano region can be mostly attributed to the second panmictic population. With SNP markers, Quartino, Ronco s. Ascona, Solduno and Verscio are grouped together and are distributed on the upper part of the Lake Maggiore. Cannobio and Melide are grouped together but are located in the two different regions. Caslano population appears more differentiated, as well as Gandria population. Manno is the most differentiated population according to the SNP structure analysis.
Discussion
The genetic analyses on Trachycarpus fortunei populations in the Southern Alps allowed us to (1) compare the two types of markers and (2) evaluate the standing genetic variation and its distribution, as well as to understand gene flows implied in the expansion process. Finally, (3) by comparing our results with the introduction historical information found in the literature, the direction of the colonisation and the dynamic in isolated populations, especially at the colonisation front, could be discussed.
Comparison of microsatellites and SNP markers
Despite differences in marker numbers and genome coverage, microsatellites and SNPs gave very similar results, namely: highly significant Fst values equal to 0.068 and 0.079, respectively; no genetic structure between the two lakes (0.000 and 0.002, respectively); similar inbreeding coefficients Fis (0.028 and 0.058, respectively); overlapping genetic diversities among populations and similar levels of variance on PCA axes for both types of markers. The two types of markers also point at the same populations for the most and least diverse samples.
These similarities open the question of the type of marker that could be used in similar genetic diversity studies. The microsatellites used here were identified on Trachycarpus fortunei for the majority of them (5/8) but also retrieved from other palm species (3/8) from a batch of 20 markers identified on Chamaerops humilis (Molecular Ecology Resources Primer Development Consortium 2013) and a batch of 18 markers identified on Phoenix dactylifera (Aberlenc-Bertossi et al. 2014; Zehdi-Azouzi et al. 2015). Therefore only 6% (1/17) of C. humilis microsatellites and 11% (2/18) of P. dactylifera microsatellites could be successfully used on our species, being polymorphic and displaying easily scored patterns. Microsatelllites are known to be highly specific and their use more or less restricted to their focal species (Zhu et al. 1999). Microsatellites can indeed degenerate or accumulate imperfect repetition of motifs (Buschiazzo and Gemmell 2006), rendering their scoring difficult. Mutations occurring in the flanking region used for primer definition may moreover produce null alleles that bias the analyses with higher levels of inbreeding than expected. In our case, no evidence was found for more null alleles in the three non-focal microsatellites compared to the focal ones and markers Pdac-11, Pdac-12 and Chum-63 gave the smallest Fis values among all (Table 2). Microsatellites are also usually much more variable (two to seven alleles versus two alleles maximum in our study) and can be more powerful at small scales than SNPs (Payseur and Cutter 2006; DeFaveri et al. 2013). Our analyses therefore suggest that having more alleles can partly compensate for a much lower number of markers than for SNPs.
GBS analyses are more costly and much complex to obtain than microsatellites overall (Puckett 2017), and might be less reproducible from one laboratory to the other as they rely on a first step of DNA digestion highly dependent on DNA quality and quantity. The use of microsatellite markers could therefore be advised when financial resources are lacking or for comparisons with other studies. Conversely the GBS analyses, as they produce markers covering the whole genome, may allow for other types of analyses such as association studies. Because of the high number of scored markers, more powerful tests can be conducted with GBS settings (see Puckett 2017 for a review).
Invasion dynamics: a high diversity and an overall panmixia
Mao et al. (2020) showed, using bi-allelic markers similar to our SNP markers, that Trachycarpus fortunei displays a high genetic structure among populations coupled with a high genetic diversity in the native species range in China. Our values of genetic diversity from SNP markers are in the same range as those obtained by Mao et al. (2020), but the genetic structure is much smaller, pointing to a lack of geographic pattern in our region. The overall genetic diversity observed in T. fortunei is higher than expected for an introduced plant but the genetic diversity among populations in the introduced range is in line with that reported in other invasive plants, like Ambrosia artemisiifolia (Asteraceae), Clidemia hirta (Melastomataceae), Kochia scoparia (Amaranthaceae), Linaria vulgaris (Plantaginaceae), Poa annua (Poaceae), Pueraria lobata (Fabaceae) and Solidago canadensis (Asteraceae; see Ward et al. 2008 for a review). As highlighted in Kang et al. (2007) and Prentis et al. (2009), multiple introductions increase the genetic diversity of invasive species in their introduced range. Our results (Table 3) identify the highest genetic diversity in T. fortunei in the upper part of the Lake Maggiore region. Therefore, the high genetic diversity observed in the large and old populations near the Lake Maggiore could be due to the different historical introductions in Europe (Gay 1861; see Appendix S2 in Walther et al. 2007) precisely in these locations, and to the important diffusion of the species through gardens (Fehr and Burga 2016). For instance, the Solduno population, which shows the highest genetic diversity with SNP markers, is the closest population to the city of Locarno and possibly among the oldest naturalised populations in the whole region. Indeed, T. fortunei was already planted there at the end of the nineteenth century (Kny 1882; Agliati and Mondada 1987; see Appendix S2 in Walther et al. 2007). The high genetic diversity and the highest allelic richness (microsatellite markers) of the Isole di Brissago population could be due to both old and recent introductions in the botanical garden settled on these islands. On the contrary, two populations (Intragna and Isola Madre) show low genetic diversity with both markers. The Isola Madre population presently shows the lowest genetic diversity, which is probably due to the sampling of closely related individuals (see also source of the invasive alien populations). For Intragna population located at the colonisation front (second highest altitude in the sampling), the low genetic diversity could be explained by its small population size in the forest and its recent formation deriving from few cultivated palms in neighbouring gardens. Manno population displays the lowest genetic diversity with SNP markers, also probably due to its isolation.
In our study, the overall high diversity is associated with a general panmixia (Table 3 and Fig. 2). Indeed, only two populations display significant heterozygote deficits with microsatellite data (Isole di Brissago and Verscio). As described for native vs alien populations of Cytisus scoparius (Fabaceae) by Kang et al. (2007), our results support the hypothesis that alien populations of T. fortunei display the same cross-pollination system as the native populations. In the southern part of the Alps, cross-pollination and the important fruit propagation therefore prevent inbreeding in naturalised populations. Conversely heterozygotes excesses were found in Intragna, Manno and Pianezzo populations which can be attributed to their location at the colonisation front and further away from the first important cultivated areas of T. fortunei according to the historical records (Kny 1882; see Appendix S2 in Walther et al. 2007). Significant signs of bottleneck events were found with microsatellite markers in Gandria, Intragna, Melano, Maccagno, and Ronco s. Ascona populations among which the first three are considered as isolated. The altitude of these three populations (Table 1), especially for Gandria and Intragna populations, is higher than that of other populations. Cultivated individuals in gardens from different genetic pools were probably concentrated at these points of colonisation, increasing the observed heterozygosity in these small populations. For Maccagno and Ronco s. Ascona populations, the explanation of bottleneck signs could be that the forest is now covering what was, in the past, a cultivated or managed area. The two populations are therefore somehow artificial, although they appeared naturalised to us during fieldwork. This would be specific to cultivated sites that were abandoned after the Second World War and that are now covered by forest (Fehr and Burga 2016) or to gardens, not sampled in our study. Finally, more than one pair of loci were found in linkage disequilibrium in five populations (Manno, Caslano and Gandria for the Lake Lugano; Cannobio and Ghiffa for the Lake Maggiore). As the pairs of loci were not always the same ones, they most probably reflect a statistical linkage disequilibrium, rather than physical linkage among loci. Processes that may cause such statistical linkage disequilibrium include genetic drift and bottleneck events (Thornsberry and Buckler 2003) as it is the case for Gandria. Similarly, Manno population is most probably the result of a founding effect leading to a high overall differentiation and isolation.
Gene flow is generally regarded as being important during the colonisation process of new areas (Lee 2002; Puth and Post 2005; Ward et al. 2008). In our case, a high gene flow was deduced from the overall low, although significant, Fst values, especially with SNPs. Dispersal mediated by humans, as well as the recent colonisation dynamics since the 1970s, are suggested as the main explanations for the lack of genetic structure. The small number of rare microsatellite alleles (Table 3) could additionally reflect the active colonisation processes. During species expansions, rare alleles can indeed become more frequent when they are located at the migration front, something known as allele surfing (Klopstein et al. 2006; Excoffier and Ray 2008). Gene flow among populations and the expansion process towards forests could therefore explain the small number of rare alleles in some naturalised populations. Kang et al. (2007) however suggested that a high genetic diversity may be more important than admixture during invasions. When genetic diversity is higher than expected in an introduced alien species, this could correlate with the species adaptability in its new environment (Parker et al. 2003). Understanding the population genetics of an emerging problematic alien species can therefore be relevant to understand and prevent future plant invasions (Lee 2002; Puth and Post 2005; Ward et al. 2008; Olivieri 2008; Hovick and Withney 2014; Gladieux et al. 2015). It might also allow early detection of the species invasive potential and provide support in the decision-making process regarding their control through spatial and reproduction management strategies or through biocontrol (Nissen et al. 1995; Ward et al. 2008). However, biocontrol appears more difficult to succeed for alien species that have been introduced multiple times from their native range to the alien range and thus display higher genetic diversity. In the case of T. fortunei, multiple introductions have already been assessed and their effect confirmed here. Only a drastic reduction of individuals that come from different genetic pools and are planted in gardens may reduce gene flow to forests, leading to a possible depletion of genetic diversity in the future.
Source of the invasive alien populations
The dispersion of T. fortunei in the Insubric region and its dynamics are the result of an intensive cultivation of the species by humans since the end of the nineteenth century (Kny 1882; Agliati and Mondada 1987; Lodari 2002; Muzzarini 2007; see Appendix S2 in Walther et al. 2007). By comparing our results with literature, we could partly trace back the source of the alien invasive populations of T. fortunei in the western side of the Insubric region. As evidenced by PCAs (Fig. 3 and Supplementary Fig. 1), all naturalised populations of T. fortunei are made of several closely related individuals, most probably originating from neighbouring gardens, and of other ones more distantly related, that might result from crosses with other genetic sources. The latter might correspond to palms that were successively planted or that came from other gardens. This pattern supports the hypothesis of a relatively recent colonisation (since the 1970s) of T. fortunei from gardens (Walther et al. 2007). Accordingly, T. fortunei populations are denser when closer to gardens (Fehr and Burga 2016). The general genetic admixture is high (Fig. 4 and Supplementary Fig. 3), certainly also due to the intensive cultivation and exchanges of individuals through horticulture. The genetic structure, especially in SNP data, seems to reflect mainly patterns in population size and degree of isolation: the larger and continuous populations in the Lake Maggiore region display less genetic structure than the smaller and geographically more isolated populations of the Lake Lugano region. While Mao et al. (2020) found a genetic structure matching geographic patterns in its native distribution range, T. fortunei does not display a well-defined genetic pattern correlated to geography in southern Switzerland and northern Italy. This might be attributed to the important admixture of cultivated individuals or could also reflect the recent invasion of this species in these areas, with populations that did not have enough time to differentiate according to geography. Some populations such as Cannobio, Gandria, Intragna, Isola Madre, Manno and Tegna are however more differentiated than the average according to structure analyses and mean pairwise Fst and Rst values (Supplementary Tables 4, 5 and 6). For five of them, their higher differentiation can be attributed to genetic drift and founder events as expressed by significant linkage disequilibrium (as in Cannobio), heterozygote excesses (Intragna, Manno), bottleneck signs (as in Gandria) or a combination of the former (Manno, Gandria, Intragna). Accordingly, Tegna, Intragna and Manno are geographically isolated, whereas Gandria is the third highest population in altitude (430 m) after Pianezzo (521 m) and Intragna (468 m). For the Isola Madre population, two out of the three sampled individuals (1IM and 2IM) are differentiated in all genetic analyses (Fig. 3 and Supplementary Figs. 1, 2, 3). These two individuals seem to have contributed little to the colonisation process and they might come from a different gene pool than the one that produced the majority of the sampled populations. This is in contradiction with the hypothesis that the dispersion started from the first exotic gardens associated to touristic areas (Schröter 1936; Pfister 1977; Walther 2000).
Conclusion
The colonisation of Trachycarpus fortunei, a problematic invasive species in southern Switzerland (Info Flora 2014) and northern Italy (Banfi and Galasso 2010; Galasso et al. 2020), results from many factors. As highlighted in previous studies, the rising winter temperatures since the seventies (Walther et al. 2007), as well as the important propagule pressure in disturbed areas (Fehr and Burga 2016), have certainly played an important role in its dispersion.
In the present study, the genetic patterns of T. fortunei in its naturalised range could be assessed. No important inbreeding signs were detected, meaning that populations do not only result from a small genetic pool with crosses among relatives. The observed general panmixia and the low genetic structure indicate that the dispersion of T. fortunei is active in its naturalised areas, and facilitated by its successful reproductive strategy through cross-pollination (Jousson et al. unpublished data on development and morpho-anatomical studies of flowers and fruits). New individuals that are widely planted in gardens near the naturalised populations are likely to increase the general admixture and diversity, therefore reducing inbreeding and increasing the ability of the species to colonise new areas. Due to heterozygotes excess and bottleneck events that are mainly found at the colonisation front (geographical points far from the historical points, e.g. into valleys, and/or at higher altitude), the direction of the colonisation could be confirmed from the lakes upwards the mountain slopes. This study is the first to have unravelled the genetic patterns of an invasive and typically ornamental species in the western side of the Insubric region. We could show that some of the historical settlements, such as Isola Madre, probably contributed little to the colonisation at the genetic level, whereas others, such as Isole di Brissago and Solduno, are found within the main genetic pool, which place them as putative source of colonisation. We hope to have provided novel and useful data to support decision-making processes for effective management strategies and measures.
Data availability
The datasets generated during and/or analysed during the current study are available on Zenodo (https://doi.org/10.5281/zenodo.6327604). This includes the list of samples, the raw reads and filtered VCF files and microsatellite data provided as a dat file. All primers’ sequences can be found in Supplementary Tables 1, 2 and 3.
References
Aberlenc-Bertossi F, Castillo K, Tranchant-Dubreuil C, Chérif E, Ballardini M, Abdoulkader S, Gros-Balthazard M, Chabrillange N, Santoni S, Mercuri A, Pintaud JC (2014) In silico mining of microsatellites in coding sequences of the date palm (Arecaceae) Genome, characterization, and transferability. Appl Plant Sci 2(1):1300058. https://doi.org/10.3732/apps.1300058
Agliati M, Mondada G (1987) Così era Locarno. Armando Dadò Editore, Locarno
Banfi E, Galasso G (2010) La flora esotica lombarda. Museo di Storia Naturale di Milano, Milano
Benjamini Y, Yekutieli D (2001) The control of false discovery rate dependency. Ann Stat 29(4):1165–1188. https://doi.org/10.1214/aos/1013699998
Berger S, Walther GR (2006) Distribution of evergreen broad-leaved woody species in Insubria in relation to bedrock and precipitation. Bot Helv 116:65–77. https://doi.org/10.1007/s00035-006-0749-8
Buschiazzo E, Gemmell NJ (2006) The rise, fall and renaissance of microsatellites in eukaryotic genomes. BioEssays 28(10):1040–1050. https://doi.org/10.1002/bies.20470
Carraro G, Gianoni P, Mossi R, Klötzli F, Walther GR (2001) Observed changes in vegetation in relation to climate warming. In: Burga CA, Kratochwil A (eds) Biomonitoring: general and applied aspects on regional and global scales. Tasks for vegetation science, vol 35. Springer, Dordrecht, pp 195–205. https://doi.org/10.1007/978-94-015-9686-2_12
Catchen J, Hohenlohe P, Bassham S, Amores A, Cresko W (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22(11):3124–3140. https://doi.org/10.1111/mec.12354
Currat M, Ruedi M, Petit RJ, Excoffier L (2008) The hidden side of invasions: massive introgression by local genes. Evolution 62(8):1908–1920. https://doi.org/10.1111/j.1558-5646.2008.00413.x
DeFaveri J, Viitaniemi H, Leder E, Merilä J (2013) Characterizing genic and nongenic molecular markers: comparison of microsatellites and SNPs. Mol Ecol Resour 13(3):377–392. https://doi.org/10.1111/1755-0998.12071
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. PNAS 91(8):3166–3170. https://doi.org/10.1073/pnas.91.8.3166
Dransfield J, Uhl NW, Asmussen BC, Baker JW, Harley MM, Lewis EC (2008) Genera palmarum: the evolution and classification of palms, 2nd edn. Royal Botanic Gardens, Kew, London
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6(5):e19379. https://doi.org/10.1371/journal.pone.0019379
Estoup A, Martin O (1996) Marqueurs microsatellites: isolement à l’aide de sondes non-radioactives, caractérisation et mise au point. Recueil technique Microsatellites, Version 1. Département des Sciences Animales de l'INA P-G. http://www2.agroparistech.fr/svs/genere/microsat/microsat.htm. Accessed 24 Sept 2021
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier L, Ray N (2008) Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23(7):347–351. https://doi.org/10.1016/j.tree.2008.04.004
Fehr V, Burga C (2016) Aspects and causes of earlier and current spread of Trachycarpus fortunei in the forests of Southern Ticino and northern Lago Maggiore (Switzerland, Italy). Palms 60(3):125–136. https://doi.org/10.5167/uzh-127489
Fehr V, Buitenwerf R, Svenning JC (2020) Non-native palms (Arecaceae) as generators of novel ecosystems: a global assessment. Divers Distrib 26(11):1523–1538. https://doi.org/10.1111/ddi.13150
Galasso G, Bartolucci F, Conti F, Martellos S, Moro A, Pennesi R, Peruzzi L, Pittao E, Nimis P (2020) L’inventario della flora spontanea italiana e il nuovo portale della flora d’Italia. In: Alessandrello A, Azuma M (eds) Al Museo per scoprire il mondo. La ricerca scientifica e le mostre temporanee. Museo di Storia Naturale di Milano 2014–2019. Natura 110(1):47–56
Gardener W (1971) Robert Fortune and the cultivation of tea in the United States. Arnoldia 31(1):1–19
Gay J (1861) Le Chamaerops excelsa Thunb., sa patrie, le climat qui lui convient, son introduction dans l’Europe occidentale, les chances qu’il a d’y être naturalisé, son fibrillitium, les usages économiques auxquelles il peut servir etc. Bull Soc bot Fr 8(7):410–430. https://doi.org/10.1080/00378941.1861.10829592
Gianoni G, Carraro G, Klötzli F (1988) Thermophile, an laurophyllen Pflanzenarten reiche Waldgesellschaften im hyperinsubrischen Seenbereich des Tessins. Ber Geobot Inst ETH Zürich 54:164–180
Gladieux P, Feurtey A, Hood ME, Snirc A, Clavel J, Dutech C, Roy M, Giraud T (2015) The population biology of fungal invasions. Mol Ecol 24(9):1969–1986. https://doi.org/10.1111/mec.13028
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate f-statistics. J Hered 86(6):485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5(1):184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp 41(2):95–98
Hallatschek O, Hersen P, Ramanathan S, Nelson DR (2007) Genetic drift at expanding frontiers promotes gene segregation. PNAS 104(50):19926–19930. https://doi.org/10.1073/pnas.0710150104
Hovick SM, Whitney KD (2014) Hybridisation is associated with increased fecundity and size in invasive taxa: meta-analytic support for the hybridisation-invasion hypothesis. Ecol Lett 17(11):1464–1477. https://doi.org/10.1111/ele.12355
Info Flora (2014) Liste Noire et Watch List (actuelle; état août 2014): Palmier chanvre. In: Néophyte envahissante: une menace pour la nature, la santé et l'économie. https://www.infoflora.ch/fr/assets/content/documents/neophytes/inva_trac_for_f.pdf. Accessed 11 Sept 2021
Jacobi K (1998) Palmen für Haus und Garten, 4th edn. BLV, München
Kang M, Buckley YM, Lowe AJ (2007) Testing the role of genetic factors across multiple independent invasions of the shrub Scoth broom (Cytisus scoparius). Mol Ecol 16(22):4662–4673. https://doi.org/10.1111/j.1365-294X.2007.03536.x
Keller SR, Fields PD, Berardi AE, Taylor DR (2014) Recent admixture generates heterozygosity–fitness correlations during the range expansion of an invading species. J Evol Biol 27(3):616–627. https://doi.org/10.1111/jeb.12330
Klopfstein S, Currat M, Excoffier L (2006) The fate of mutations surfing on the wave of a range expansion. Mol Biol Evol 23(3):482–490. https://doi.org/10.1093/molbev/msj057
Klötzli F (1988) On the global position of the evergreen broad-leaved (non-ombrophilous) forest in the subtropical and temperate zone. Veröff Geobot Inst ETH Zürich 98:169–196. https://doi.org/10.5169/seals-308900
Klötzli F, Walther GR, Carraro G, Grundmann A (1996) Anlaufender Biomwandel in Insubrien. Verh Ges F Ökol Zürich 26:537–550
Kny L (1882) Die Gärten des Lago Maggiore, Garten-Zeitung. Parey, Berlin
Koike F (2001) Plant traits as predictors of woody species dominance in climax forest communities. J Veg Sci 12(3):327–336. https://doi.org/10.2307/3236846
Lavergne S, Molofsky J (2007) Increased genetic variation and evolutionary potential drive the success of an invasive grass. PNAS 104(10):3883–3888. https://doi.org/10.1073/pnas.0607324104
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evol 17(8):386–391. https://doi.org/10.1016/S0169-5347(02)02554-5
Lodari R (2002) Giardini e ville del Lago Maggiore. Un paesaggio culturale tra Ottocento e Novecento. Editore Regione Piemonte, Torino
Luikart G, Allendorf FW, Cornuet JM, Sherwin WB (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89(3):238–247. https://doi.org/10.1093/jhered/89.3.238
Mao YX, Lu YT, Hu ZJ, Zhao Y (2020) Genetic diversity of Trachycarpus fortunei (Hook.) H. Wendl germplasm resources in Guizhou by SRAP molecular markers analysis. J Southern Agric 51(1):27–35. https://doi.org/10.3969/j.issn.2095-1191.2020.01.004
Maruyama T (1970) Effective number of alleles in a subdivided population. Theor Popul Biol 1(3):273–306. https://doi.org/10.1016/0040-5809(70)90047-X
MeteoSwiss (2021) Climate normals (1981–2010). https://www.meteoswiss.admin.ch/home/climate/swiss-climate-in-detail/climate-normals/normal-values-per-measured-parameter.html Accessed 11 Oct 2021
Molecular Ecology Resources Primer Development Consortium et al (2013) Permanent genetic resources added to molecular ecology database. 1 December 2012–31 January 2013. Mol Ecol Resour 13(3):546–549. https://doi.org/10.1111/1755-0998.12095
Muzzarini S (2007) Verbania, nelle cartoline d’epoca (1895–1950). Alberti Libraio Editore, Verbania
Nissen SJ, Masters RA, Lee DJ, Rowe ML (1995) DNA-based marker systems to determine genetic diversity of weedy species and their application to biocontrol. Weed Sci 43(3):504–513. https://doi.org/10.1017/S0043174500081546
Olivieri I (2008) Alternative mechanisms of range expansion are associated with different changes of evolutionary potential. Trends Ecol Evol 24(6):289–292. https://doi.org/10.1016/j.tree.2009.01.015
Parker IM, Rodriguez J, Loik ME (2003) An evolutionary approach to understanding the biology of invasions: local adaptation and general-purpose genotypes in the weed Verbascum thapsus. Conserv Biol 17(1):59–72. https://doi.org/10.1046/j.1523-1739.2003.02019.x
Patterson N, Price AL, Reich D (2006) Population structure and eigenanalysis. PLoS Genet 2(12):e190. https://doi.org/10.1371/journal.pgen.0020190
Pauli H, Gottfried M, Reiter K, Klettner C, Grabherr G (2007) Signals of range expansions and contractions of vascular plants in the high Alps: observations (1994–2004) at the GLORIA master site Schrankogel, Tyrol. Austria Glob Change Biol 13(1):147–156. https://doi.org/10.1111/j.1365-2486.2006.01282.x
Payseur BA, Cutter AD (2006) Integrating patterns of polymorphism at SNPs and STRs. Trends Genet 22(8):424–429. https://doi.org/10.1016/j.tig.2006.06.009
Pfister M (1977) Sonnenstube Tessin. Romantisches Ferienparadies im Süden der Schweiz. Ringier, Zürich
Piry S, Luikart G, Cornuet JM (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Heredity 90(4):502–503. https://doi.org/10.1093/jhered/90.4.502
Prentis PJ, Sigg DP, Raghu S, Dhileepan K, Pavasovic A, Lowe AJ (2009) Understanding invasion history: genetic structure and diversity of two globally invasive plants and implications for their management. Divers Distrib 15(5):822–830. https://doi.org/10.1111/j.1472-4642.2009.00592.x
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. https://doi.org/10.1093/genetics/155.2.945
Puckett EE (2017) Variability in total project and per sample genotyping costs under varying study designs including with microsatellites or SNPs to answer conservation genetic questions. Conserv Genet Resour 9:289–304. https://doi.org/10.1007/s12686-016-0643-7
Puth LM, Post DM (2005) Studying invasion: have we missed the boat? Ecol Lett 8:715–721. https://doi.org/10.1111/j.1461-0248.2005.00774.x
R Core Team (2021) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Raj A, Stephens M, Pritchard JK (2014) fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197(2):573–589. https://doi.org/10.1534/genetics.114.164350
Reynolds J, Weir BS, Cockerham CC (1983) Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics 105(3):767–779. https://doi.org/10.1093/genetics/105.3.767
Richardson D, Rejmánek M (2011) Trees and shrubs as invasive alien species: a global review. Divers Distrib 17(5):788–809. https://doi.org/10.1111/j.1472-4642.2011.00782.x
Schmid E (1956) Flora des Südens 2nd. Rascher, Zürich
Schoenenberger N, Rötlisberger J, Carraro G (2014) La flora esotica nel Cantone Ticino. Boll Soc Ticin Sci Nat 102:13–30
Schröter C (1936) Flora des Südens. Rascher, Zürich
Siebold PF (1856) Catalogue raisonné et prix-courant des plantes et graines du Japon cultivées dans l'établissement de von Siebold & Comp. à Leide. Bonn, Henry & Cohen, Leiden
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139(1):457–462. https://doi.org/10.1093/genetics/139.1.457
Smith AL, Hodkinson TR, Villellas J, Catford JA, Csergö AM, Blomberg SP, Crone EE, Ehrlén J, Garcia MB, Laine AL, Roach DA, Salguero-Gómez R, Wardle GM, Childs DZ, Elderd BD, Finn A, Munné-Bosch S, Baudraz MEA, Bódis J, Brearley FQ, Bucharova A, Caruso CM, Duncan RP, Dwyer JM, Gooden B, Groenteman R, Hamre LN, Helm A, Kelly R, Laanisto L, Lonati M, Moore JL, Morales M, Olsen SL, Pärtel M, Petry WK, Ramula S, Rasmussen PU, Enri SR, Roeder A, Roscher C, Saastamoinen M, Tack AJM, Töpper JP, Vose GE, Wandrag EM, Wingler A, Buckley YM (2020) Global gene flow releases invasive plants from environmental constraints on genetic diversity. PNAS 117(8):4218–4227. https://doi.org/10.1073/pnas.1915848117
Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374. https://doi.org/10.1146/annurev.arplant.54.031902.134907
Walther GR (2000) Laurophyllisation in Switzerland. Dissertation Doctoral Thesis, ETH Zürich
Walther GR (2001) Laurophyllisation – a sign of a changing climate? In: Burga CA, Kratochwil A (eds) Biomonitoring: general and applied aspects on regional and global scales. Tasks Vegetation Sci 35:207–223. https://doi.org/10.1007/978-94-015-9686-2_13
Walther GR (2003) Plants in a warmer world. Perspect Plant Ecol Evol Syst 6(3):169–185. https://doi.org/10.1078/1433-8319-00076
Walther GR, Gritti SE, Berger S, Hickler T, Tang Z, Sykes TM (2007) Palms tracking climate change. Glob Ecol Biogeogr 16(6):801–809. https://doi.org/10.1111/j.1466-8238.2007.00328.x
Ward SM, Gaskin JF, Wilson LM (2008) Ecological genetics of plant invasion: what do we know? Invasive Plant Sci Manag 1(1):98–109. https://doi.org/10.1614/IPSM-07-022.1
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer-Verlag, New York
Wolf JB, Brodie ED, Wade MJ (2000) Epistasis and the evolutionary process. Oxford University Press, Oxford
Zehdi-Azouzi S, Cherif E, Moussouni S, Gros-Balthazard M, Abbas Naqvi S, Ludeña B, Castillo K, Chabrillange N, Bouguedoura N, Bennaceur M, Si-Dehbi F, Abdoulkader S, Daher A, Terral J-F, Santoni S, Ballardini M, Mercuri A, Bensalah M, Kadri K, Othmani A, Littardi C, Salhi-Hannachi A, Pintaud JC, Aberlenc-Bertossi F (2015) Genetic structure of the date palm (Phoenix dactylifera L.) in the Old World reveals a strong differentiation between eastern and western populations. Ann Bot 116(1):101–112. https://doi.org/10.1093/aob/mcv132
Zhu Y, Queller DC, Strassman JE (1999) A phylogenetic perspective on sequence evolution in microsatellite loci. J Mol Evol 50(4):324–338. https://doi.org/10.1007/s002399910036
Zuber RK (1979) Untersuchungen über die Vegetation und die Wiederbewaldung einer Brandfläche bei Locarno (Kanton Tessin). Beih Schweiz Z Forstwes 65
Acknowledgements
For the laboratory work and for the genetic analyses, we would like to thank all those that have helped us, especially Régine Niba, Luisa Carvalho and Mathieu Perret (CJBG) as well as Karl Perron (University of Geneva) who kindly provided us the pUC19 digested with smaI. For the visit on the Isola Madre, we would like to thank Gianfranco Giustina and Simone Colongo. For their precise advice on the species and information, we would like to thank Gian-Reto Walther (FOEN), Boris Pezzatti and Marco Conedera (WSL), Vincent Fehr (University of Aarhus) and Phaedra Sys. For the financial support, we would also like to thank the Augustin Lombard from the SPHN of Geneva for fieldwork and microsatellite development and the Academic Society of Geneva for GBS analyses.
Funding
Open access funding provided by University of Geneva. This work was supported by an Augustin Lombard Fund attributed to AJ by the Société de Physique et d'Histoire Naturelle of Geneva for fieldwork and microsatellite development and by a fund attributed to YN by the Academic Society of Geneva for GBS analyses.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation and data collection was performed by Antoine Jousson, Yamama Naciri, Fred Stauffer, Brigitte Marazzi and Guido Maspoli. The laboratory work was carried out by Antoine Jousson and supervised by Yamama Naciri. Data generation and analysis was conducted by Antoine Jousson with the assistance of Yamama Naciri, Camille Christe and Frédérique Aberlenc. The first draft of the manuscript was written by Antoine Jousson and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest. The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10530_2022_2874_MOESM1_ESM.tiff
Supplementary Fig. 1 Genetic analyses in Trachycarpus fortunei: PCA Plots on microsatellites (A) and SNP markers (B). The individuals are named according to their population (abbreviations) and coloured according to the two regions (Lake Maggiore in blue and Lake Lugano in purple).
10530_2022_2874_MOESM2_ESM.tiff
Supplementary Fig. 2 Genetic analyses in Trachycarpus fortunei: MDS Plots with microsatellites (A; stress value = 0.198) and SNP markers (B; stress value = 0.167) on the 21 analysed populations. The populations are coloured according to the two regions (Lake Maggiore in blue and Lake Lugano in purple).
10530_2022_2874_MOESM3_ESM.tiff
Supplementary Fig. 3 Genetic analyses in Trachycarpus fortunei: structure plots with microsatellites (A; K = 2) and SNP markers (B; K = 7) on 200 and 199 individuals, respectively.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jousson, A., Christe, C., Stauffer, F. et al. Panmixia and active colonisation of the invasive palm Trachycarpus fortunei (Arecaceae) in Southern Switzerland and Northern Italy as inferred by microsatellites and SNP markers. Biol Invasions 24, 3737–3756 (2022). https://doi.org/10.1007/s10530-022-02874-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-022-02874-8