
Abstract
Knowledge of the introduction history and spread dynamics of invasive species can provide important insights for management (Ens et al. in Environ Rev (in press), 2022), however such information is often unavailable for accidental introductions. Here we infer how the European shore crab, Carcinus maenas, and its congener, the Mediterranean shore crab, C. aestuarii, were introduced to and spread within South Africa. We do this using nuclear microsatellite data and Bayesian assignment tests and Approximate Bayesian Computation (ABC) modelling that included samples from the native and other invasive ranges of these two species. We also compared the genetic diversity and structure of one of the South African populations during and after intensive management, with that of another, unmanaged, population. South African populations had higher genetic diversity than invasive Carcinus populations from elsewhere in the world. Moreover, the ABC analyses suggest that South African populations originated from an admixture event between individuals of C. maenas from a population in the native range and an invasive population from Canada. We also identified instances of hybridisation between Carcinus maenas and C. aestuarii in South Africa. South African populations showed no genetic structure, suggesting either extensive migration between them or that populations arose from the same initial introduction. Management of Carcinus did not affect genetic diversity or structure, and we suspect that the management duration was insufficient to target a full generation of crabs. Together these results suggest multiple introductions and/or high propagule pressure to South Africa, crab (larval or adult) movement between existing populations, and some hybridisation. For eradication from South Africa to be achieved, management would need to concurrently target all known invasive populations and clearly establish that new introductions could be prevented.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Understanding invasion histories and post-introduction demographic dynamics of invasive species are important considerations for the development of effective management approaches (Johnston et al. 2009; Le Roux and Wieczorek 2009; Wilson et al. 2017). For taxa that have been deliberately introduced, these aspects can sometimes be quantified through historical introduction records (Richardson 1998). However, such information is rarely available for accidental introductions, as is the case for most marine invasions (Hewitt and Campbell 2007; Mead et al. 2011). In addition, many marine species have pelagic life stages in highly connected ecosystems, potentially diluting pathway signatures (Johnston et al. 2009). Molecular approaches can, however, provide insights into historical and contemporary population-level processes (Le Roux and Wieczorek 2009; Chown et al. 2015; Sherpa and Després 2021). Estimates such as the number of founding individuals, introduction frequency, the occurrence of admixture and hybridisation, can all be reliably inferred from molecular data (Rius et al. 2012; Rius and Darling 2014; Le Roux 2021).
Shore crabs in the genus Carcinus are some of the world’s most widespread marine invaders. For instance, the European shore crab, Carcinus maenas, has established invasive populations in four continents: Africa, Australia, North America and South America (Carlton and Cohen 2003). Its congener, the Mediterranean shore crab, C. aestuarii, is also invasive in Japan (Sakai 1986). Such widespread invasion success suggests that these crabs are easily transported over long distances, perhaps because of their pelagic larval life stages (Queiroga 1996). Yet, except for invasions in Japan and South Africa, these two species rarely co-invade (Geller et al. 1997). Carcinus crabs are considered high-impact invaders across many of their invasive ranges (for reviews on C. maenas see Behrens Yamada 2001; Walton et al. 2002; Chen et al. 2004; Ens et al. 2022). In regions such as Canada, multiple introductions of C. maenas have led to genetically diverse invasive populations (Roman 2006) while, in others, such as on the Pacific coast of North America, secondary introductions founded populations with limited genetic diversity (Darling et al. 2008). The latter population, covering approximately 1,000 km, stemmed from a single introduction event from the Atlantic coast (Darling et al. 2008; Tepolt et al. 2009).
Carcinus maenas was first detected in South Africa in 1983 (Joska and Branch 1986). Previous genetic studies identified both C. maenas and C. aestuarii in South Africa (Geller et al. 1997; Darling et al. 2008). Hybridisation between the two species has also been detected in Japan (Darling 2011). It has been suggested that the Japanese population likely resulted from a single introduction event from a hybrid zone in the native range (Darling 2011). Genetic studies on South African Carcinus populations suggest a complex introduction history characterised by multiple introductions (Darling et al. 2008). Subsequent morphometric analyses of thousands of individuals, however, found no evidence of C. aestuarii in the country (Robinson et al. 2005). In South Africa, Carcinus is largely restricted to two harbours (Table Bay and Hout Bay) (Mabin et al. 2017). Table Bay harbour is Cape Town’s largest commercial harbour with substantial international traffic. Hout Bay harbour, located approximately 30 km to the south of Table Bay harbour, is relatively small and used by commercial fishing boats and recreational watercraft. A 2013 study estimated that these two harbours contained approximately 164,000 and 7,000 Carcinus crabs, respectively (Jooste 2013).
In recognition of the negative impacts of Carcinus crabs elsewhere in the world (e.g. Grosholz et al. 2000, Matheson et al. 2016, Ens et al. 2022), and in support of authorities charged with managing biological invasions in South Africa, a management trial was implemented during 2014 and 2015 in Hout Bay harbour (Mabin et al. 2020). The primary aim of that project was to assess the feasibility of eradicating Carcinus crabs from South Africa. However, approximately five times more crabs (36,244 individuals) were caught than the previously estimated total population size and extirpation was not achieved (Mabin et al. 2020). This management trial also highlighted the need to understand the movement of individuals within and between populations that are targeted for management. In the context of the South African Carcinus invasion, three important questions arose that would need to be addressed to assess the feasibility of a nationwide eradication attempt. First, is there significant movement of crabs between Table Bay and Hout Bay harbours? If so, eradication attempts need to be strictly concurrent between harbours. Second, to what degree did the management trial in Hout Bay Harbour reduce the genetic diversity of the population? And third, if both crab populations could be extirpated, is reintroduction likely to occur from elsewhere in the world?
Using a population genetic approach, this study aimed to provide insights to these management questions by establishing: (1) whether the two South African populations of Carcinus have similar levels of genetic diversity and structure and how these attributes compare to other invasive and native populations, (2) the most likely scenarios that explain the colonisation history of Carcinus in South Africa, (3) whether there is any genetic evidence for C. maenas and C. aestuarii co-invading South Africa and, if so, whether they hybridise, and (4) whether management efforts have affected the genetic diversity and structure of the Carcinus population in Hout Bay harbour. We hypothesised that the two South African populations would have similar genetic diversity and no structure, in line with the attributes observed in invasive Carcinus populations globally. Support for this hypothesis will indicate frequent movement between the two harbours and multiple introductions, implying a high likelihood of reintroduction if both populations were successfully extirpated. We also hypothesised that no C. aestuarii individuals or Carcinus hybrids would be present in South Africa (Robinson et al. 2005). Lastly, we hypothesised that a Carcinus trial management programme would reduce the genetic diversity and structure of the Hout Bay harbour population, compared with the unmanaged Table Bay population (Hampton et al. 2004; Abdelkrim et al. 2007).
Materials and methods
Sample collection and microsatellite genotyping
Samples were collected from native and invasive populations of Carcinus maenas and C. aestuarii around the world in 2015 (Table 1). South African samples were collected at Hout Bay and Table Bay harbours in 2014, 2015 and 2016, i.e. before, during and after the management trial in Hout Bay, with Table Bay as a control (Mabin et al. 2020).
In South Africa, Carcinus individuals were collected using baited traps containing 200 g of crushed sardines in a muslin bag with a soak time of two hours. Captured crabs were euthanised by freezing at −20 °C and frozen gill tissue was removed for DNA extractions. International samples were provided as gill tissue, except for samples from the USA, which consisted of chelae tissue. All tissue samples were stored in 90% ethanol until DNA extraction. DNA was extracted from all samples using a QIAGEN DNeasy Blood and Tissue Kit (QIAGEN, California, USA, supplied by Whitehead Scientific, Cape Town, South Africa) according to the manufacturer’s protocol, with the following modifications. After the addition of Buffer AW2, the solution was centrifuged for 5 min at 13,300 rpm. The DNeasy mini spin column was then removed from the collection tube and centrifuged for a further 3 min at 13,300 rpm in a clean collection tube to remove excess ethanol. The final elution step was modified using 50 µL Buffer AE to increase final DNA concentrations. DNA quality and concentrations were determined using the Nanodrop spectrophotometer (Infinite 200 PRO NanoQuant, Tecan Group Ltd, Männedorf, Switzerland) and all samples were diluted to a final concentration of 20 ng/µl.
Polymerase chain reaction (PCR) was used to test the amplification of 23 microsatellite loci using primers previously developed for C. maenas and C. aestuarii (Tepolt et al. 2006; Marino et al. 2008). After PCR optimisation, successful amplification was consistently achieved for eight loci (Table S1). PCR amplifications for these loci were performed in two multiplex reactions using the QIAGEN Multiplex PCR kit (multiplex 1) and the KAPA2G Fast Multiplex PCR kit (multiplex 2). PCR reactions were performed in 96 well plates (with nine technical replicate samples and two water samples per plate as positive and negative controls, respectively). For multiplex 1, PCR reactions contained 1.5 µL x 0.2 µM primer mix (primers mixed in equal concentrations), 7.5 µL QIAGEN multiplex mix, 1.5 µL Q solution, 3 µL water and 1.6 µL DNA template. For multiplex 2, PCR reactions contained 4.5 µL water, 7.5 µL 2x Kapa2G Fast Multiplex mix, 1.5 µL × 2 µM primer mix, and 1.6 µL template DNA. PCR cycling conditions for multiplex 1 consisted of 15 min initial denaturation at 95 °C, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 52.7 °C for 90 s and extension at 72 °C for 60 s, before a final extension for 30 min at 60 °C. PCR cycling conditions for multiplex 2 consisted of denaturation at 95 °C for 3 min, followed by 30 cycles of denaturation at 95 °C for 15 s, annealing at 54.4 °C for 30 s and extension at 72 °C for 25 s, and a final extension for 1 min at 72 °C. Purified PCR fragments were separated on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, California, USA), using GENESCAN TM-500 (-250) as an internal size standard (Applied Biosystems). Allele sizes were visualised and scored using GENE-MARKER version 1.95; SoftGenetics LLC, State College, Pennsylvania, USA). Samples that failed to amplify at more than four loci were excluded from the final dataset, resulting in a total of 718 usable genotypes.
Genetic diversity
The number of alleles per locus were calculated using the ‘NB’ package in the R statistical environment (Hui 2014; R core team, 2016). Observed heterozygosity (HO), expected heterozygosity (HE), allelic richness (AR), and the inbreeding coefficient (FIS), and significant departures of allele frequencies from Hardy Weinberg equilibrium (HWE) expectations were calculated in the ‘diveRsity’ package (Keenan et al. 2013).
Population genetic diversity metrics (AR, HO, HE and FIS) were compared between invasive- and native-range C. maenas populations using student t-tests. We only included data from 2016 for South African samples (highest sample size) in these analyses to avoid pseudo replication.
Global population genetic structure
The final global dataset (including South Africa) was assessed for the presence of null alleles using the FreeNA software (Chapuis and Estoup 2007). Pairwise fixation indices (FST) were calculated in FreeNA, with and without null allele correction (Chapuis and Estoup 2007). To compute 95% confidence intervals for FST values, 10,000 simulations were performed. FST values for each population were then used to test whether the presence of null alleles significantly influenced the results using a Kruskal-Wallis test.
The genetic structure of all sampled crab populations was estimated using Bayesian assignment tests implemented in the STRUCTURE version 2.3.4 software (Pritchard et al. 2000). STRUCTURE models were run on all populations of Carcinus (n = 14). Including both Carcinus species in our models allowed us to infer instances of hybridisation between them. The number of genetic clusters (i.e. K-values) used for each analysis was between one and the number of populations included in each analysis. Twenty model iterations were run for each value of K. Each run consisted of 500,000 generations of which the first 100,000 were discarded as burnin. The admixture model was applied with correlated allele frequencies as hybrids between C. maenas and C. aestuarii were suspected to be present in the dataset (Darling et al. 2008). Optimal K values were determined using the method described by Evanno et al. (2005) and as implemented in STRUCTURE Harvester (Earl and von Holdt 2012). The programme CLUMPP (version 1.1.2; Jakobsson and Rosenberg 2007) was used to merge the results from STRUCTURE, utilising the greedy option method with 10,000 repeats. The resultant output file was visualised in DISTRUCT (version 1.1; Rosenberg 2004).
Inferring putative the source(s) of Carcinus introductions to South Africa
The potential source(s) of Carcinus populations in South Africa was inferred using an Approximate Bayesian Computation (ABC) approach and the DIYABC software (version 2.1.0; Cornuet et al. 2008, Cornuet et al. 2014). This approach allows the simulation of a large number of genetic datasets for different potential introduction scenarios that are compared with the observed dataset to identify the most likely scenario (Beaumont et al. 2002). A hierarchical approach was adopted as this reduced the number of scenarios and thus computational power required and has been used successfully in previous studies (Boissin et al. 2012; Stone et al. 2017; Wauters et al. 2018). In total, 78 DIYABC analyses were conducted, with several variations to the biological and historical parameters (results not shown). These variations included the size, source and founding date of all populations considered, the presence and strength of potential bottlenecks, rate of admixture, mutation models and rates. The three best-performing analyses are detailed below.
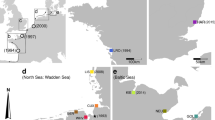
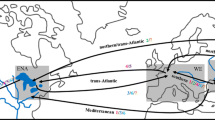
Analysis 1 involved simulations that considered all potential sources of Carcinus. The probabilities of three invasion scenarios were estimated, including a direct introduction from the ranges of C. maenas or C. aestuarii and an admixture event between these two ranges. These ranges included three native and three invasive C. maenas populations and one native and one invasive C. aestuarii population (Fig. 1). Various scenarios were analysed, including those with ghost populations (i.e. unsampled populations) and strong population genetic bottlenecks. However, these were not included in the final analyses due to low support (see Results). The complete global dataset was used for this analysis, excluding the 2014 and 2015 South African samples to avoid pseudo replication while maximising sample sizes.
Analysis 2 was conducted at higher resolution, with different invasion scenarios developed using the inferred results from Analysis 1. This led to more detailed modelling to infer the contribution of native and invasive C. maenas populations to South African invasions. Scenario 2.1 involved a direct introduction from the native range of C. maenas while Scenario 2.2 represented a direct introduction from the invasive range of C. maenas to South Africa. Scenario 2.3 modelled an admixture event between a native and invasive C. maenas population. Scenario 2.4 and 2.5 simulated an introduction to South Africa from a ghost population in the C. maenas native and invasive range respectively.
Analysis 3 was based on the results from analysis 2 and aimed to identify the most likely invasive range source of invasive populations in South Africa: Canada (Scenario 3.1), the USA (Scenario 3.2), and Australia (Scenario 3.3), noting in each case that the invasive population in South Africa appears to have been the result of admixture of other invasive populations with populations from the native range.
All ABC models were parameterised by drawing on the biology of Carcinus and presence data in invaded areas. It was assumed that the two congeners shared demographic parameters where data were unavailable for C. aestuarii (the least studied of the two species). The timing of events was measured in terms of generations, which was one year in this case, based on the life history characteristics of C. maenas (Behrens Yamada 2001). The values estimated for the temporal priors were assumed from historical shipping and detection records (Joska and Branch 1986; Carlton and Cohen 2003; Mead et al. 2011). The prior parameters (estimated with uniform distributions) for temporal events, population sizes and bottleneck reductions are detailed in Table S2. Loci were estimated to have an allelic range between 40 and 72. The minimum value of 40 represented the DIYABC default value, which is greater than recorded in this study, but allowed for the inclusion of unsampled alleles. The final model utilised a single-step mutation model due to improved performance over the default generalised stepwise mutation model. Microsatellite mutation rates have not been determined for crustaceans, however for mammals, rates are between 1 × 10− 3 and 1 × 10− 5 (Dallas 1992; Ellegren 1995) while in arthropods (e.g. Drosophila melanogaster), mutation rates of 6 × 10− 6 have been observed (Schlötterer et al. 1998). Accordingly, rates in this study ranged from 1 × 10− 2 to 1 × 10− 6. The DIYABC default rates of admixture were used, i.e., 0.001–0.999. The summary statistics used to differentiate between the different scenarios modelled included the mean number of alleles and the mean genic diversity (Nei 1987; as measures of the diversity among the sampled populations), in addition to the fixation index (FST) (Weir and Cockerham 1984) and genetic distance (dµ2; Goldstein et al. 1995) to distinguish between populations. Datasets were simulated 1 × 106 times for each scenario. The pre-evaluation analysis allowed a visual comparison between the simulated and observed datasets of each scenario in a principal coordinate analysis (PCA) as well as a ranked comparison between the summary statistics of each dataset. The posterior probabilities were then compared in a Linear Discriminant Analysis using a logistic regression approach to select the 1% closest simulated dataset to that observed, with non-overlapping 95% confidence intervals. The accuracy of the posterior distributions of the model parameters were evaluated using the relative bias. In order to evaluate the confidence in scenario choice, various scenario-parameter combinations were analysed in the prior and posterior distributions. Type I and II errors were calculated using 100 pseudo-observed datasets (pods) for each scenario. Then for each analysis, type I errors were estimated by determining the number of times that the correct analysis was not selected, over all 100 pods. Type II errors were calculated based on the proportion of times the correct scenario was selected when it did not have the highest posterior probability.
Genetic demographic impacts of management trials of Carcinus in South Africa
Allelic richness (AR) of the three sampling periods in Table Bay and Hout Bay was calculated using the ‘diveRsity’ (Keenan et al. 2013) package in R with a rarefaction approach to account for unequal sample sizes. AR was analysed with respect to harbour (2 levels: Table Bay and Hout Bay) and year (3 levels: 2014, 2015 and 2016), using a two-factor generalised linear model (GLM) with a quassipoisson error distribution. The Genalex (version 6.5; Peakall and Smouse 2012) programme was used to calculate the number of private alleles (PA) and these were analysed with respect to harbour and year using a Pearson’s Chi-Squared test.
Temporal changes in the genetic structure of South African populations were evaluated using Bayesian assignment tests in STRUCTURE (version 2.3.4; Pritchard et al. 2000). This methodology was identical to that of the global population structure analysis described above, except that K values between 1 and 6 were tested for the South African-only analysis. Assignment values for the South African populations were analysed with respect to harbour (2 levels: Table Bay and Hout Bay) and year (3 levels: 2014, 2015 and 2016) and the interactions between the factors, using a two-factor generalised linear model (GLM) with a quassipoisson error distribution.
Mitochondrial DNA analyses
Fourteen South African samples (collected in 2014 and 2015) were identified as potential hybrids in our STRUCTURE analysis (i.e. being admixed), using an approach adopted in previous studies (Le Roux et al. 2015, Castillo et al. 2021). To confirm the maternal identities of these genotypes, a portion of the CO1 mtDNA gene was sequenced. PCR amplification was done using the primers COIF-PR115: TCW ACN AAY CAY AAR GAY ATT GG and COIR-PR114: ACY TCN GGR TGN CCR AAR ARY CA (Darling et al. 2008). Each PCR reaction contained 6 µL template DNA, 3 µL of each primer (5 pmol), 0.6 µL dNTP’s, 3 µL 10 x Buffer, 1.8 µL MgCl2 (25 mM), 0.6 µL BSA (10 mg/ml), 0.6 µL Supertherm Taq and 11.4 µL distilled water. PCR cycling conditions followed initial denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 60 s and extension at 72 °C for 60 s, before a final extension at 72 °C for 15 min.
An additional forty-eight South African samples previously sequenced in a 2013 study were included (Jooste 2013). For these samples, DNA was extracted using the CTAB method (Doyle and Doyle 1990) and a portion of the COI gene amplified with the forward primer CarcF1 (5’ CCR GGR ACY TTA ATY G 3’) and the reverse primer jgHCO2198 (5’ TAI ACY TCI GGR TGI CCR AAR AAY CA 3’) (Geller et al. 2013). Each PCR reaction contained 3 µL template DNA, 1.25 µL of each primer (5 µM), 2.5 µL 10 x Buffer, 2.5 µL MgCl2, 0.1 µL Super Isotherm Taq, and 13.4 µL distilled water. PCR cycling conditions were: initial denaturation at 94 °C for 2 min, 30 denaturation cycles at 94 °C for 1 min, annealing at 50 °C for 60 s, and a final elongation step at 72 °C for 15 min.
DNA sequences were aligned and edited in BIOEDIT (version 7.0.5, Hall 1999) along with Carcinus sequences from Genbank (http://www.ncbi.nlm.nih.gov, FJ159008 to FJ159102). Haplotype networks were constructed using sequence data in TCS (version 1.21; Clement et al. 2000) and visualised using the Population Analysis with Reticulate Trees (PopART, version 3; Leigh and Bryant 2015) software.
Results
Global genetic diversity
Null alleles were detected in our dataset but at a low frequency, with a mean of 0.036 (SD: 0.056) across all loci. Seven of the microsatellite loci were identified as polymorphic in all populations included, with a maximum of 36 alleles detected per locus. One locus was monomorphic in six of the 14 populations but had a total of five alleles across all populations. Thirty-one (28%) loci by population combinations showed significant departures (p < 0.05) from HWE in the 14 populations.
Native C. maenas populations had significantly higher high levels of allelic richness (AR of 10.8 to 11.3, Table S3) compared to most invasive populations (AR between 4.5 and 7.3; t = -2.448, df = 6, p < 0.05). It is noteworthy, however, that South African populations had high levels of AR (mean = 10.0). Across all populations, observed heterozygosity (HO) was lower than the expected heterozygosity HE (Table S3). Levels of HE were similar between native and invasive populations (t = -2.167, df = 6, p > 0.05). HO was significantly higher in native populations ranging between 0.65 and 0.67, while in invasive populations ranging between 0.58 and 0.64 (t = -3.796, df = 6, p = 0.009). The number of private alleles (PA) in native range populations varied from four to seven; in invasive populations there were fewer than three per population), with the exception of South Africa (mean = 4). Inbreeding (FIS) estimates were higher in invasive (mean = 0.067) than in native populations (mean = 0.05) (Table S3), however, this difference was non-significant (t = 1.219, df = 6, p > 0.05).
Global population genetic structure
There was no significant difference between pairwise FST values when using data that were uncorrected or corrected for the presence of null alleles (Table S4 and S5 respectively, H = 0.394, p = 0.530), therefore subsequent analyses were conducted using the uncorrected dataset. Among global Carcinus populations, excluding South African populations, pairwise FST distances ranged from zero between Portugal and Ireland, to 0.2473 between the USA and Japan (Table S4). Unsurprisingly, the highest differentiation was observed between C. maenas and C. aestuarii populations. STRUCTURE analyses identified four genetic clusters (K = 4) within the global range of Carcinus (Fig. 2a, Fig. S1). These consisted of a distinct C. aestuarii cluster (including native Italian and invasive Japanese populations), a North American C. maenas cluster (comprised of the USA and Canadian populations), a C. maenas cluster (including native European populations) and a less distinct C. maenas cluster, that corresponded mostly to populations from Australia.
Putative source(s) of Carcinus in South Africa
The first DIYABC analysis (which assessed the likelihood of a direct introduction from the C. maenas or C. aestuarii native and invasive ranges as well as an admixture event between these two ranges) indicated that a C. maenas population most likely founded the South African population, i.e. Scenario 1.1 (Fig. 1, Table S6). This scenario had the highest posterior probability with 95% confidence intervals that did not overlap with the other tested scenarios (P = 0.6800, 95%CI = 0.6690–0.6910). The type I error associated with the selection of this scenario was 0.07 using the direct approach and 0.08 for the logistic approach. The mean type II errors calculated under the direct and logistic approach were 0.12 (ranging from 0 to 0.24) and 0.06 (with a range of 0 to 0.11) respectively. The next most likely scenario inferred was that the South African population was an admixture between C. maenas and C. aestuarii, i.e. Scenario 1.3 (P = 0.3143, 95%CI = 0.3034–0.3253). The last scenario (which tested that the founding population in South Africa came from a C. aestuarii population) was not well supported by the model (P = 0.0057, 95%CI = 0.0008–0.0105).
The second analysis (which involved scenarios focusing on C. maenas populations only, grouped as native or invasive) strongly supported the scenario of South African populations originating from an admixture event between populations in the native and invasive range (i.e. Scenario 2.3) (P = 0.6886, 95%CI = 0.6791–0.6982) (Fig. 1, Table S6). Type I errors associated with the selection of Scenario 2.3 were 0.4 using the direct approach and 0.2 for the logistic approach. The mean type II errors associated with the false acceptance of Scenario 2.3 under the direct and logistic approach were 0.05 (ranging from 0.03 to 0.06) and 0.19 (with a range from 0 to 0.4) respectively. The next most probable scenario for the genetic patterns observed in South Africa was that the colonists originated from a native-range population of C. maenas (P = 0.135, 95%CI = 0.129–0.142).
The third analysis (which addressed the C. maenas invasive populations at a country level, in combination with populations from the native range) indicated that Canada [instead of invasive populations from the USA (P = 0.197, 95%CI = 0.190–0.204) or Australia (P = 0.287, 95%CI = 0.279–0.295)] was the likely source of South African populations that subsequently admixed with a native range population (P = 0.516, 95%CI = 0.508–0.525) (Fig. 1, Table S6). Type I errors associated with this most likely scenario were 0.24 and 0.25 (for the direct and logistic approaches respectively). The mean type II errors associated with the false acceptance of this scenario under the direct approach were 0.15 (ranging from 0.13 to 0.17) and 0.22 (with a range from 0.16 to 0.27) for the logistic approach.
The effective population size (i.e. the number of individuals that are reproductive in a population) of the South African Carcinus populations according to the posterior distributions of the Bayesian model parameters in analysis 1 was 3,410 (95% CI: 720–8,840) while analysis 2 estimated the populations to comprise 6,570 individuals (95% CI: 2,520–9,780) and analysis 3 estimated this population to contain 7,870 (95% CI: 4,210–9,890) (Table S6). The most-supported invasion scenario in analysis 1 estimated that Carcinus arrived in South Africa in 1895 (95% CI: 1683–1979). Analysis 2 estimated that the crab was introduced in 1864 (95% CI: 1660 and 1962), while Analysis 3 supported an introduction occurring in 1843 (95% CI 1488 and 1995).
Demographic impacts of trial management interventions against Carcinus in South Africa
Within South African populations, there was no main effect of harbour or year (or an interaction between them) on AR (p > 0.05) (Fig. 3a). The AR of the Hout Bay population did not change through time because of management or stochastic loss via genetic drift. The Hout Bay population had higher PA (18 alleles) compared with Table Bay (6 alleles) (χ2 = 14, df = 5, p = 0.016) but this did not change over time [χ2 = 6, df = 6, p = 0.423, (Fig. 3b)].
Among South African populations, genetic differentiation was extremely low, ranging from 0 to 0.0076. STRUCTURE analyses of these populations assigned individuals to three genetic clusters (K = 3), but with all individuals assigned with equal probabilities to all three clusters (Fig. S2). Such complete and equal admixture indicates the true value of K is more likely to be one, i.e., a panmictic population (Le Roux et al. 2008).
There was no main effect of year or site on the Bayesian assignment values for South African populations (q1-4) (Table S7, Fig. 4) when considering data from the full global model (p > 0.05). None of the interactions between the two factors were significant, except for q2, where there was a significant interaction between site and year (F = 5.559, df = 2, p = 0.004).
Mitochondrial DNA diversity of Carcinus in South Africa
Of the 62 samples sequenced, only one C. aestuarii individual was identified. This specimen was collected during a minor sampling event in 2013, rather than the more extensive sampling of 2014 and 2015. The haplotype network (Fig. 2b) showed considerable genetic distance between the congeners, with 33 mutational steps separating C. maenas and C. aestuarii haplotypes. The network clearly identified one South African individual that has a haplotype that closely resembles C. aestuarii, in contrast to the majority of samples collected in South Africa that were identified as C. maenas.
Discussion
Reintroductions and movement of individuals between invasive populations of an alien species that is the focus of management action can undermine control efforts. As such, understanding these demographic aspects can help to identify incursions that can be effectively and efficiently managed. Following a trial to manage invasive Carcinus crabs in South Africa, this study used a genetic approach to consider introductions and within-region movement in relation to two invasive populations to inform future management. It was found that the two harbour populations of Carcinus in South Africa had high and very similar genetic diversity and no structure. This most likely resulted from multiple independent introductions to both sites and/or ongoing gene flow between the two harbours. Extensive sampling during this study confirmed that C. maenas is the predominant species in South Africa and there is limited evidence of hybridisation with C. aestuarii. Management action in Hout Bay harbour did not affect the genetic diversity of that population. If both harbour populations were successfully extirpated in future, these findings suggest that reintroduction is likely to occur from elsewhere in the world.
Except for South African populations, invasive Carcinus populations harbour significantly lower genetic diversity than native-range populations, indicative of lower propagule pressure and possible founder effects in these regions. These findings fit the general observation that introduced populations often exhibit reduced genetic diversity compared to their native range source populations (Dlugosch and Parker 2008; Le Roux 2021). By contrast invasive South African populations had remarkably high levels of genetic diversity, comparable to that found in native range populations. Carcinus populations in Hout Bay and Table Bay harbours represent a panmictic population suggesting that these they likely stemmed from the same introduction and/or experience frequent gene flow. Our approximate Bayesian computation estimated that Carcinus was introduced to South Africa earlier than previously thought (Joska and Branch 1986), in 1843. One of the earliest recorded sightings of C. maenas outside its native range was in the Red Sea in approximately 1817, before the Suez Canal opened in the 1860s (Carlton and Cohen 2003). Therefore, this introduction may have occurred via a shipping route from the native range, around the tip of Africa (Carlton and Cohen 2003). In North America, lag phases of between two and four years have been documented for C. maenas (Medcof and Dickie 1955; Cohen et al. 1995) but other marine crustaceans have been known to have experienced lag phases of up to 50 years (Witte et al. 2010). However, there is a wide confidence interval around the introduction dates estimated in this study and considering that the crab is highly conspicuous, it is unlikely to have gone undetected for so long in such a well-studied region (Awad et al. 2002). The discharge of ballast water in South African waters began in the late 1800s and completely replaced the use of dry ballast by the 1950s (Griffiths et al. 2009). However, the first detection of an alien species that was thought to have arrived in South Africa via ballast water occurred in the 1940s (Griffiths et al. 2009). Therefore, it is plausible that the upper range of introduction dates estimated by our ABC analyses are closer to the true arrival date of the crab.
In addition to multiple introductions from global Carcinus populations, the similarities in the genetic diversity and structure of the Carcinus populations in South Africa indicates frequent secondary dispersal within the country. This might have occurred through the natural or human-mediated transfer of propagules. It is possible that coastal advective currents flowing South (Nelson and Polito 1987), enabled larval dispersal from Table Bay to Hout Bay. Oceanographic processes were identified as a possible factor contributing to the invasion of C. maenas on the pacific coast of Canada (Brasseale et al. 2019). However, an experimental study in South Africa found that adult C. maenas are unable to effectively grip rocky substrata under high wave action (Hampton and Griffiths 2007) that typifies much of the intertidal coastline between Hout Bay and Table Bay. As such, environmental conditions might limit the establishment of adults outside protected harbour areas (Mabin et al. 2017). Alternatively, predatory driven biotic resistance might restrict the spread of the crabs (Mabin 2018). In terms of human-mediated spread, the most plausible vectors between the two harbours are pelagic fishing vessels and recreational yachts. Such transfer by coastal vessels has been documented for C. maenas in Canada (Blakeslee et al. 2010). However, in the absence of clear evidence, it is not possible to determine the vector(s) responsible for the initial or secondary introductions and maintenance of genetic diversity found in South African populations.
In agreement with previous reports (Geller et al. 1997; Darling et al. 2008), our STRUCTURE analyses identified 13 admixed genotypes (based on microsatellites) between the two Carcinus species. Yet, almost all of these admixed genotypes had mtDNA haplotypes corresponding to C. maenas, suggesting that C. maenas is the predominant invader in South Africa and that C. aestuarii is likely no longer present in the country. The latter is supported given the absence of ‘pure’ C. aestuarii multi-locus microsatellite genotypes in South African populations. The most parsimonious introduction scenario inferred through the ABC approach also suggests that current South African Carcinus populations are the result of at least one admixture event between native and non-native North American Carcinus populations. This seems more likely when considering that most marine taxa introduced to South Africa came from Europe and North America (Faulkner et al. 2017). However, to date, no evidence exists for the presence of C. aestuarii or Carcinus hybrids in Canada (Darling et al. 2008). Therefore, the presence of admixed genotypes in South Africa may have arisen under various circumstances. First, individuals of both species could have been directly introduced to South Africa from their native range where they co-occur and hybridise. The assignment analysis found evidence for C. aestuarii genotypes in the native range of C. maenas. An oceanographic mixing zone between Atlantic and Mediterranean waters, off the coast of Spain in the Gibraltar Strait (Tintore et al. 1988) has been proposed as an important phylogeographic break for several marine organisms such as the Mediterranean mussel, Mytilus galloprovincialis (Quesada et al. 1995) and the European flat oyster, Ostrea edulis (Saavreda et al. 1995) and possibly the genus Carcinus (Geller et al. 1997). This region might therefore have acted as the source of South African Carcinus populations. The port of Gibraltar is an important contributor to contemporary and historical global species flow within this zone as a result of its position at the entrance to the Mediterranean Sea (Xu et al. 2014). Alternatively, it is possible that larvae of both species were collected in ballast water in multiple ports by international vessels, prior to discharge in South Africa. The third possibility is that there were multiple introductions and that the two species were introduced on different occasions in South Africa, C. maenas in high numbers and C. aestuarii in low numbers. The absence of C. aestuarii individuals from the extensive sampling of 2014 and 2015 suggests the Mediterranean shore crab has not established in South Africa.
Tracking temporal changes in genetic diversity can be useful in assessing the demographic responses of invasive species to disturbances (Mairal et al. 2021) and management interventions (Abdelkrim et al. 2007; Vardien et al. 2013). For example, three years after a ten-year-long management programme against Spartina alterniflora in California, significant changes in the genetic diversity of the species were observed (Ort and Thornton 2016). In the current study, similar and high levels of genetic diversity in Carcinus populations were maintained over three years in both Table Bay (unmanaged) and Hout Bay (managed) harbours. There are three possible explanations for the apparent stability of the Hout Bay population through time. First, the diversity and structure trends simply reflect the presence of juveniles in the population who were below the minimum catchable size during the period of management (Mabin et al. 2020) that have since grown to a sufficient size to be retained in the traps. These juveniles would carry most, if not all, standing genetic variation from the previous generation. Second, it could be that continued natural or human-mediated spread from Table Bay during management maintained the genetic diversity of the Hout Bay population. Lastly, and most likely, our management duration may have been insufficient to lower the Hout Bay harbour population size to levels that would affect its genetic diversity.
Considering the limited resources that are available to address threats to biodiversity (Echols et al. 2019), it is vital that management of invasive species is effective and efficient (Loureiro et al. 2020). Key to supporting such an outcome is scientific feasibility. While this can relate to the susceptibility of the target taxa to control techniques (Panetta 2015) or the biology of the species (Dana et al. 2019), reintroductions together with movement of individuals from within the invaded range into management areas, can also moderate management success (Williams and Grosholz 2008). While tracking such movement can be challenging (especially in aquatic systems), this study used genetic approaches to investigate the role it may have played in a management trial that was unsuccessful at removing a population of Carcinus maenas. It seems likely that there have been multiple introductions from multiple ranges into South Africa, possibly earlier than previously reported. Thus, we would predict a high likelihood of re-colonisation even if removal was feasible. As such, eradication is unlikely to be achieved unless the vectors of this crab are correctly identified and managed. In addition, there seems to be extensive gene flow between the harbours. While it is not clear how frequently dispersal occurs, this suggests that any attempts at eradication would need to be concurrent between all invasive populations and that invasive populations are likely to establish in other harbours along the South African coast. In conclusion, understanding inter- and intra-regional dispersal of target alien species can offer invaluable insights into the feasibility of management actions.

Sequential ABC analyses conducted to assess the likely origin of South African Carcinus populations. Analysis 1: Scenario (1.1) A direct introduction from the native or invasive range of C. maenas to South Africa; Scenario (1.2) A direct introduction from the native or invasive range of C. aestuarii; Scenario (1.3) An admixture event between C. maenas and C. aestuarii leads to the founding of the South African population. DIYABC output: Posterior probability comparison using a logistic regression approach identifies Scenario 1.1 as the most parsimonious. Analysis 2: Scenario (2.1) Direct introduction from the native range of C. maenas to South Africa; Scenario (2.2) Direct introduction from the invasive range of C. maenas to South Africa; Scenario (2.3) The South African population formed as a result of admixture between native and globally invasive C. maenas; Scenario (2.4) A ghost population in the C. maenas native range founds the South African population; and Scenario (2.5) A ghost population in the C. maenas invasive range founds the South African population. DIYABC output: A comparison of the posterior probabilities inferred that Scenario 2.3 is the most likely. Analysis 3: The South African populations is the result of admixture between populations in the C. maenas native range and invasive populations from Scenario (3.1) Canada; Scenario (3.2) the USA; or Scenario (3.3) Australia. DIYABC output: Posterior probability comparison indicated that Scenario 3.1 is the most likely

a STRUCTURE barplots where vertical axes illustrate the proportional assignment of individual genomes to the inferred genetic clusters where K = 4 for global Carcinus populations sampled, including both native and invasive regions. This analysis included South African samples collected in 2014, 2015 and 2016. b Haplotype network based on the mtDNA sequences analysed using POPART (including samples from 2013). The size of circles indicates the number of samples with those haplotypes. Lengths of connections are not to scale. C.m_native = C. maenas native range populations; C.a_native = C. aestuarii native range populations

Temporal changes in a allelic richness (AR) and b the number of private alleles (PA) observed in the South African Carcinus populations. There were no significant differences (p > 0.05)

Temporal changes (from 2014–2016) to Bayesian assignment values in the Hout Bay and Table Bay populations with respect to four identified genetic clusters: a q1 (predominantly Australian cluster); b q2 (C. aestuarii cluster); c q3 (North American cluster); and d q4 (C. maenas native cluster)
Data Availability
(all data will be deposited into the Dryad online repository upon acceptance of the manuscript)
Code Availability
Not applicable.
References
Abdelkrim J, Pascal M, Samadi S (2007) Establishing causes of eradication failure based on genetics: case study of ship rat eradication in Ste. Anne archipelago. Conserv Biol 21:719–730
Awad AA, Griffiths CL, Turpie JK (2002) Distribution of South African marine benthic invertebrates applied to the selection of priority conservation areas. Divers Distrib 8:129–145
Beaumont MA, Zhang W, Balding DJ (2002) Approximate Bayesian computation in population genetics. Genetics 162:2025–2035
Behrens Yamada S (2001) Global invader: the European green crab Oregon Sea Grant, Oregon State University. 123 pp
Blakeslee AM, McKenzie CH, Darling JA, Byers JE, Pringle JM, Roman J (2010) A hitchhiker’s guide to the maritimes: anthropogenic transport facilitates long-distance dispersal of an invasive marine crab to Newfoundland. Divers Distrib 16:879–891
Boissin E, Hurley B, Wingfield MJ, Vasaitis R, Stenlid J, Davis C, De Groot P, Ahumada R, Carnegie A, Goldarazena A, Klasmer P (2012) Retracing the routes of introduction of invasive species: the case of the Sirex noctilio woodwasp. Mol Ecol 21:5728–5744
Brasseale E, Grason EW, McDonald PS, Adams J, MacCready P (2019) Larval transport modeling support for identifying population sources of european green crab in the Salish Sea. Estuaries Coasts 42:1586–1599
Carlton JT, Cohen AN (2003) Episodic global dispersal in shallow water marine organisms: the case history of the European shore crabs Carcinus maenas and C. aestuarii. J Biogeogr 30:1809–1820
Castillo ML, Schaffner U, Van Wilgen BW, Montaño NM, Bustamante RO, Cosacov A, Mathese MJ, Le Roux JJ (2021) Genetic insights into the globally invasive and taxonomically problematic tree genus Prosopis. AoB Plants 13:plaa069
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chown SL, Hodgins KA, Griffin PC, Oakeshott JG, Byrne M, Hoffmann AA (2015) Biological invasions, climate change and genomics. Evol Appl 8:23–46
Clement M, Posada D, Crandall K (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 10:1657–1660
Cohen AN, Carlton JT, Fountain MC (1995) Introduction, dispersal and potential impacts of the green crab Carcinus maenas in San Francisco Bay, California. Mar Biol 122:225–237
Cornuet JM, Santos F, Beaumont MA, Robert CP, Marin JM, Balding DJ, Guillemaud T, Estoup A (2008) Inferring population history with DIY ABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics 24:2713–2719
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin JM, Estoup A (2014) DIYABC v2. 0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30:1187–1189
Dallas JF (1992) Estimation of microsatellite mutation rates in recombinant inbred strains of mouse. Mamm Genome 3:452–456
Dana ED, Garcia-de-Lomas J, Verloove F, Vila M (2019) Common deficiencies of actions for managing invasive alien species: a decision-support checklist. NeoBiota 48:97–112
Darling JA (2011) Interspecific hybridization and mitochondrial introgression in invasive Carcinus shore crabs. PLoS One 6:e17828
Darling JA, Bagley MJ, Roman J, Tepolt CK, Gellers JB (2008) Genetic patterns across multiple introductions of the globally invasive crab genus Carcinus. Mol Ecol 17:4992–5007
Darling JA, Tsai YH, Blakeslee AM, Roman J (2014) Are genes faster than crabs? Mitochondrial introgression exceeds larval dispersal during population expansion of the invasive crab Carcinus maenas. Royal Soc open Sci 1:140202
Dlugosch KM, Parker I (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Doyle J, Doyle J (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Earl DA, Von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Echols A, Front A, Cummins J (2019) Broadening conservation funding. Wildl Soc Bull 43:372–381
Ellegren H (1995) Mutation rates at porcine microsatellite loci. Mamm Genome 6:376–377
Ens NJ, Harvey B, Davies MM, Thomson HM, Meyers KJ, Yakimishyn J, Lee LC, McCord ME, Gerwing TG (2022) The green wave: reviewing the environmental impacts of the invasive European green crab (Carcinus maenas) and potential management approaches. Environ Rev 30: 306–322.
Evanno F, Regneut S, Groudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Faulkner KT, Robertson MP, Rouget M, Wilson JRU (2017) Prioritising surveillance for alien organisms transported as stowaways on ships travelling to South Africa. PLoS One 12:e0173340. https://doi.org/10.1371/journal.pone.0173340
Geller JB, Walton ED, Grosholz ED, Ruiz GM (1997) Cryptic invasion of the crab Carcinus detected by molecular phylogeny. Mol Ecol 6:901–906
Goldstein DB, Linares AR, Cavalli-Sforza LL, Feldman MW (1995) An evaluation of genetic distances for use with microsatellite loci. Genetics 139:463–471
Griffiths CL, Mead A, Robinson TB (2009) A brief history of marine bio-invasions in South Africa. Afr Zool 44:241–247
Grosholz ED, Ruiz GM, Dean CA, Shirley KA, Maron JL, Connors PG (2000) The impacts of nonindigenous marine predator in California bay. Ecology 81:1206–1224
Hampton S, Griffiths CL (2007) Why Carcinus maenas cannot get a grip on South Africa’s wave exposed coastline. Afr J Mar Sci 9:123–126
Hampton JO, Spencer PBS, Alpers DL, Twigg LE, Woolnough AP, Doust J, Higgs T, Pluske J (2004) Molecular techniques, wildlife management and the importance of genetic population structure and dispersal: a case study with feral pigs. J Appl Ecol 41:735–743
Hassan M, Harmelin-Vivien M, Bonhomme F (2003) Lessepsian invasion without bottleneck: example of two rabbitfish species (Siganus rivulatus and Siganus luridus). J Exp Mar Biol Ecol 291:219–232
Hauser L, Carvalho GR (2008) Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fish 9:333–362
Hewitt CL, Campbell ML (2007) Mechanisms for the prevention of marine bioinvasions for better biosecurity. Mar Pollut Bull 55:395–401
Holland BS (2000) Genetics of marine bioinvasions. Hydrobiologia 420:63–71
Holland BS (2001) Invasion without a bottleneck: microsatellite variation in natural and invasive populations of the brown mussel Perna perna (L). Mar Biotechnol 3:407–415
Hui TY(2014) NB: Maximum Likelihood method in estimating effective population size from genetic data.R package version0.9
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Johnston EL, Piola RF, Clark GF (2009) The role of propagule pressure in invasion success. In: Rilov G, Crooks JA (eds) Biological invasions in marine ecosystems. Ecological Studies (Analysis and Synthesis), vol 204. Springer, Berlin, Heidelberg, pp 133–151
Jooste M (2013) The genetic status, spread and population size of the invasive crab, Carcinus, in South Africa. Dissertation. Stellenbosch University
Joska MA, Branch GM (1986) The European shore crab – another invader? Afr Wildl 40:63–64
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788
Le Roux JJ (2021) The Evolutionary Ecology of Invasive Species. Elsevier Chap 8, p 165
Le Roux JJ, Wieczorek AM (2009) Molecular systematics and population genetics of biological invasions: towards a better understanding of invasive species management. Ann Appl Biol 154:1–17
Le Roux JJ, Wieczorek AM, Meyer JY (2008) Genetic diversity and structure of the invasive tree Miconia calvescens in Pacific islands. Divers Distrib 14:935–948
Le Roux JJ, Wieczorek AM, Tran CT, Vorsino AE (2010) Disentangling the dynamics of invasive fireweed (Senecio madagascariensis Poir. species complex) in the Hawaiian Islands. Biol Invasions 12:1–16
Le Roux JJ, Foxcroft LC, Herbst M, MacFadyen S (2015) Genetic analysis shows low levels of hybridization between African wildcats (Felis silvestris lybica) and domestic cats (F. s. catus) in South Africa. Ecol Evol 5:288–299
Leigh JW, Bryant (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Loureiro TG, Peters K, Robinson TB (2021) Dropping plates to pick up aliens: towards a standardised approach for monitoring alien fouling species. Afr J Mar Sci 43:1–15
Mabin CA (2018) Carcinus maenas in South Africa: status, impacts and management. Dissertation. Stellenbosch University
Mabin CA, Wilson JRU, Le Roux JJ, Robinson TB (2017) Reassessing the invasion of South African waters by the European shore-crab Carcinus maenas. Afr J Mar Sci 39:259–267
Mabin CA, Wilson JR, Le Roux JJ, Majiedt P, Robinson TB (2020) The first management of a marine invader in Africa: the importance of trials prior to setting long-term management goals. J Environ Manag 261:110213
Mairal M, Chown SL, Shaw J, Chala D, Chau JH, Hui C, Kalwij JM, Münzbergová Z, van Vuuren BJ, Le Roux JJ (2021) Human activity strongly influences genetic dynamics of the most widespread invasive plant in the sub-Antarctic. https://doi.org/10.1111/mec.16045. Molecular Ecology Accepted Author Manuscript
Marino IA, Barbisan F, Gennari M, Bisol P, Zane L (2008) PERMANENT GENETIC RESOURCES: isolation and characterization of microsatellite loci in the Mediterranean shore crab Carcinus aestuarii (Decapoda, Portunidae). Mol Ecol Resour 8:370–372
Matheson K, McKenzie CH, Gregory RS, Robichaud DA, Bradbury IR, Snelgrove PV, Rose GA (2016) Linking eelgrass decline and impacts on associated fish communities to European green crab Carcinus maenas invasion. Mar Ecol Prog Ser 548:31–45
Mead A, Carlton JT, Griffiths CL, Rius M (2011) Revealing the scale of marine bio-invasions in developing regions: a South African re-assessment. Biol Invasions 13:1991–1200
Medcof JC, Dickie LM (1955) Watch for the green crab - a new clam enemy. Fisheries Res Board Can 26:316–320
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York, p 512
Nelson G, Polito A (1987) Information on currents in the peninsula area, South Africa. In: Payne AIL, Gulland JA, Brink KH (eds) The Benguela and Comparable Ecosystems. South Afr J Marine Sci 5: 287–304
Ort BS, Thornton WJ (2016) Changes in the population genetics of an invasive Spartina after 10 years of management. Biol Invasions 18:2267–2281
Panetta FD (2015) Weed eradication feasibility: lessons of the 21st century. Weed Res 55:226–238
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research. Bioinformatics 28:2537–2539
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:170–181
Queiroga H (1996) Distribution and drift of the crab Carcinus maenas (L.) (Decapoda, Portunidae) larvae over the continental shelf off northern Portugal in April 1991. J Plankton Res 18:1981–2000
Quesada H, Beynon CM, Skibinski DO (1995) A mitochondrial DNA discontinuity in the mussel Mytilus galloprovincialis Lmk: pleistocene vicariance biogeography and secondary intergradation. Mol Biol Evol 12:521–524
R core team (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
Rambaud A(2016) FigTree version 1.4.3, Edinburgh, United Kingdom. http://tree.bio.ed.ac.uk/
Richardson DM (1998) Forestry trees as invasive aliens. Conserv Biol 12:18–26
Rius M, Darling JA (2014) How important is intraspecific genetic admixture to the success of colonising populations? Trends in Ecology and Evolution 29:233–242
Rius M, Turon X, Ordóñez V, Pascual M (2012) Tracking invasion histories in the sea: facing complex scenarios using multilocus data. PLoS One 7:e35815
Robinson TB, Griffiths CL, McQuaid CD, Andrius M (2005) Marine alien species of South Africa — status and impacts. Afr J Mar Sci 27:297–306
Roman J (2006) Diluting the founder effect: cryptic invasions expand a marine invader’s range Proceedings of the Royal Society of London B: Biological Sciences 273: 2453–2459
Rong BC, Seiichi W, Masashi Y (2004) Feeding habits of an exotic species the Mediterranean green crab Carcinus aestuarii in Tokyo Bay. Fisheries Sci 70(3) 430–435 https://doi.org/10.1111/j.1444-2906.2004.00822.x
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Saavedra C, Zapata C, Alvarez G (1995) Geographical patterns of variability at allozyme loci in the European oyster Ostrea edulis. Mar Biol 122:95–104
Sakai T (1986) Rare species and their genus of crabs in Japan. Res Crustacea 15:1–4
Schlötterer C, Ritter R, Harr B, Brem G (1998) High mutation rate of a long microsatellite allele in Drosophila melanogaster provides evidence for allele-specific mutation rates. Mol Biol Evol 15:1269–1274
Sherpa S, Després L(2021) The evolutionary dynamics of biological invasions: a multi-approach perspective. Evolutionary Applications. DOI: https://doi.org/10.1111/eva.13215. In Press
Stone GN, White SC, Csóka G, Melika G, Mutun S, Pénzes Z, Sadeghi SE, Schönrogge K, Tavavkoli M, Nicholls JA (2017) Tournament ABC analysis of the western Palaearctic population history of an oak gall wasp, Synergus umbraculus. Molecural Ecol 26:6685–6703
Tepolt CK, Bagley MJ, Geller JB, Blum MJ (2006) Characterization of microsatellite loci in the European green crab (Carcinus maenas). Mol Ecol Resour 6:343–345
Tepolt CK, Darling JA, Bagley MJ, Geller JB, Blum MJ, Grosholz ED (2009) European green crabs (Carcinus maenas) in the northeastern Pacific: genetic evidence for high population connectivity and current-mediated expansion from a single introduced source population. Divers Distrib 15:997–1009
Tintore JP, Laviolette E, Blade I, Cruzado A (1988) A study of an intense density front in the eastern Alboran sea: the Almeria-Oran front. J Phys Oceanogr 18:1384–1397
Vardien W, Richardson DM, Foxcroft LC, Wilson JRU, Le Roux JJ (2013) Management history determines gene flow in a prominent invader. Ecography 36:1032–1041
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Wauters N, Dekoninck W, Fournier D (2018) Introduction history and genetic diversity of the invasive ant Solenopsis germinata in the Galápagos Islands. Biol Invasions 20:3207–3226
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Williams SL, Grosholz ED (2008) The invasive species challenge in estuarine and coastal environments: marrying management and science. Estuaries Coasts 31:3–20
Wilson JR, Panetta FD, Lindgren C (2017) Detecting and responding to alien plant incursions. Cambridge University Press, Cambridge p, p 286
Witte S, Buschbaum C, van Beusekom JE, Reise K (2010) Does climatic warming explain why an introduced barnacle finally takes over after a lag of more than 50 years? Biol Invasions 12:3579–3589
William CW, Craig M, Laura FR, Craig P, Gregory R (2002) Effect of an invasive crab upon a marine fishery: green crab Carcinus maenas predation upon a venerid clam Katelysia scalarina in Tasmania (Australia). J of Exp Mar Biol and Ecol 272(2) 171–189 https://doi.org/10.1016/S0022-0981(02)00127-2
Xu J, Wickramarathne TL, Chawla NV, Grey EK, Steinhaeuser K, Keller RP, Drake JM, Lodge DM(2014) Improving management of aquatic invasions by integrating shipping network, ecological, and environmental data: data mining for social good. Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining 2014 Aug 24 pp. 1699–1708
Zeller M, Reusch TB, Lampert W (2008) Small effective population sizes in two planktonic freshwater copepod species (Eudiaptomus) with apparently large census sizes. J Evol Biol 21:1755–1762
Acknowledgements
This study was made possible by funding from the then South African Department of Environmental Affairs through the South African National Biodiversity Institute (SANBI) Marine Programme. CM acknowledges the DSI-NRF Centre of Excellence for Invasion Biology for bursary support. JRUW thanks the South African Department of Forestry, Fisheries and the Environment (DFFE) for funding, noting that this publication does not necessarily represent the views or opinions of DFFE or its employees. Several international researchers assisted with the collection of genetic samples, including Dr Ilaria Marino, Julie Gonzalez, Kiley Best, Dr Danny O’Neill, Dr Antony Knight, Prof. Henrique Queiroga, Dr Brett Howard, Dr Masashi Yokota and Rene Campbell. This work would not have been possible without the assistance of many in the Molecular Ecology laboratory with special thanks owed to Megan Mathese and Dr Jan-Hendrik Keet. MJ would like to thank Prof. Sophie von der Heyden for supervising the genetic analysis of data for her Honours thesis. JLR acknowledges support from ProBraaiTM.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This study was funded by several organisations, including the then South African Department of Environmental Affairs through the South African National Biodiversity Institute (SANBI) Marine Programme and the South African Department of Forestry, Fisheries and the Environment (DFFE). The DSI-NRF Centre of Excellence for Invasion Biology provided bursary support for the first author.
Author information
Authors and Affiliations
Contributions
Not applicable.
Corresponding author
Ethics declarations
Conflict of interest
Not applicable.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mabin, C.A., Robinson, T.B., Wilson, J.R.U. et al. Molecular insights into the invasion dynamics of Carcinus crabs in South Africa. Biol Invasions 24, 3597–3613 (2022). https://doi.org/10.1007/s10530-022-02865-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-022-02865-9