
Abstract
Background
COVID-19 has proved to be a fatal disease of the year 2020, due to which thousands of people globally have lost their lives, and still, the infection cases are at a high rate. Experimental studies suggested that SARS-CoV-2 interacts with various microorganisms, and this coinfection is accountable for the augmentation of infection severity.
Methods and results
In this study, we have designed a multi-pathogen vaccine by involving the immunogenic proteins from S. pneumonia, H. influenza, and M. tuberculosis, as they are dominantly associated with SARS-CoV-2. A total of 8 antigenic protein sequences were selected to predict B-cell, HTL, and CTL epitopes restricted to the most prevalent HLA alleles. The selected epitopes were antigenic, non-allergenic, and non-toxic and were linked with adjuvant and linkers to make the vaccine protein more immunogenic, stable, and flexible. The tertiary structure, Ramachandran plot, and discontinuous B-cell epitopes were predicted. Docking and MD simulation study has shown efficient binding of the chimeric vaccine with the TLR4 receptor.
Conclusion
The in silico immune simulation analysis has shown a high level of cytokines and IgG after a three-dose injection. Hence, this strategy could be a better way to decrease the disease's severity and could be used as a weapon to prevent this pandemic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The year 2020 has proved quite fatal due to the pandemic COVID-19 disease, caused by life-threatening SARS-CoV-2 (wild-type strain), severe acute respiratory syndrome coronavirus 2) of the Coronaviridae family (genus- Betacoronavirus), and is spreading its impression all over the world. Wuhan, China, reported the first case of this infectious disease, and then this deadly disease spread rapidly worldwide. As of September 2, 2022, ~ 608 million confirmed cases of COVID-19, out of which ~ 6.5 million deaths were reported by the World Health Organization (https://www.who.int/). The wild-type strain of SARS-CoV-2 majorly affects all age groups and, in severe cases, harshly infects the lungs, causing complications like pneumonia and ARDS (acute respiratory distress syndrome), and bronchitis necessitates hospitalization (Hu et al. 2021). Several national and international organizations are working together to develop therapeutics against this severe infection, and as of now, 11 vaccinations have received emergency authorization. These vaccines are produced using a variety of vaccine types, including protein subunit vaccines, RNA vaccines, vaccinations based on non-replicating viral vectors, and inactivated vaccines. Two protein subunit vaccines developed against COVID-19 are COVOVAX and Nuvaxovid by Novavax, produced by Serum Institute of India and Novavax, respectively. Compared to Nuvaxovid, which has been approved in 39 countries and has 20 trials across 13 nations, Covovax has four approved trials in two countries, and five approved this vaccine. The two most popular RNA vaccines are Spikevax by Moderna (authorized in 88 countries with 68 trials in 24 countries) and Comirnaty by Pfizer/BioNTech (approved in 149 countries with the approval of 90 trials in 29 countries) (https://covid19.trackvaccines.org/vaccines/).
Most COVID-19 vaccines are developed using non-replicating virus-based vaccines. Covishield (Oxford/AstraZeneca formulation) received permission in 40 countries with four trials in a single country. Three vaccines- Covaxin from Bharat Biotech, Covilo from Sinopharm (Beijing), and CoronaVac from Sinovac are listed as possessing emergency authorization approval under the COVID-19 vaccine category for inactivated vaccines. Covilo has been approved in 93 countries, with 38 trials throughout 16 countries. CoronaVac gets approval for trial in 10 countries with 40 trials and is currently being used in 56 countries, whereas Covaxin has been authorized in 14 countries with 14 trials in two counties (McGill-University 2022; WHO 2022).
COVID-19 and other respiratory ailments range from acute to chronic, instigated by various microorganisms. Recent studies demonstrated that SARS-CoV-2 infection is associated with many antibiotic resistance bacteria (Vaillancourt and Jorth 2020; Zhou et al. 2020). The rigorous usage of antibiotics throughout the SARS-CoV-2 pandemic led to upsurges in the pervasiveness of multidrug-resistant bacteria. Several antibiotic-resistant bacteria are known to be associated with SARS-CoV-2 patients, such as S. aureus, P. aeruginosa, Klebsiella spp., S. pneumonia, etc. (Zhou et al. 2020).
In a multicenter study of 476 patients (suffering from COVID-19) inclusive of three groups, such as moderately ill, severely ill, and critically ill patients, it was found that critically ill patients have the maximum number of bacterial coinfections associated with SARS-CoV-2 compared to severely and moderately ill patients (Feng et al. 2020). Another study of COVID-19 detection, using real-time PCR, suggested that 243 patients have shown coinfection with at least 1 of 39 different pathogens; among the bacterial, fungal, and viral coinfection, the former was leading (Zhu et al. 2020).
Wang et al. and colleagues reported that 5.8% of patients who tested positive for COVID-19 had been diagnosed with multiple pathogens coinfection (Massey et al. 2020). Later, Feldman et. al., in a review, concluded that bacterial coinfections (having pneumonia-like symptoms) and secondary infections associated with COVID-19-affected patients seem to be allied with the severity of COVID-19 infection and poor consequences (Feldman and Anderson 2021). Zhu et al. stated that 39 respiratory illness-related pathogens (bacteria, viruses, and fungus) were associated with SARS-CoV-2 infection in humans and are accountable for the augmentation of infection severity (Zhu et al. 2020). These microbes share common symptoms such as a runny nose, coughing, sneezing, fever, nasal and chest congestion, etc. The microbial coinfection of Streptococcus pneumonia, Hemophilus influenza, and Mycobacterium tuberculosis was predominantly associated with SARS-CoV-2, and coinfections primarily arose in 1–4 days of the beginning of COVID-19 (Zhu et al. 2020). These pathogens interact with each other in the nasopharynx to occupy the same habitat for their survival and spread the host's infection by developing the microbial community. These pathogenic organism spectacles the commensal relationship, in which one organism acquire benefits from the other without harming it. These commensal organisms' direct or indirect interaction helps each other penetrate the host cell to cause severe lower and upper respiratory tract infections (Bosch et al. 2013). Coinfection with antibiotic-resistant bacteria could make the COVID-19 treatment more challenging and result in septic shock in patients (Lai, et al. 2020a).
Some observational studies showed that the countries like India, with a compulsory BCG vaccination, had shown fewer critical COVID-19 cases than the BCG vaccine nonobligatory countries (TB non-endemic) like Europe and USA (Mohapatra et al. 2021). In South Africa, BCG vaccination (Bacilli Calmette-Guérin), an FDA-approved vaccine for M. tuberculosis, has been registered, is recruiting healthcare workers, and has successfully reached phase 3 of a clinical trial (ClinicalTrials.gov Identifier: NCT04379336). The in vivo studies have shown that BCG vaccination can provide non-specific protective effects against many other respiratory tract infections. It is believed that the BCG protective immunity in humans is mediated by heterologous activation of lymphocytes and hence induces the immune memory cells, which may be responsible for the clearance of SARS-CoV-2 (Mohapatra et al. 2021). Around 23 BCG vaccination trials against SARS-CoV-2 have been registered as per clinicaltrials.gov data (https://clinicaltrials.gov/ct2/results?cond=Covid19&term=BCG&cntry=&state=&city=&dist =) (ClinicalTrials.gov 2022). However, these studies demand substantial evidence to get a recommendation from the World Health Organization.
Vaccination is the only preventive measure as it has proved to be safer and more efficacious than therapeutic medicines and could prevent infections from spreading worldwide and provide long-lasting immunity. A conjugate multi-pathogen vaccine could be effective against this disastrous COVID-19 and its associated common pneumococcal coinfections. Here, in this study, we have framed a multi-pathogen-based multiepitope probable vaccine candidate composed of antigenic and highly pathogenic proteins from each pathogen, interacting with SARS-CoV-2; this microbial coinfection leads to the prognosis of the disease (Chen, et al. 2020; Ojha et al. 2019). The utilization of immunogenic epitopes (B-cell, HTL, and CTL) would generate probable cell-mediated and humoral immune responses. The involvement of HLA (Human leukocyte antigen) restricted T cell epitopes would be specific and cover the worldwide population. This strategy could be a better way to decrease the disease's severity and can be used as a weapon to prevent this pandemic.
Methodology
Investigation of multi-pathogen protein sequences for vaccine designing
Here, in this study, we have targeted four pathogens and two proteins from each different pathogen. Like, from the SARS-Cov-2- Spike glycoprotein (QIC53213.1) and Nucleocapsid protein (QIC53221.1) (Dutta et al. 2020), from Mycobacterium tuberculosis (H37Rv)- Rv2608 (PPE family protein PPE42_KBF82311 (Hatherill et al. 2020) and Rv1813 (NP_216329) (Coler, et al. 2018), from Streptococcus pneumonia- PhtD (pneumococcal histidine triad protein WP_061818224.1) and PCPA (choline-binding protein CAB04758.1) (Pichichero et al. 2016) whereas from Hemophilus influenzae type B- Protein D (AAA24998.1) (Forsgren and Riesbeck 2008) and Protein E (TWU89946.1) (Singh et al. 2010) were selected. These selected proteins have already been reported as potential vaccine candidates. Sequences of proteins were retrieved from NCBI (National Center for Biotechnology Information- https://www.ncbi.nlm.nih.gov/) database. Next, we looked into important parameters like antigenicity, allergenicity, and toxicity for the selected protein sequences with VaxiJen, AllerTop, and ToxinPred servers.
Prediction of immunogenic epitopes for multi-pathogen vaccine designing
B-cell epitopes prediction
B-cell epitopes are vital for instigating humoral immunity, i.e., secretion of antibodies; for this, they must be recognized by the B-cell receptors (BCR). Here, in this study, the prediction of immunogenic B-cell epitopes from the antigenic protein sequences was made with the assistance of the ABCpred server (http://crdd.osdd.net/raghava/abcpred/). The server's highest prediction accuracy means specificity and sensitivity to predict an epitope was the only rationale behind selecting the ABCPred server. This server utilizes the ANN (artificial neural network) and machine learning language. The server extracted the B-cell epitopes from BCIPEP (B-cell epitope database), and this data set consists of experimentally evidenced immunogenic (1617) and immunodominant (654) epitopes from a pathogenic microorganism (bacteria, virus, parasite, and fungi); the dataset later screened for immunogenic epitopes from the non-immunogenic ones (Saha and Raghava 2006).
HTL (Class II MHC molecules) epitopes prediction
T-lymphocytes are the dominant warriors involved in the induction of cell-mediated and humoral immune responses against pathogens. As the name suggests, Helper T-lymphocytes HTL signals the immune system's other cells to generate immune responses. To acquire the potential antigenic epitopes, we have utilized IEDB, the immune epitope database (https://www.iedb.org/) server, to predict MHC-II binding epitopes. This easily offered online server encompasses the data on experimentally approved immune epitopes of species like primates and humans about various infectious and inflammatory diseases (Fleri et al. 2017). The MHC-II binding prediction tool (epitope analysis resource) accepted all the selected sequences in FASTA format, and the prediction was made using the human allele reference data set.
CTL (Class I MHC molecules) epitopes prediction
The class I MHC molecules presented the antigens on their surface are responsible for activating cytotoxic T cells. To identify immunogenic cytotoxic T cell epitopes from the multi-pathogen protein sequences, we have utilized the NetCTL 1.2 (http://www.cbs.dtu.dk/services/NetCTL/) server. The reliability, specificity, and epitope prediction accuracy of this server are higher than others, and it is so because of TAP transport efficiency, MHC class I affinity, and proteasomal cleavage (Larsen et al. 2007). Apart from these abilities, the server consists of 12 standard HLA (Human leukocyte antigen) supertypes associated with the potent peptide binding specificities distributed widely among the world population. All the parameters at the time of epitope prediction were set as default except supertypes.
Antigenicity, allergenicity, and toxicity analysis of elected epitopes
Solitary epitope prediction is not sufficient for vaccine designing; they should be antigenic enough to bind with the immune cell receptors (TCRs and BCRs) to engender specific immune responses. The antigenicity of each sorted epitope was predicted via the VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) server. This server is based on ACC (auto cross-covariance). It is also an advanced platform for predicting protective antigens from the various target organisms, scilicet viruses, bacteria, parasites, tumors, parasites, and fungi compared to the sequence alignment method (Doytchinova and D.R.J.B.b. Flower 2007).
Our next parameter was allergenicity and Toxicity prediction of each antigenic epitope, which was done individually with AllerTop and ToxinPred servers' aid. AllerTop (https://www.ddg-pharmfac.net/AllerTOP/) prediction tool is also based on ACC (auto cross-covariance) and applied the k-nearest neighbor algorithm to finalize the output from the training dataset, which consists of 2427 allergens and the same number of non-allergens from many species (Dimitrov et al. 2014). The ToxinPred (http://crdd.osdd.net/raghava/toxinpred/) server scans the entire protein to identify the non-toxic and toxic portion of the same function for the peptides (Gupta et al. 2013).
Framing of multi-pathogen-based multiepitope vaccine candidate with optimal TLR4 agonist as adjuvant
After sorting and analyzing antigenic B-cell, HTL, and CTL epitopes from the pathogenic protein sequences, the epitopes were linked with each other with the linkers' help. KK (Lys-Lys), AAY (Ala-Ala), and GPGPG (Gly-Pro-Gly-Pro-Gly) spacer sequences were exploited to fuse the B-cell, HTL, and CTL epitopes, respectively. For the vaccine to drive competently by generating a robust immune response, we have added adjuvants (TLR-4 agonist) to the N-terminal part of the designed vaccine protein sequences. In this study, three optimal TLR-4 agonist adjuvants were selected, namely TR-433 (TLR4 derived peptide adjuvant ranges from 433 to 452 amino acid residues), 50S ribosomal (Accession no.- P9WHE3), and HABA (Heparin-binding hemagglutinin; Accession no.- AGV15514.1). The details have been discussed in the result section.
Evaluation of antigenic vaccine candidate factors and physiochemical property assessment
Valuation of various factors such as antigenicity, allergenicity, and toxicity of the framed vaccine candidate was done with the same servers: VaxiJen, AllerTop, and ToxinPred, respectively (the details of the servers have already been discussed). Physicochemical property assessment of the vaccine candidates was achieved with the ProtParam (https://web.expasy.org/protparam/) server (Gasteiger 2005). The ExPASy web tool calculates the various protein parameters, for instance, molecular weight, theoretical pI, instability index, extinction coefficient, GRAVY, aliphatic index, and so forth.
Tertiary structure prediction of vaccine candidates and the validation described above
For the prediction of tertiary structure, Robetta- The Baker lab server (https://robetta.bakerlab.org/) was utilized and uninterruptedly evaluated by CAMEO (continuous automated model evaluation) for the assessment of the predictions. The server can perform comparative modeling (if the input protein sequence has homologs), ab initio modeling (when the input protein sequence has no homologs), and domain prediction in which each protein domain can be modeled independently. Validation of the predicted tertiary structure was shown in the Ramachandran plot, and the same was predicted by the RAMPAGE server (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php). This server defines the input tertiary model as allowed, favored, and disallowed regions. The regions were predicted by analyzing the rotations of phi and psi angles (Lovell et al. 2003).
Molecular docking analysis of vaccine candidate and TLR4 receptor
The ClusPro server (https://cluspro.org/login.php) was used to perform the molecular docking between the vaccine construct and the TLR4 receptor (Chouhan et al. 2022). This server performs the rigid docking, RMSD clustering, and energy minimization of the docking complex by generating dozens of confirmations with the lowest energy. The server utilized a Fast Fourier Transform approach-based PIPER program to perform the rigid docking between the protein and ligand. The server selected the models with the best desolvation and electrostatic free energies for the clustering process by the server, and the finest docked complex was selected based on cluster members and the lowest energy score (Kozakov et al. 2017; Sharma et al. 2022).
Immune simulation study of the designed vaccine candidate
The in silico immune stimulation is vital in studying the immune system response generated by proposed vaccine candidates. For evaluating the immune reaction profile generated from vaccine protein in this study, we have utilized the C-ImmSim server (https:/kraken.iac.rm.cnr.it/C-IMMSIM/server). This server is an agent-based immune simulator and is freely accessible to all users. The C-ImmSim simulator utilizes the position to define the scoring matrix (PSSM) and machine learning techniques to predict immune responses (Rapin et al. 2010). Three injections for the designed vaccine profile for a preventative multi-pathogen vaccine are simulated at intervals of 4 weeks.
Consequently, vaccine construct injections have been tested with conserved HLA alleles at four weeks and a time stage of 1, 84, and 168 (each step equals eight real-life hours, and phase 1 injection at time = 0). The volume and steps for simulation were set to 100 and 1050. Simulation parameters were set by default (random seed = 12,345, what to inject = vaccine (no LPS), adjuvants = 100, and amount Ag to inject = 1000).
Molecular dynamics studies of designed vaccine candidate and TLR-4 receptor
Molecular dynamics simulation was usually performed to analyze the stability of the complex macromolecules. Here in this study, the protein complex (vaccine protein and TLR4 immune receptor) simulation was conducted with the GROMCS (GROningen MAchine for Chemical Dynamics) package by using the Gromos96 43a1 force field (Abraham et al. 2015). Next, the topology generation was achieved, which is the foremost step, as this contains all the information related to nonbonded and bonded parameters essential to characterize the docked complex inside a simulation. A Rhombic dodecahedron boundary box in the system was used to mimic the natural environment along with a protein complex, water (solvent; SPC water model), and ions added (Singh 2022; Singh and Prajapati 2022). Further, the complex was energy minimized to reduce steric clashes and weak interactions. Later, the system was stabilized with NVT and NPT at 100 ps time frames. Finally, the MD simulation was carried out for 100 ns to calculate RMSD (Root Mean Square Deviation) and RMSF (Root Mean Square Fluctuation) for the protein backbone and side chains, respectively (Singh 2022; Singh and Prajapati 2022).
Result and discussion
Selection of pathogens and their antigenic protein sequences
Recently, it was reported that the diagnostic samples taken from COVID-19-infected patients had shown the presence of a variety of microorganisms, and this coinfection is directly or indirectly involved in the augmentation of disease severity. The highest coinfection rates were seen in people aged 15–44, whereas people under 15 had the lowest number of coinfection cases (Zhu et al. 2020). Due to the unavailability of precise treatment, in this study, we have designed the multi-pathogen-based multiepitope vaccine by using pathogens that have some physiobiological interaction with SARS-CoV-2 and somewhat resemblance in the symptoms with each other. The pathogens and proteins selected were identified through a literature survey and have already been reported as promising vaccine candidates. The role of each protein has been mentioned in Fig. 1.

Diagrammatic representation of multiple pathogens involved in SARS-CoV-2 coinfection along with the role of selected proteins used for multiepitope vaccine designing
Prediction of immunogenic epitopes-B-cell, HTL, and CTL
Continuous B-cell epitopes from the ABCPred server were predicted and selected based on their highest score. The higher score and rank of the epitopes represent the probability of an epitope being antigenic. Hence, all the selected protein sequences were submitted to the server for obtaining the probable B-cell epitopes. One B-cell epitope was selected from each protein sequence; hence, this study chose eight top antigenic epitopes based on the highest score obtained (Table 1).
Further, all the sequences were subjected to the IEDB server individually to predict the HTL epitopes. The lowest percentile rank denotes that the epitope will have the highest binding affinity with the MHC class II molecules to generate robust immune responses. For each epitope, IC50 (inhibitory concentration) value lower than 50 was selected. The subsequent norm was the allele selection, and the allele-specific epitopes will be beneficial in developing a highly polymorphic vaccine (Table 2).
The binding and presentation of antigen on MHC molecules are diverse for each antigen, and each allele defines the expression of MHC class I and II molecules differently; therefore, in this study, those types of HLA alleles were selected which have the highest allelic frequency and covers approx. 90% of the world population is at broad-spectrum, and the same was achieved with the help of the Allele frequency database (http://www.allelefrequencies.net/hla6006a.asp) (Fig. 2) (Wang et al. 2010).

Graphical presentation of HLA with the geographical region and allelic frequency selected for the prediction of HTL epitopes
MHC class I CTL epitopes were predicted via NetCTL server for all the protein sequences, and this was done by taking A2, A3, and B7 supertypes into contemplation, because the HLA supertype restricted epitopes obtained by using this will mutually cover a broad range of human population (nearly 86%) (Vejbaesya et al. 2015). After the supertype selection, the epitopes were sorted based on their combined score, and the highest score denotes the excellent binding efficiency of epitopes (Sharma, et al. 2022) (Table 3).
Antigenicity, allergenicity, and toxicity analysis of chosen epitopes
The analysis of allergenicity, antigenicity, and toxicity are the crucial aspects of examining the epitope eminence, that is, whether the selected epitopes would be able to generate a tremendous immune response or not against the pathogen. Following this, the antigenicity of each epitope (B-cell, HTL, and CTL) was predicted by VaxiJen by setting the threshold filter as 0.4 (default). The result showed that all the epitopes were probable antigenic, with scores between 0.5 and 1. Allergenicity prediction (via AllerTop) affirmed that all epitopes were probable non-allergen; the antigen would not induce inflammatory responses or hypersensitivity reactions inside the host. The final aspect, toxicity prediction of each epitope, concluded that all the selected epitopes were non-toxic (Tables 1, 2, and 3).
Multi-pathogen-based multiepitope vaccine candidate framing with optimal TLR4 agonist adjuvant
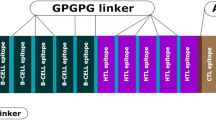
All the carefully chosen epitopes were linked with each other with the help of spacer sequences or linkers. Apposite linkers are essential for the fusion of epitopes and provide flexibility and stability to the protein sequences. For the multiepitope vaccine design, the linkers should be insusceptible to the degradation via host proteases, as sometimes they can be proved as the target for degradation. Here, in this study, we have utilized KK (for B-cell epitopes), GPGPG (for HTL epitopes), and AAY spacer sequences (for CTL epitopes) (Yano et al. 2005; Livingston et al. 2002; Ojha et al. 2020).
Epitopes alone could not be sufficient for initiating likely immune responses, so we have added an adjuvant to the designed vaccine candidate (Fig. 3 A). TLR4 is mainly responsible for mediating the immune response against bacterial pathogens by recognizing LPS (Lipopolysaccharide). However, when we designed this study and looked at the literature, we found that the SARS-CoV-2 infection triggered an anti-bacterial-like response at the early stage of infection via the TLR4 receptor (Zhao et al. 2021). It has also been reported that the spike protein of SARS-CoV 2 is mainly involved in initiating the TLR 4 signaling (Choudhury and Mukherjee 2020). Here, in this study, for the generation of a robust immune response, we have exploited three different TLR4 agonists as adjuvants for framing the vaccine construct- (1) TR-433, a synthetic and non-toxic adjuvant, possesses pro-inflammatory and self-assembling characteristics (Tandon et al. 2018). (2) The second adjuvant was from Mycobacterium tuberculosis (Rv0652); that is, 50S ribosomal L7/L12 protein, a natural adjuvant, has been shown to induce dendritic cell maturation, which activates the naïve T cells to instigate cell-mediated immune responses. The adjuvant is also known to enhance Tumor Necrotic Factor-alpha production, interleukin-1beta, and interleukin-6 pro-inflammatory cytokines (Lee et al. 2014). (3) The last one was HABA, a natural adjuvant from M. tuberculosis, which is proficient enough to activate the TLR4 signaling, which activates the CD4 + T-cells (Hasan et al. 2019; Gupta et al. 2020). These selected adjuvants were linked with the help of the EAAAK linker, a helix forming linker, and a total of six vaccine candidates were designed based on epitopes rearrangement; the details of each has given in the supplementary file as supplementary Fig. 1 (VC-1–6).

A Designed vaccine construct arrangement in order of pathogens and their selected proteins. B Tertiary structure modeling. 3D model prediction with the help of Robetta server (A) showing, Vaccine candidate 2 (blue color) and C Vaccine candidate 5 (red color)
Assessment of physicochemical properties and other aspects of designed exogenous vaccine candidates
The designed vaccine candidates were assessed for their physicochemical parameters (evaluating the characteristics of protein-based in vitro or in vivo environment), antigenicity, and allergenicity. Out of 6 designed vaccine constructs, we have filtered out 2 VC (vaccine candidates) that are VC2 and VC5 (having 50S ribosomal L7/L12 adjuvant) based on various parameters such as antigenicity, half-life, instability index, and aliphatic index, etc. (Supplementary Table 1). Construct 2 and 5 showed similarity between various physiochemical parameters (Supplementary Table 2), so we have performed 3D modeling followed by discontinuous B-cell epitope screening for both.
Tertiary structure prediction validation and refinement of the vaccine candidate
Tertiary structure prediction for the selected VC 2 and VC 5 was made with the Robetta server's help. By analyzing the data, the confidence score (GDT score) for both the predicted models was found to be the same, i.e., 0.11; hence, it denotes that the predicted 3D model has average model quality (Fig. 3 B and C). Further, with the help of Ramachandran plot assessment, we have found that VC2 has 98.6% of residues in the favored regions, whereas 1.2% (8) and 0.2% (1) residues were suspected in allowed and outlier regions, respectively. However, the Ramachandran plot for VC5 suggested that 98.8% (653) of residues were found to be in the favored region, and 1.1% (7) were in allowed regions, whereas 0.2% (1) of residues appeared in the outlier region (Supplementary Fig. 2).
Additionally, with the DiscoTope server's help (http://www.cbs.dtu.dk/services/DiscoTope/) (Haste Andersen et al. 2006), the discontinuous B-cell epitopes were predicted for the two selected vaccine candidates as this would help in the generation of the specific antibodies. This comparative analysis found that VC5 has a higher propensity (78 B-cell epitopes among 653 residues) for the generation of antibodies compared to VC2 (72 B-cell epitopes among 653 residues). Considering the Ramachandran plot and discontinuous B-cell epitopes result, it was concluded that VC5 was the finest and could be used for further study.
Protein–Protein interaction between vaccine candidate and TLR4 receptor
In this multi-pathogen vaccine study, we have exploited key receptor TLR4, which has been triggered by LPS (lipopolysaccharides) in the case of infectious diseases (bacterial and viral) and leads to the induction of inflammatory and immune responses by the secretion of various cytokines (Duthie et al. 2011). By performing the molecular docking of vaccine candidates with the TLR4 receptor, a total of 30 models were generated, along with their energy scores. With the help of obtained data, it was found that among 30 models, cluster_23 has a significantly lower binding energy score, i.e., − 1143.8 concerning the higher number of interacting residues between VC and TLR-4, having cluster size 10, and center energy -1138.1; henceforth appraised as the best-docked complex. The interaction of amino acid residues between the TLR4 and VC was generated with the help of the PDBSUM server (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html) (Fig. 4). This interaction map found that Chains B and D of TLR-4 positively and efficiently interacted with the designed VC5. Finally, the Prodigy server calculates the binding affinity (ΔG) and dissociation constant (Kd) between the VC5 and Chain B and Chain D of TLR4 receptor to be − 19.2 kcal mol-1 and 7.7E-15 M at 25.0 °C.

Docked complex analysis. A Vaccine candidate (VC) and TLR4 docked complex, Chain 1 representing the VC (red), whereas Chain A (purple), B (yellow), C (blue), and D (cyan) represent the chains of TLR4 receptor. The red and yellow globular dots show the interface or interacting residues between chain 1 of VC and chain B of TLR4 receptor, respectively. In contrast, correspondingly, blue and cyan color globular dots represent the interacting residues between chain 1 of VC and chain D of TLR4 receptor. B showing the types of interaction between the VC and chains of TLR4 receptor. C Interacting residues and types of interaction between VC (chain 1) and TLR4 (chain B), whereas D show the interacting residues between chain 1 of VC and chain D of TLR4 receptor
Immune simulation to check the immune responses obtained by the designed vaccine candidate
Results section
The immune simulation study was performed using the C-ImmSim server, which provides a possible map for the immune response upon immunization. The immune response graph plots were obtained using the server's default settings. It was observed that the vaccine candidate could elicit the B and T cell response. The simulation results have been obtained upon three injections of the vaccine candidates and the data observed concludes that every injection potentially elicited the immune response. Also, the secondary and tertiary immune response was greater than the primary immune response. Because an increased level of IgM + IgG, IgM, IgG1 + IgG1, and IgG2 populations/peaks reflected the presence of a secondary response, all of which appear to peak in the middle of 50–100 days at high titer (Fig. 5A). In the case of the humoral immune response, the B cell population was also at a peak between 50 and 100 days (Fig. 5B&C). Likewise, T-cell-mediated immunity, i.e., Tc and Th cells, was also higher at 30–100 days. The IFN‐γ level was high and almost similar at all exposures. This tells us that the vaccine candidate developed in this study elicited a robust immune response at first injection, which was consequently increased (Shepard et al. 1978; Rajput, et al. 2021; Singh, et al. 2020) (Fig. 5).

In silico immune simulation response predicted after three consecutive doses of injections of the proposed vaccine as an antigen. A Antigen and Immunoglobins, B B-cell population, C B-cell population per state, D Helper T-cell population per state, E Helper T-cell population, F Cytotoxic T-cell population, G Cytotoxic T-cell population per state, H Macrophages population per state, I Dendritic cell population per state, and J Cytokine production
A molecular dynamics simulation study of the docked complex
To check the docked complex's stability (vaccine protein and TLR4 receptor) obtained from ClusPro was achieved by performing the GROMACS MD simulation. In the initial step, the docked complex's energy minimization was performed, and the obtained average potential energy of the complex was − 1.12333e + 07, which denotes that the complex was energetically minimized and ready for the MD simulation. Next, the solvent and ions surrounding the protein must be equilibrated to begin the simulation. The two-step equilibration process was achieved by calculating the NVT (constant Number Volume Temperature) at 300 K temperature and NPT (constant Number Pressure Temperature) at a constant 1 bar pressure 100-ps. Both NVT and NPT plots represent that the system was stable throughout the MD simulation. Afterward, MD simulation was executed at 100 ns, and the RMSD and RMSF plots were generated concerning the protein backbone. The RMSD plot (Fig. 6A) suggested that the docked complex was stabilized after 40 ns, whereas RMS fluctuation (Fig. 6B) of the residues was higher at 160, 325, and 600 residue positions, which denotes the presence of flexible regions. With this, it can be concluded that the docked complex was stable, flexible, and had fewer deformed regions.

Molecular dynamics simulation between vaccine protein and immune receptor TLR4 obtained via GROMACS. A RMSD (Root Mean Square Deviation) for 100 ns and B RMSF (Root Mean Square Fluctuation)
Conclusion
This is utterly true that our planet is facing dark times, and there is no exit solution/strategy to this crisis unless there is a suitable vaccine for this newly dubbed continuously dangerous unseen creature SARS-CoV-2. The infection of COVID-19, which is different from its other SARS cousins in severity, will not vanish from the globe until science takes us to the vaccine. The secondary & tertiary waves of infection will threaten us long-term and create havoc worldwide. Science lost interest after previous coronavirus outbreaks; if we had made the successful vaccine even after the SARS and MERS outbreaks, it would have been used for the recurrent coronavirus outbreaks. We must not be at the same fault again and be intelligently prepared to work towards a universal vaccine of a broad range for coronavirus and its associated coinfections. It is petrifying that, in some cases, the person infected with SARS-CoV-2 is not showing the symptoms; this makes them asymptomatic carriers. However, many infected persons show symptoms related to respiratory illnesses or pneumonia-like diseases (Lai et al. 2020b). From several other recent ongoing studies, the SARS-CoV-2 infection accompanies other coinfections that could be viral or bacterial, which intensifies the disease's severity. The study has developed a multi-pathogen subunit vaccine, which is efficaciously antigenic, non-toxic, and non-allergenic. This involves epitopes from SARS-CoV-2, S. pneumonia, H. influenza, and M. tuberculosis since the last three microorganisms are recently known to be dominantly associated with COVID-19 infection. It has also been seen that old age people having cardiovascular and other comorbidities are more prone to bacterial coinfection that has been clinically identified and also has the probability of multiple organ failures.
Therefore, in this study, we identified the immunogenic B-cell epitopes, HTL and CTL, and further analyzed the epitopes' eminence to generate the immune response, toxicity, antigenicity, and allergenicity of all the predicted epitopes were analyzed. For the enhancement of the immunogenicity, three TLR agonists (adjuvants) were tested, and six vaccine constructs were framed with suitable linkers. Based on physicochemical parameters, vaccine constructs VC2 and VC5 (both having 50S ribosomal L7/L12 adjuvant) were selected for further studies. Tertiary structures of both constructs were predicted, followed by their refinement and validation via plotting the Ramachandran curve to analyze the percentage of favored region residues.
Furthermore, the discontinuous B-cell epitopes were also predicted for VC2 and VC5. Based on results obtained from the Ramachandran plot and discontinuous B-cell, VC5 was best used for downstream studies. The binding specificity and active binding residues were assessed by molecular docking interaction studies where the lowest energy score of clusters_23, i.e., -1143.8, showed the best-docked complex/strong interaction between the TLR4 binding groove and VC5. The in silico immune response simulation studies of the VC5 revealed an excellent immune response for brief exposures and enhanced immunity even after repetitive exposures. Finally, the docked complex was simulated, and the RMSD and RMSF results suggested that the developed vaccine candidate, VC5 with human TLR4 receptor, is stable and flexible and has less deformed regions. This study gives insights into an innovative chimeric/universal vaccine for SARS-CoV-2 and its associated coinfections and encourages developing a universal vaccine for other emerging viral and bacterial infections. The vaccine candidate would prevent not only the SARS-CoV-2 infection but also the infection of those pathogens which interact with SARS-CoV-2 and generate a similar type of disease symptoms, enhancing disease severity in the host. The epitopes from each pathogen proved to be highly antigenic, cytokine-producing, non-toxic, non-allergenic, and stable and could possess the ability to generate the defined immune response. So, combining different pathogens conclusively allows protection against several diseases at once and can generate a broad immune response against the pathogen encounter solely.
References
Abraham MJ et al (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25
Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, Bogaert D (2013) Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog 9(1):e1003057
Chen X, Liao B, Cheng L, Peng X, Xu X, Li Y, Hu T, Li J, Zhou X, Ren B (2020) The microbial coinfection in COVID-19. Appl Microbiol Biotechnol 104:7777–7785
Choudhury A, Mukherjee S (2020) In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol 92(10):2105–2113
Chouhan P, Singh S, Sharma V, Prajapati VK (2022) Anti-IL-10 antibody humanization by SDR grafting with enhanced affinity to neutralize the adverse response of interleukin-10. Int J Pept Res Ther 28(5):148
ClinicalTrials.gov (2022). ClinicalTrials.gov is a database of privately and publicly funded clinical studies conducted around the world.
Coler RN, Day TA, Ellis R, Piazza FM, Beckmann AM, Vergara J, Rolf T, Lu L, Alter G, Hokey D, Jayashankar L (2018) The TLR-4 agonist adjuvant, GLA-SE, improves magnitude and quality of immune responses elicited by the ID93 tuberculosis vaccine: first-in-human trial. Npj Vaccines 3(1):34
Dimitrov, I., I. Bangov, D. R. Flower and I. Doytchinova (2014). AllerTOP v 2—a server for in silico prediction of allergens. Journal of molecular modeling, 20(6): 1–6.
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8(1):1–7
Duthie MS, Windish HP, Fox CB, Reed SG (2011) Use of defined TLR ligands as adjuvants within human vaccines. Immunol Rev 239(1):178–196
Dutta NK, Mazumdar K, Gordy JT (2020) The nucleocapsid protein of SARS–CoV-2: a target for vaccine development. J Virol 94(13):e00647-e720
Feldman C, Anderson R (2021) The role of co-infections and secondary infections in patients with COVID-19. Pneumonia 13:1–15
Feng Y, Ling Y, Bai T, Xie Y, Huang J, Li J, Xiong W, Yang D, Chen R, Lu F, Lu Y, Liu X, Chen Y, Li X, Li Y, Summah HD, Lin H, Yan J, Zhou M, Lu H, Qu J (2020) COVID-19 with different severities: a multicenter study of clinical features. Am J Respir Crit Care Med 201(11):1380–1388
Fleri W, Paul S, Dhanda SK, Mahajan S, Xu X, Peters B, Sette A (2017) The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front Immunol 8:278
Forsgren A, Riesbeck K (2008) Protein D of Haemophilus influenzae: a protective nontypeable H. influenzae antigen and a carrier for pneumococcal conjugate vaccines. Clin Infect Diseases 46(5):726–731
Gasteiger E, Hoogland C, Gattiker A, Duvaud SE, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The Proteomics Protocols Handbook. Humana press, Totowa, pp 571–607
Gupta N, Regar H, Verma VK, Prusty D, Mishra A, Prajapati VK (2020) Receptor-ligand based molecular interaction to discover adjuvant for immune cell TLRs to develop next-generation vaccine. Int J Biol Macromol 152:535–545
Gupta, S., Kapoor, P., Chaudhary, K., Gautam, A., Kumar, R., Open Source Drug Discovery Consortium and Raghava, G.P (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8(9):e73957
Hasan M, Azim KF, Begum A, Khan NA, Shammi TS, Imran AS, Chowdhury IM, Urme SRA (2019) Vaccinomics strategy for developing a unique multi-epitope monovalent vaccine against Marburg marburgvirus. Infect Genet Evol 70:140–157
Haste Andersen P, Nielsen M, Lund OLE (2006) Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci 15(11):2558–2567
Hatherill M, White RG, Hawn TR (2020) Clinical development of new TB vaccines: recent advances and next steps. Front Microbiol 10:3154
Hu B, Guo H, Zhou P, Shi ZL (2021) Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 19(3):141–154
Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S (2017) The ClusPro web server for protein–protein docking. Nat Protoc 12(2):255–278
Lai CC, Liu YH, Wang CY, Wang YH, Hsueh SC, Yen MY, Ko WC, Hsueh PR (2020a) Asymptomatic carrier state, acute respiratory disease, and pneumonia due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): Facts and myths. J Microbiol Immunol Infect 53(3):404–412
Lai CC, Wang CY, Hsueh PR (2020b) Co-infections among patients with COVID-19: The need for combination therapy with non-anti-SARS-CoV-2 agents? J Microbiol Immunol Infect 53(4):505–512
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 8:1–12
Lee SJ, Shin SJ, Lee MH, Lee MG, Kang TH, Park WS, Soh BY, Park JH, Shin YK, Kim HW, Yun CH (2014) A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS ONE 9(8):e104351
Livingston B, Crimi C, Newman M, Higashimoto Y, Appella E, Sidney J, Sette A (2002) A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J Immunol 168(11):5499–5506
Lovell SC, Davis IW, Arendall WB III, De Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins 50(3):437–450
Massey BW, Jayathilake K, Meltzer HY (2020) Respiratory microbial co-infection with SARS-CoV-2. Front Microbiol 11:2079
McGill-University (2022). "VIPER Group COVID-19 Vaccine Tracker Team."
Mohapatra PR, Mishra B, Behera B (2021) BCG vaccination induced protection from COVID-19. Indian J Tuberculosis 68(1):119–124
Ojha R, Pandey RK, Prajapati VK (2020) Vaccinomics strategy to concoct a promising subunit vaccine for visceral leishmaniasis targeting sandfly and leishmania antigens. Int J Biol Macromol 156:548–557
Ojha R, Pareek A, Pandey RK, Prusty D, Prajapati VK (2019) Strategic development of a next-generation multi-epitope vaccine to prevent Nipah virus zoonotic infection. ACS Omega 4(8):13069–13079
Pichichero ME, Khan MN, Xu Q (2016) Next generation protein based Streptococcus pneumoniae vaccines. Hum Vaccin Immunother 12(1):194–205
Rajput VS, Sharma R, Kumari A, Vyas N, Prajapati V, Grover A (2022) Engineering a multi epitope vaccine against SARS-CoV-2 by exploiting its non structural and structural proteins. J Biomol Struct Dyn 40(19):9096–9113
Saha S, Raghava GPS (2006) Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65(1):40–48
Sharma V, Singh S, Ratnakar TS, Prajapati VK (2022) Chapter 29 - Immunoinformatics and reverse vaccinology methods to design peptide-based vaccines. In: Tripathi T, Dubey VK (eds) Advances in Protein Molecular and Structural Biology Methods. Academic Press, Cambridge, pp 477–487
Shepard CC, Walker LL, Van Landingham RM (1978) Immunity to Mycobacterium leprae infections induced in mice by BCG vaccination at different times before or after challenge. Infect Immun 19(2):391–394
Singh A, Thakur M, Sharma LK, Chandra K (2020) Designing a multi-epitope peptide based vaccine against SARS-CoV-2. Sci Rep 10(1):16219
Singh B, Brant M, Kilian M, Hallström B, Riesbeck K (2010) Protein E of Haemophilus influenzae is a ubiquitous highly conserved adhesin. J Infect Dis 201(3):414–419
Singh S, Kumar K, Panda M, Srivastava A, Mishra A, Prajapati VK (2022) High-throughput virtual screening of small-molecule inhibitors targeting immune cell checkpoints to discover new immunotherapeutics for human diseases. Mol Div. https://doi.org/10.1007/s11030-022-10452-2
Singh S, Prajapati VK (2022) Exploring actinomycetes natural products to identify potential multi-target inhibitors against Leishmania donovani. Biotech 12(9):235
Tandon A, Pathak M, Harioudh MK, Ahmad S, Sayeed M, Afshan T, Siddiqi MI, Mitra K, Bhattacharya SM, Ghosh JK (2018) A TLR4-derived non-cytotoxic, self-assembling peptide functions as a vaccine adjuvant in mice. J Biol Chem 293(51):19874–19885
Vaillancourt M, Jorth P (2020) The unrecognized threat of secondary bacterial infections with COVID-19. Mbio 11(4):e01806-e1820
Vejbaesya S, Thongpradit R, Kalayanarooj S, Luangtrakool K, Luangtrakool P, Gibbons RV, Srinak D, Ngammthaworn S, Apisawes K, Yoon IK, Thomas SJ (2015) HLA class I supertype associations with clinical outcome of secondary dengue virus infections in ethnic Thais. J Infect Dis 212(6):939–947
Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B (2010) Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics 11:1–12
WHO (2022). "WHO recommendation Serum Institute of India Pvt. Ltd. (SIIPL) – COVID-19 vaccine (SARS-CoV-2 rS Protein Nanoparticle [Recombinant]) - COVOVAX™."
Yano A, Onozuka A, Asahi-Ozaki Y, Imai S, Hanada N, Miwa Y, Nisizawa T (2005) An ingenious design for peptide vaccines. Vaccine 23(17–18):2322–2326
Zhao Y, Kuang M, Li J, Zhu L, Jia Z, Guo X, Hu Y, Kong J, Yin H, Wang X, You F (2021) SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res 31(7):818–820
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet 395(10229):1054–1062
Zhu X, Ge Y, Wu T, Zhao K, Chen Y, Wu B, Zhu F, Zhu B, Cui L (2020) Co-infection with respiratory pathogens among COVID-2019 cases. Virus Res 285:198005
Acknowledgements
RO, SS is thankful to the Central University of Rajasthan for providing a research fellowship. VKP is thankful to the Central University of Rajasthan for providing the computational facility.
Funding
The authors have no relevant financial or non-financial interests to disclose. No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
Rupal Ojha: Conceptualization, Data curation, Software analysis, and Validation, Writing original draft. Satyendra Singh: Data curation, Software analysis, and Validation Writing original draft. Nidhi Gupta: Writing—review & editing, Visualization. Ketan Kumar: Writing—review & editing, Visualization. Aditya K. Padhi: Writing—review & editing, Visualization. Vijay Kumar Prajapati: Conceptualization, Supervision, Writing original draft, and finalizing draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared no competing interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ojha, R., Singh, S., Gupta, N. et al. Multi-pathogen based chimeric vaccine to fight against COVID-19 and concomitant coinfections. Biotechnol Lett 45, 779–797 (2023). https://doi.org/10.1007/s10529-023-03380-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-023-03380-0