
Abstract
Serological assays for SARS-CoV-2 are being utilized at an exponential rate for surveillance programs. This enterprise was designed to develop and validate a qualitative immunochromatographic test, via the Lateral Flow Assay (LFA), for detection of immunoglobulins M and G (IgM and IgG) against both nucleocapsid (N) and the receptor-binding domain (RBD) of the spike protein of SARS-CoV-2. Both targeted proteins were cloned and expressed in baculovirus expression system utilizing insect cells Sf9. The recombinant RBD and N proteins were purified and conjugated with gold nanoparticles (AuNPs) to set up the coating antigens pad. Both anti-human IgG and IgM were dispensed on nitrocellulose membrane to capture human antibodies in serum samples. A home-made dispensing system was developed to draw identical test and control lines. The validity of the developed LFA was verified by testing serum samples from 103 convalescent COVID-19 patients who were PCR positive for SARS-CoV-2 along with 28 control serum samples. The developed strips showed distinctive bands for IgM and IgG of both proteins (RBD and N) in positive samples. The sensitivity of RBD-based LFA was 70.9% and 39.8% for IgG and IgM, respectively, with a specificity of 100% for both. The N-based LFA exhibited a sensitivity of 73.8% and 35.9% for IgG and IgM, respectively, while its specificity was 75% and 100% for IgG and IgM, respectively. Our developed LFA could afford a tool for surveillance programs in low-resource countries. Moreover, it might be functional for rapid and inexpensive monitoring of the anti-SARS-CoV-2 antibodies in the sera of vaccinated individuals.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a highly contagious virus transmitted through human airborne droplets and may trigger systematic and respiratory symptoms although, on some occasions may develop asymptomatic infection (up to 41% of SARS-CoV-2 infections have been caused by asymptomatic cases) (Wang et al. 2020; World Health Organization 2020). Through the fight against the COVID-19 pandemic, the diagnostic performance of different assay approaches for SARS-CoV-2 has been crucial (Marca et al. 2020). Laboratory procedures that shorten the time between testing and results are essential for reducing onward transmission. In addition, PCR technology could be negative after 15 days of infection, and as an antigen-based test, it cannot detect the antibodies which are generated in the circulation against SARS-CoV-2 in the early phase of infection. Moreover, taking swabs require skillfully trained individuals to minimize false-negative results (Crozier et al. 2021).
SARS-CoV-2 genome encodes a number of structural and non-structural proteins. The structural proteins, like spike (S), nucleocapsid (N), membrane (M), and envelope (E), are mainly responsible for genome maintenance, cell membrane attachment, immune evasion, antibodies neutralization, and viral pathogenesis. While, the non-structural proteins, such as RNA-dependent RNA polymerase (RdRp) and protease (3CL), account for translation, transcription, replication, and viral assembly (Fehr and Perlman 2015).
Meanwhile, spike protein has the fundamental role in the stimulation of immune responses and initiation of antibody neutralization process. It has the most divergent mutations in the non-conserved regions which contribute to the antigenic variations in various SARS-CoV-2 genotypes. Regarding the role of protein N in viral infection, it is the most abundant and highly immunogenic expressed protein that has the ability to elicit the antibodies response (Ahmed et al. 2020; Ying et al. 2003). The rapid growth of SARS-CoV-2 infections worldwide with a variety of clinical signs has been attributed to the emergence of mutations with several related variants. These mutations were reported mostly at loci on the receptor-binding domain (RBD) of spike gene and in part on the N gene (Ziegler et al. 2020). Therefore, studying those proteins is the central target for each scientist to assist control of the current COVID-19 pandemic.
Serological approaches such as Enzyme-linked immunosorbent assay (ELISA) and Lateral Flow Assay (LFA) have a fundamental role in the detection of antibodies against pathogens. Serological approaches, such as LFA, have many limitations with lower sensitivity compared to PCR technology. However, they are based on serum sample, which is less painful to apply and does not need skillful operators (Li et al. 2021). It has been reported that some emerging SARS-CoV-2 mutations had a variable impact on the performance of the molecular diagnostic assays depending on nucleotide mismatch and position on the targeted genomic sequence (Ziegler et al. 2020). Hence, subsequent efforts were to be conducted to verify how far the rising SARS-CoV-2 mutations would impact the performance of such assays and to ensure their diagnostic utility.
Seroconversion to SARS-CoV-2 is manifested by the onset of specific antibodies IgM (~ 3–6 days) and IgG (~ 14 days) post-infection (Guo et al. 2020). Additionally, a study has demonstrated that seroconversion of IgM and IgG may be detectable after a few days (2–23 days) as clinical symptoms start (Long et al. 2020).
In this endeavor, we intended to develop and validate a colloidal gold-based immuno-chromatographic strip using SARS-CoV-2 recombinant N and RBD proteins, cloned and expressed in the baculovirus-insect cells expression system. Moreover, the developed LFA was verified for its utility in serodiagnosis of SARS-CoV-2 infected patients by specific detection of early IgM and late IgG antibodies in serum samples.
Materials and methods
Serum samples
A total of 103 convalescent Egyptian cases with recorded positive RT-PCR for SARS-CoV-2 infection, from a private accredited Medical Laboratory, were enrolled in this retrospective study. All serum samples were collected after written informed consent from the patients, from November 2020 to January 2021. Patients’ clinical history was collected in accordance with the Declaration of Helsinki and was approved by the Research Ethics Committee, Faculty of Medicine, Suez Canal University with reference number #4597.
Of the 103 cases, female (n = 45; 43.6%) and male (n = 58; 56.3%) patients, 16 cases suffered from severe illness (SpO2 < 94% on room air, a respiratory rate > 30 breaths/min, and lung infiltrates > 50%); whereas, the other 87 cases went through mild to moderate symptoms of COVID-19. All sera were collected at 14-days after the onset of clinical symptoms (up to 50 days in severe cases).
As a control group, 28 sera have been collected from healthy female (n = 11) and male (n = 17) cases prior to the pandemic, from November 2018 till June 2019, were used. Samples from both SARS-CoV-2 convalescent and healthy cases were collected following firm exclusion criteria were applied: ages of ˂10 years and > 80 years; positive cases for Hepatitis C/B and HIV, along with immunocompromised patients.
All samples were heat-inactivated at 56 °C for 30 min and kept at -20 °C to be tested. Handling of samples and assays were performed in a Biological Safety Level 3, as needed, according to the guidance of the Center for Disease Control and Prevention (Sick 2020).
Generation of recombinant SARS-CoV-2N and RBD antigens
Designing, synthesis, and cloning of the N and RBD genes
The open reading frames encoding for the N and RBD proteins of SARS-CoV-2, derived from the nucleotide sequence of the reference isolate Wuhan-Hu-1 (GenBank accession number: NC_045512.2), were cloned, expressed, and purified according to our lab’s established and modified protocols (Salem et al. 2019a, 2020, 2022).
The corresponding nucleotide sequences (N and RBD) were subjected to codon usage optimization to better matched expression profile of Spodoptera frugiperda (the surrogate cells used for expression). The coding sequence of 6-Histidine residues and an enterokinase recognition sequence were inserted at the 3´ end of N and RBD coding sequences. The synthesized sequences were cloned into the BssHII/PstI sites of the pFastBac cloning vector and expression was derived by the Ppol promoter (Salem et al. 2019b; Elmenofy et al. 2020; Sheikhzadeh et al. 2020).
Generation of recombinant baculoviruses
To generate recombinant baculoviruses harboring the N and RBD genes, the hybrid pFastBac1-N and pFastBac1-RBD plasmids were transformed into DH10Bac E. coli cells following the manufacturer's instructions (Thermo-Fisher). In brief, the constructed hybrid plasmids were individually transformed into DH10Bac cells. This allows the cassettes carrying the N or RBD genes between Tn7R and Tn7L sequences in pFastBac1 to be transferred into the bacmid in DH10Bac cells through site-specific transposition. Recombinant baculoviruses were then isolated, screened, amplified, and titrated according to the standard methods provided with the Bac-to-Bac baculovirus system (Thermo-Fisher). The N and RBD recombinant proteins were produced via infection of Sf9 cells, with the generated recombinant baculoviruses. The insect cell line Sf9 (Thermo-Fisher) was grown and maintained at 27 °C using ExCell-420 Serum-Free medium (Sigma-Aldrich), supplemented with 10 µg Gentamycin. It was used in all procedures of baculovirus expression of SARS-CoV-2 N and RBD genes, preparation of recombinant baculovirus stocks and production of the recombinant proteins (Scholz and Suppmann 2017).
Infected cells were cultured in T-flasks for small-scale production, and culture supernatants, as well as infected cells, were harvested daily at 3–7 days post-infection (DPI). Each harvest was subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analyses (El-Gaied et al. 2017; Salem et al. 2021), to verify integrity and immunoreactivity of the expressed proteins, as well as their peak-expression times.
Purification of N and RBD proteins
Five days post-infection, infected Sf9 cells using the recombinant baculoviruses were collected and total protein was estimated. The culture supernatants were tested for secretory N and RBD recombinant proteins by SDS-PAGE. The N and RBD polypeptides fused with N-terminal 6xHis-tag were purified from the re-suspended cell pellets using Ni–NTA agarose resin (Qiagen, Germany). Finally, the recombinant N and RBD containing imidazole were dialyzed in 1 × phosphate-buffered saline (PBS) (pH 7.4) for overnight at 4 °C, and the concentrations were determined using the Bradford protocol (Kielkopf and Bauer 2020).
Mice immunization
Two female BALB/c mice, 21 days old were obtained from Theodor Bilharz Research Institute, Giza, Egypt, and treated in compliance with the principles and policies of the American Physiological Society's Guiding Principles in the Care and Use of Animals and after the approval of research ethics committee, Faculty of Medicine, Suez Canal University. Mice were injected with the recombinant N and RBD proteins; according to the mentioned injection schedule. The primary immune response was initiated by intraperitoneal injection of mice with 50 μg of recombinant protein (N or RBD) emulsified in complete Freund’s adjuvant, followed by 4 (each/week) subsequent intravenous boosters, each with 100 μg protein emulsified in incomplete Freund’s adjuvant that was excluded from the last booster. Seven days post the 5th injection, sera were collected for assessment of anti- N and RBD seroconversion; then mice were euthanized and spleens were gathered for isolation.
Preparation of colloid gold nanoparticle
Gold nanoparticles (AuNPs) with a diameter of about 30 nm were prepared by citrate-based reduction method according to Turkevich Method (Dong et al. 2020). Briefly, 1 mL of 1% sodium citrate was added to a 99 mL of 0.01% stirred boiling HAuCl4 solution. After vigorous stirring with continuous boiling, till the solution’s color has turned from purple to red wine (approximately for 3–5 min). With no longer changes in color, the solution was allowed to gradually cool down at room temperature. The colloid AuNPs concentration was checked by Spectrophotometer at a wavelength of 525 nm (Beckman DU 530 UV–Vis, USA) and their particle size was confirmed by transmission electron microscopy (TEM, Jeol JEM-1400, Japan), as has been described (Zhang et al. 2009).
Preparation and optimization of protein-gold conjugate
For preparing the protein-AuNPs conjugate, serial pH values of gold solution were prepared by 0.2 M potassium carbonate solution. In addition, several protein concentrations were prepared and added to the colloidal gold solutions (pH adjusted) (Zhang et al. 2009). After 30 min of incubation, a 125 µl of 1 M sodium chloride (NaCl) was added to the reaction to induce the aggregation effect on the gold particles. The optimal amount of protein required for gold stabilization was determined by spectrophotometer at 530–650 nm (Zhang et al. 2009; Busch et al. 2019).
To block the surface sites on AuNPs, increasing the colloidal AuNPs stability and avoiding unspecific interactions, the optimized concentrations of dialyzed recombinant proteins N and RBD were separately added to 1 ml of adjusted pH gold solution and incubated for 1 h with shaking, then, a 125 µl of 10% bovine serum albumin (BSA) was added. After extra 15 min of incubation with stirring, the prepared AuNPs were washed with 250 µl of BSA (1%) for three times then, centrifuged for 30 min at 15,000 rpm and 4 °C. Eventually, the supernatants were discarded and the pellets (containing antigen-gold conjugate) were distinctly re-suspended in a 100 µl of conjugation buffer (0.5% BSA, 50 mM Tris–HCL, 0.5% Tween 20, 2% sucrose) and kept at 4 °C (Zhang et al. 2009).
Assembly and evaluation of the Lateral Flow strips
The structural design of the developed strips is demonstrated in (Fig. 1). The components of the Lateral Flow strip were assembled into an adhesive-backing card including: nitrocellulose membrane (3 × 10 cm), absorbent pad (2.5 × 10 cm), conjugate pad (1.5 × 10 cm), sample pad (1.5 × 10 cm) and adhesive double taps. Briefly, the conjugate pad was dipped in the conjugate solution containing 20 mM sodium borate (pH 8.5), BSA (2%), sucrose (3%), NaCl (0.6 M), Tween 20 (0.2%), and sodium azide (0.1%). It was then left for 30 min at 50 °C to dry. To prepare the sample pad, it was treated with PBS (pH 7.2) containing NaCl (0.1 M), Tween 20 (0.2%), and sodium azide (0.1%), then left to dry for 30 min at 50 °C (Zhang et al. 2009, 2006). As test lines, Goat anti-human IgG (Cat.#: 31,130, 2.4 mg/mL), and Goat anti-human IgM (Cat.#: A18843, 2.5 mg/mL) were dispensed onto the nitrocellulose membrane, whereas, the control lines were contained purified mice IgGs against recombinant N and RBD to SARS-CoV-2. The space between each line was ~ 0.5 cm. The blotted membrane was blocked with BSA (1%) and allowed to dry at 37 °C for 2 h.

The basic structure of the developed AuNPs-Lateral Flow Assay. A Principle structure of AuNPs-Lateral Flow strip. B Positive reaction of the developed strip. C Negative reaction. D Invalid reaction
For assembly, firstly; the blotted membrane was stamped in the middle followed by an adsorption pad with 0.5 cm over-crossing at the adsorption pad end. Both sample and conjugate pads were affixed with 0.5 cm overlapping at the end of the conjugate pad. Then, pads were fixed next to the blotted membrane with 0.5 cm overlapping at the end of the conjugate pad. The strips were then sealed and stored at 4 °C till used.
For evaluation, fifty µl of infected serum sample was directly loaded into the sample pad on the strip, followed by 50 µl of PBS. Detection was occurred by providing a strong and reliable band on test and control lines. Lateral flow for IgM and IgG sensitivity and specificity as 95% confidence intervals were estimated, and Cohen’s Kappa was calculated (Landis and Koch 1977).
Results and discussion
Production and purification of N and RBD proteins
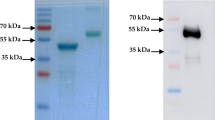
The successful generation of recombinant baculoviruses, harboring the proper nucleotide sequences encoding for the N and RBD cloned genes of SARS-CoV-2, was verified by specific PCR (data not shown). Expression of both recombinant proteins (N and RBD) in Sf9 insect cells infected with the recombinant baculoviruses were demonstrated via SDS-PAGE and western blot. As shown by SDS-PAGE (Fig. 2A), discrete protein bands at the size of ~ 46 KDa and ~ 21 KDa were evident for the expressed N and RBD recombinant proteins, respectively, in the Sf9 cell lysates infected with the corresponding recombinant baculovirus, where were absent in lysates from non-infected Sf9 cells. Furthermore, the recombinant N and RBD proteins fused to His-tag were purified using Ni–NTA affinity chromatography. The identities and immunoreactivity of these polypeptides were also confirmed by western blotting (Fig. 2B).

A SDS-PAGE showing the expression and purification of recombinant RBD and N proteins. 1: protein ladder; 2: negative control (total protein lysates from non-infected Sf9 cells); 3 and 4: showing the expression of RBD and N, respectively in the Sf9 cell lysates infected with the corresponding recombinant baculovirus; 5 and 6: purified RBD and N, respectively, purified using Ni–NTA affinity chromatography. B Confirming the identities of the SDS-PAGE polypeptides by western blotting
Characterization of gold nanoparticles
Uniformity, homogeny, morphology, and distribution of the prepared AuNPs were characterized by Spectrophotometry and TEM imaging. Results shown in (Fig. 3A) indicated obtaining the optimal size of prepared AuNPs as the maximum optical density (OD = 1) noticed at the wavelength of 525 nm (Zhang et al. 2009). Regarding TEM images, the gold particles were spherical with uniformity and sound-dispersion in a good approximation. The average size of particles was approximately 25–30 nm as shown in (Fig. 3B).

A Optimal size for the prepared gold nanoparticles (AuNPs) as the maximum optical density (OD = 1) noticed at the wavelength of 525 nm. B TEM imaging showing the uniformity, homogeny, morphology, and distribution of the prepared AuNPs
Conjugation of prepared AuNPs with recombinant proteins
The conjugation of AuNPs with recombinant proteins was conducted by the physical electrostatic interaction. Basically, in this type of conjugation, the pH value is a critical point and should be in a range of ± 0.5 around the protein's isoelectric point. Thus, a range of pH values (6.0, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, 9.5, and 10) were set. We found that the optimal pH value for conjugation was 9 (as the wine-red color was formed). Conversely, the other pH values exhibited a purple or white–gray color, which indicating the aggregation and destructive effect for gold particles.
Furthermore, to add the optimal protein’s amount for conjugation, different concentrations of the recombinant proteins 1, 3, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, and 110 µg/ml were prepared using the adjusted pH gold solution (pH 9). We found that a 70 to 80 µg/ml and 60–70 µg/ml of recombinant N and RBD, respectively were the optimal concentrations for conjugation.
Optimization of the lateral flow strips
For accurate dispensing of the used antigens and antibodies on the lateral flow membranes, we developed a home-made system (Fig. 4) to make precise lines (test and control). For control lines, the mice purified IgG (raised against recombinant proteins) was tested in different concentrations 0.5, 1, 1.5, 1.8, and 2 mg/ml. Results showed that the optimal IgG concentration 2 mg/ml and 1.8 mg/ml for N and RBD strip, respectively. Regarding the test lines, the most favorable concentrations were 1.0 and 1.2 mg/ml for Anti- IgG and IgM, respectively.

Home-made developed dispensing system
To demonstrate clear control and test lines, avoiding non-specific binding, and eliminating the false-positive results, 1% of bovine serum albumin (BSA) was used to block the nitrocellulose membrane and this is consistent with previous studies (Huang et al. 2020; Wen et al. 2020). Notably, the strips that were blocked with 2% or 0.5% or 0.0% of BSA exhibited unreliable bands and a deepened red background with false-positive bands in the control sera.
In addition, for sampling, various dilutions of serum samples 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, and 1:8 in PBS (pH 7.2) were prepared. The test bands revealed shortcomings except the dilution 1:4 which demonstrated a slightly clear result which is in a contrary with Huang et al. 2020. However, direct loading of sample (50 µl) onto the strip, followed by a 50 µl of PBS resulted in strong and trustworthy bands to distinguish. Notably, less than 20 µl of the direct sample loading provided negligible bands and thus leading to unreliable results.
Sensitivity and specificity of the lateral flow strips
To evaluate the developed Lateral Flow strips, human serum samples of 103 convalescent COVID-19 patients (previously proven by PCR) were tested. These patients were of median age of 41.5 years; a group with severe illness (n = 16) and a group with mild-to-moderate illness (n = 87), as (IQR: 22-62ys), 46 years (IQR: 22-65ys); respectively. For the severe illness group, the median period was 32 days from symptoms onset to samples withdrawal, while the median period for the mild-to-moderate symptoms group was 23 days. In addition, 28 serum samples that had been collected prior to the pandemic were used as negative controls.
Each sample was tested in duplicate and results were observed by the naked eye. Data in (Table 1) provided the sensitivity and specificity of LFA that developed based on IgG against N and RBD proteins calculated upon the PCR results. RBD-based LFA IgG sensitivity was 70.9% and specificity was 100%. While, N-based LFA IgG sensitivity was 73.8% and 75% specificity. A value of 0.51 of Cohen’s Kappa was obtained revealing a moderate agreement between RBD-based LFA IgG and PCR; while, a fair agreement with a value of 0.39 was obtained for N-based LFA IgG.
While, Table 2 provided the sensitivity and specificity of LFA based on IgM against N and RBD proteins calculated upon the PCR results. PCR and RBD-based LFA IgM sensitivity was 39.8%, while specificity was 100%. N-based LFA IgG sensitivity was 35.9% with 100% specificity and a value of 0.22 of Cohen’s Kappa for RBD-based LFA IgG, which is a fair agreement. While a slight agreement with a value of 0.19 was obtained for N-based LFA IgG.
Table 2 RBD and N IgM lateral flow assay (LFA) test compared to positive SARS-CoV-2 reverse transcription polymerase chain reaction (RT-PCR) from nasopharyngeal swab (103 patients and 28 control).
Both molecular and serological methods could give false results because of incorrect sampling, insufficient viral material, improper RNA extraction, non-specific cross-reactivity with other viruses, contamination or technical issues. Although the serological tool could enhance identification capacity, particularly during pandemics, and they provide rapid and affordable screening programs to control or mitigate the infection. The relatively low sensitivity and specificity is a major limitation. Possible improvement strategy focusing on identifying novel signal augmentation approaches and quantification systems warrant further study. So, our next step is to develop an ELISA with a quantitative or semi-quantitative approach in which enzymatic reactions could be used to improve the sensitivities and specificities.
Here, we developed the first Egyptian Lateral Flow platform to evaluate the IgG and IgM antibodies against SARS-CoV-2 infection in human serum. PCR-based technology was performed on virus-containing specimens with extreme risk, requiring exceptional biosafety precautions, intensive labor with well-trained skills to mitigate the invalid results. Furthermore, its limited and costive testing capacity, the exposure time to the virus, the onset of symptoms and the optimal time of viral detection are critical elements to detect the infection by PCR. Alternatively, the point of care test such as LFA is an easy-to-handle tool, could boost the identification capacity and provide the result within 20 min. The developed strips provide sensitivity 70.9% and 73.8% for RBD-based IgG and N-based IgG, respectively. In a previous study, the gold nanoparticle-Lateral Flow strip (AuNPs-LF) was developed with a sensitivity value of 69.1% (Wen et al. 2020). Also, Zeng et al. (Zeng et al. 2020) demonstrated that the positive rate of the single IgG-RBD was 61.76% for the AuNPs-LF strip. In the current study, the level of agreement between developed RBD-based LFA IgG and the PCR tool was 0.51 (Cohen’s Kappa), compared to 0.39 for N-based LFA IgG. This might be due to the highly conserved N-terminal domain of the N protein of beta-coronaviruses compared to RBD-protein which is only specific to SARS-CoV-2.
Interestingly, the IgM antibodies arise gradually during the first week of infection and may decline rapidly within two weeks. Unlike IgM, the IgG is elicited after 10–14 days of exposure and persist in sera (over 48 days) for an extended period of time (Guo et al. 2020; Hou et al. 2020).
In this study, the median time for detection of the SARS-CoV-2 antibodies in serum samples was 32 days from the onset of symptoms. This might explain why the patients in this study were seropositive for IgG more than IgM. Besides, this would clarify why sensitivity for the RBD-IgM and N-IgM, in this study, was 39.8% and 35.9%, respectively. Consequently, IgM serological assay is potentially accounting for poor sensitivity after one month of infection. Thereby, it was recommended to combine the IgG and IgM in a diagnostic test designed to detect SARS-CoV-2 infection rather than using a single antibody testing (Li et al. 2020).
Conclusion
During the pandemic, the need for a test that is easy, rapid, portable and affordable, was raised for screening and controlling the spread of infection. LFA is a promising preliminary serological test. Although, it is fast, affordable, and easy to use, it should only be used for serological surveillance. A clinical correlation and confirmatory test should be the basis for starting treatment. The generated LFA will be used in a future study for monitoring the antibodies against SARS-CoV-2 after vaccination and in quality control assessment for developed vaccines. The developed assay initiate a potential for future upgrading of COVID-19 routine diagnostic agenda as it could be deployed in low-economy countries or regions at risk. Rapid, accurate, simple and inexpensive diagnostic tests are advantageous as they provide better patient triage and treatment, lessening the spread of outbreaks by fast recognition of infected persons, appropriate to lab facilities at regular clinics without the need for a skilled practitioner, and lower testing costs. The developed LFA utilizes a complete single-use, room thermo-stable test cartridge, which is appropriate for early (IgM) and late (IgG) rapid serodiagnosis of suspected SARS-CoV-2 infected patients, and might be a useful tool for quality control of vaccines and sero-monitoring of vaccinates as well.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Ahmed SF, Quadeer AA, McKay MR (2020) Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 12:254. https://doi.org/10.3390/v12030254
Busch RT, Karim F, Weis J, Sun Y, Zhao C, Vasquez ES (2019) Optimization and structural stability of gold nanoparticle-antibody bioconjugates. ACS Omega 4(12):15269–15279. https://doi.org/10.1021/acsomega.9b02276
Crozier A, Rajan S, Buchan I, McKee M (2021) Put to the test: use of rapid testing technologies for covid-19. BMJ 372:n208. https://doi.org/10.1136/bmj.n208
Dong J, Carpinone PL, Pyrgiotakis G, Demokritou P, Moudgil BM (2020) Synthesis of precision gold nanoparticles using Turkevich method. Kona 37:224–232
El-Gaied L, Salem R, Elmenofy W (2017) Expression of tomato yellow leaf curl virus coat protein using baculovirus expression system and evaluation of its utility as a viral antigen. 3 Biotech 7:269. https://doi.org/10.1007/s13205-017-0893-4
Elmenofy W, Mohamed I, El-Gaied L, Salem R, Osman G, Ibrahim M (2020) Expression of 1B capsid protein of Foot-and-mouth disease virus (FMDV) using baculovirus expression system and its validation in detecting SAT 2- specific antisera. PeerJ 8:e8946. https://doi.org/10.7717/peerj.8946
Fehr AR, Perlman S (2015) Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 1282:1–23. https://doi.org/10.1007/978-1-4939-2438-7_1
Guo L, Ren L, Yang S et al (2020) Profiling early humoral response to diagnose novel coronavirus disease (COVID-19). Clin Infect Dis 71:778–785. https://doi.org/10.1093/cid/ciaa310
Hou H, Wang T, Zhang B et al (2020) Detection of IgM and IgG antibodies in patients with coronavirus disease 2019. Clin Transl Immunology 9:e01136. https://doi.org/10.1002/cti2.1136
Huang C, Wen T, Shi FJ, Zeng XY, Jiao YJ (2020) Rapid detection of IgM antibodies against the SARS-CoV-2 virus via colloidal gold nanoparticle-based lateral-flow assay. ACS Omega 5(21):12550–12556. https://doi.org/10.1021/acsomega.0c01554
Kielkopf CL, Bauer W, Urbatsch IL (2020) Bradford assay for determining protein concentration. Cold Spring HarbProtoc 4:102269. https://doi.org/10.1101/pdb.prot102269
La Marca A, Capuzzo M, Paglia T et al (2020) Testing for SARS-CoV-2 (COVID-19): a systematic review and clinical guide to molecular and serological in-vitro diagnostic assays. Reprod Biomed Online 41:483–499. https://doi.org/10.1016/j.rbmo.2020.06.001
Landis JR, Koch GG (1977) The measurement of observer agreement for categorical data. Biometrics 33:159–174. https://doi.org/10.2307/2529310
Li Y, He Q, Yu R et al (2021) Highlighted prospects of an IgM/IgG antibodies test in identifying individuals with asymptomatic severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) infection. Arch Pathol Lab Med 145:39–45. https://doi.org/10.5858/arpa.2020-0310-SA
Li Z, Yi Y, Luo X et al (2020) Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. J Med Virol 92:1518–1524. https://doi.org/10.1002/jmv.25727
Long QX, Liu BZ, Deng HJ et al (2020) Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med 26:845–848. https://doi.org/10.1038/s41591-020-0897-1
Salem R, El-Kholy AA, Ibrahim M (2019a) Eight novel single chain antibody fragments recognising VP2 of foot-and-mouth disease virus serotypes A, O, and SAT 2. Virology 533:145–154. https://doi.org/10.1016/j.virol.2019.05.012
Salem R, El-Kholy AA, Omar OA et al (2019b) Construction, expression and evaluation of recombinant VP2 protein for serotype independent detection of FMDV seropositive animals in Egypt. Sci Rep 9:10135. https://doi.org/10.1038/s41598-019-46596-9
Salem R, Assem KS, Omar AO et al (2020) Expressing the immunodominant projection domain of infectious bursal disease virus fused to the fragment crystallizable of chicken IgY in yellow maize for a prospective edible vaccine. Mol Immunol 118:132–141. https://doi.org/10.1016/j.molimm.2019.12.015
Salem R, El-Kholy AA, Waly FR, Khaled R, Elmenofy W (2021) Removal of 3C protease from the 3ABC improves expression, solubility, and purification of the recombinant 3AB of foot-and-mouth disease virus. Virus Genes 57:72–82. https://doi.org/10.1007/s11262-020-01815-8
Salem R, El-Kholy AA, Waly FR, Ayman D, Sakr A, Hussein M (2022) Generation and utility of a single-chain fragment variable monoclonal antibody platform against a baculovirus expressed recombinant receptor binding domain of SARS-CoV-2 spike protein. Mol Immunol 141:287–296. https://doi.org/10.1016/j.molimm.2021.12.006
Scholz J, Suppmann S (2017) A new single-step protocol for rapid baculovirus-driven protein production in insect cells. BMC Biotechnol 17:83. https://doi.org/10.1186/s12896-017-0400-3
Sheikhzadeh E, Eissa S, Ismail A, Zourob M (2020) Diagnostic techniques for COVID-19 and new developments. Talanta 220:121392. https://doi.org/10.1016/j.talanta.2020.121392
Sick PG. (2020) Interim laboratory biosafety guidelines for handling and processing specimens associated with coronavirus disease 2019 (COVID-19). Atlanta, GA: Centers for Disease Control and Prevention. https://www.cdc.gov/coronavirus/2019-nCoV/lab/lab-biosafety-guidelines.html
Wang W, Xu Y, Gao R et al (2020) Detection of SARS-CoV-2 in different types of clinical specimens. JAMA 323:1843–1844. https://doi.org/10.1001/jama.2020.3786
Wen T, Huang C, Shi FJ et al (2020) Development of a lateral flow immunoassay strip for rapid detection of IgG antibody against SARS-CoV-2 virus. Analyst 145:5345–5352. https://doi.org/10.1039/d0an00629g
World Health Organization (2020) Antigen-detection in the diagnosis of SARS-CoV-2 infection using rapid immunoassays: interim guidance, 11 September 2020. World Health Organization https://apps.who.int/iris/handle/10665/334253. License: CC BY-NC-SA 3.0 IGO
Ying LI, Xu SH, Yang RF et al (2003) Identification of an epitope of SARS-coronavirus nucleocapsid protein. Cell Res 13:141–145. https://doi.org/10.1038/sj.cr.7290158
Zeng L, Li Y, Liu J et al (2020) Rapid, ultrasensitive and highly specific biosensor for the diagnosis of SARS-CoV-2 in clinical blood samples. Mater Chem Front 4:2000–2005. https://doi.org/10.1039/D0QM00294A
Zhang GP, Wang XN, Yang JF et al (2006) Development of an immunochromatographic lateral flow test strip for detection of β-adrenergic agonist Clenbuterol residues. J Immunol Methods 312:27–33. https://doi.org/10.1016/j.jim.2006.02.017
Zhang G, Guo J, Wang X (2009) Immunochromatographic lateral flow strip tests. In: Biosensors and biodetection. Humana Press, Totowa, pp 169–183
Ziegler K, Steininger P, Ziegler R, Steinmann J, Korn K, Ensser A (2020) SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Euro Surveill 25:2001650. https://doi.org/10.2807/1560-7917.ES.2020.25.39.2001650
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This study was financially supported by Science, Technology & innovation Funding Authority (STIFA), Egypt; Emergency Targeted Program; COVID -19 Emergency call, Project ID: 43798.
Author information
Authors and Affiliations
Contributions
All authors have contributed to the manuscript in significant way, have reviewed and agreed upon the manuscript content. RS: conceptualization, funding acquisition, laboratory work, recombinant antigens generation and supervision, reviewing final draft. AME: sample collection, lab work, data curation, and reviewing final draft. NK: writing original draft, data curation, and reviewing final draft. SY: data analysis and reviewing final draft. OM: methodology, and reviewing final draft. FW: recombinant antigens’ generation, and reviewing final draft. AAE: conceptualization, laboratory work, and reviewing final draft. WE: recombinant antigens’ generation, and reviewing final draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Ethical approval
All data was collected in accordance with the Declaration of Helsinki and was approved by the Research Ethics Committee, Faculty of Medicine, Suez Canal University with reference number #4597. Immunized mice were treated in compliance with the principles and policies of the American Physiological Society's Guiding Principles in the Care and Use of Animals and after the approval of research ethics committee, Faculty of Medicine, Suez Canal University.
Consent to participate
All serum samples were collected after written informed consent from the patients, from November 2020 to January 2021. The consent was approved by the Research Ethics Committee, Faculty of Medicine, Suez Canal University.
Consent for publication
In case of acceptance, the authors give a consent for the publication of the above article, including figures and details within the text to be published in Gold Bulletin Journal.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salem, R., Elshamy, A.M., Kamel, N. et al. A gold nanoparticles-based lateral flow assay utilizing baculovirus expressed recombinant nucleocapsid and receptor binding domain proteins for serodetection of IgG and IgM against SARS-CoV-2. Biotechnol Lett 44, 1507–1517 (2022). https://doi.org/10.1007/s10529-022-03316-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-022-03316-0