
Abstract
Seven overlapping truncated forms of the C subunit of porcine aminopeptidase N (pAPN-C) were expressed in Escherichia coli. By western blotting and ELISA test, all recombinant proteins were recognized by the antibody against native porcine aminopeptidase N. Recombinant proteins, rpAPN-C2 (aa 623–722) and rpAPN-C3 (aa 673–772), had the highest binding activity with swine transmissible gastroenteritis virus among the truncated pAPN-C recombinant proteins. The overlapping region (aa 673–722) between rpAPN-C2 and rpAPN-C3 is indicated to play a key role in viral binding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porcine aminopeptidase N (pAPN) is a cellular receptor of swine coronavirus transmissible gastroenteritis virus (TGEV) (Delmas et al. 1992), which causes severe diarrhea and a high mortality rate of up to 90% in piglets (Reynolds and Garwes 1979). pAPN is highly expressed on the surface of pig intestinal epithelial cells (Kenny and Maroux 1982), where it is involved in digestion of N-terminal amino acids from biologically active peptides (Semenza 1986; Delmas et al. 1994). Generally, native pAPN is a mixed protein which consists of the full-length pAPN protein (aa 1–963, 150 kDa), the B subunit of pAPN (aa 1–572, 95 kDa) and the C subunit of pAPN (aa 573–963, 50 kDa), because of trypsin cleavage at the potential site Arg572 of the pAPN molecule (Delmas et al. 1994). In the full-length pAPN, the TGEV binding site is mainly located in the C subunit of pAPN (pAPN-C) and the catalytic site of pAPN is located in the B subunit (Jongeneel et al. 1989). APN enzymatic activity is not, however, involved in the process of TGEV entry into the host cells (Delmas et al. 1994).
TGEV is a member of the genus Coronavirus, family Coronaviridae, order Nidovirales. Of several TGEV structural proteins, the S protein is a major structural protein involved in virus entry (Egberink et al. 1988; Delmas et al. 1990; Schwegmann–Wessels et al. 2011). Interaction of the S protein with pAPN and sialic acid residues plays a significant part in the process of adsorption and penetration, thus initiating virus infection (Gebauer et al. 1991; Ren et al. 2008). pAPN and the S protein of TGEV have become interesting targets for research into the pathogenesis and prevention of TGEV infection (Sun et al. 2007; 2008). Recombinant pAPN protein and its antiserum can block TGEV infection in vitro (Liu et al. 2009) and the amino acid regions, aa 36–223, aa 349–591 and aa 592–963, in pAPN are major virus-binding regions for TGEV (Ren et al. 2010). However, the precise mechanism involved in virus binding to these regions of pAPN-C has not yet been fully elucidated.
In this study, we constructed a series of prokaryotic expression vectors for generating recombinant proteins of the truncated overlapping forms of pAPN-C. The binding competence of all recombinant proteins to antibodies was detected using a polyclonal antibody against native pAPN on western blot and ELISA. Moreover, an ELISA was carried out to analyze the TGEV-binding activity of these truncated recombinant proteins covering the whole pAPN-C region. The aim of the present study was to provide some information about the potential virus-binding activity of the truncated forms of pAPN-C.
Materials and methods
Virus, plasmid and serum
The swine transmissible gastroenteritis virus (TGEV) strain H was kindly provided by the Division of Swine Infectious Diseases, National Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute of the Chinese Academy of Agricultural Sciences, and propagated using DMEM medium in swine testicular (ST) cells. A previously constructed recombinant plasmid (pMD18-T-pAPN) containing the full-length pAPN gene was used (GenBank accession no. HQ824547) (Sun et al. 2011). Antiserum against the native pAPN (Sigma-Aldrich, St Louis, MO, USA) was prepared previously in BALB/c mice and stored at −80°C.
Construction of expression vectors
Seven pairs of primers were designed based on the nucleotide sequence of the pAPN gene (GenBank accession no. HQ824547). All primers and the location of their amplicons are shown in Supplementary Table 1. The seven overlapping truncated forms, covering the entire pAPN-C gene (1719–2889 bp), were amplified using the plasmid DNA encoding full-length pAPN (pMD18-T-pAPN) as a common template. The PCR reaction was performed in 25 μl containing 100 nM of each forward and reverse primer, 1 μl pMD18-T-pAPN plasmid DNA, 0.25 mM dNTP mixture (each), and 0.25 U ExTaq DNA polymerase (Takara, Dalian, China). The initial reaction was at 94°C for 5 min, and the following cycle was repeated 30 times: 94°C for 30 s, 60°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 10 min. The amplicons were cloned into the restriction sites BamHI and XhoI of the pET-32a vector with a His tag (Novagen, Madison, WI, USA), resulting in the expression plasmids of the truncated forms of pAPN-C.
Expression and purification of the truncated forms of pAPN-C
The expression plasmids of the truncated forms of pAPN-C described above were transformed into Escherichia coli Rosetta (DE3) plysS cells, Gold strain, and the pET-32a empty vector was used as the transformation control. Recombinant bacteria were induced with 0.8 mM IPTG at 37°C for 4 h. Protein expression of all induced recombinant bacteria was analyzed by 12% (v/v) SDS-PAGE.
Inclusion bodies of each truncated pAPN-C recombinant protein were extracted from the lysate of the IPTG-induced host bacteria processed by sonication. The extracted inclusion bodies were washed three times using phosphate buffered saline (PBS, pH 7.4) by repeated centrifugation at 10,000×g. After treating the washed inclusion bodies with 2 × SDS loading buffer, 12% SDS-PAGE was performed to isolate each truncated pAPN-C recombinant protein. After electrophoresis, the gel was stained with 0.3 M KCl until the protein of interest in the gel appeared white. These recombinant pAPN-C proteins were collected from the stained gel by cutting out the band. The excised band was ground into small particles, and then soaked in 1 ml PBS overnight at 4°C. After centrifugation at 15,000×g, the supernatant containing the purified pAPN-C recombinant proteins was collected, and refolded by dialysis in TE buffer (10 mM Tris/HCl, pH 7.5, 1 mM EDTA). The His-tagged protein used as a control was also prepared and purified by the same procedure.
ELISA
To analyze the binding competence of the truncated pAPN-C recombinant proteins to antibodies against native pAPN, an indirect ELISA assay was carried out using antibody against native pAPN. Briefly, wells of an ELISA plate were coated using 1 mg purified recombinant protein l−1 at 4°C for 12 h, then blocked with 50 g skimmed milk l−1 at 37°C for 1 h. After washing three times with PBST (0.5% (v/v) Tween 20, PBS, pH 7.4), 100 μl of antiserum against the native pAPN (1:100 dilution in PBST) was added to the wells, and incubated at 37°C for 1 h. After washing three times with PBST, the plates were incubated with 100 μl HRP-conjugated sheep anti-mouse IgG (1:8,000 dilution in PBST) at 37°C for 1 h. Color development was carried out using TMB solution as the substrate, and the reaction was stopped with 50 μl 2 M H2SO4. The absorbance at 450 nm was measured. The purified His-tag protein was used as the negative control. A sample A450 value/negative control A450 value (S/N) >2 was used as the positive standard.
Western blot
To verify the binding competence of the expressed proteins to antibodies against native pAPN, the truncated pAPN-C recombinant proteins were also subjected to western blot analysis. Briefly, the original purified recombinant proteins were separated by SDS-PAGE on a 15% gel (v/v). After electrophoresis the separated proteins were transferred onto a nitrocellulose (NC) membrane. The NC membranes were blocked with 50 g skimmed milk l−1 in PBST at 37°C for 1 h, then incubated with 100 μl antiserum against native pAPN (1:100 dilution in PBST). After washing three times with PBST, the NC membrane was incubated with 100 μl IRDye 700DX-conjugated affinity purified anti-mouse IgG (H&L) (Goat) (1:5,000 dilution in PBST) at 37°C for 1 h. The NC membrane was washed three times with PBST and then protein bands in the NC membrane were detected using the ODYSSEY Infrared Imaging System (Li-Cor Biosciences).
Binding of the truncated pAPN-C proteins to TGEV
To analyze the virus-binding activity of the truncated pAPN-C recombinant proteins, an indirect ELISA assay was performed using TGEV strain H as the coating antigen. The ELISA procedure was as follows: TGEV particles purified by sucrose density gradient ultracentrifugation were used to coat the ELISA plate wells. After blocking with 50 g skimmed milk l−1, the purified recombinant pAPN-C proteins (1 mg l−1) were added to the wells and incubated at 37°C for 1 h. After washing three times with PBST, 100 μl mouse monoclonal antibody against the His 6 tag (1:1,000 dilution in PBST) was added to the wells and incubated at 37°C for 1 h. The plates were washed three times and then incubated with 100 μl HRP-conjugated sheep anti-mouse IgG (1:8000 dilution in PBST) at 37°C for 1 h. Color development was carried out using TMB solution as the substrate and the reaction was stopped with 50 μl 2 M H2SO4. The A450 was measured. The purified His-tag protein and native pAPN protein were used as negative and positive control, respectively. The sample OD450 value/negative control OD450 value (S/N) >2 was determined as the positive standard.
Results
Expression and purification of the truncated forms of pAPN-C
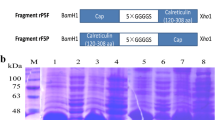
The seven overlapping truncated forms of the pAPN-C gene were generated by PCR, using the plasmid DNA (pMD18-T-pAPN) encoding the entire pAPN gene as a template. The seven truncated forms of the pAPN-C gene were cloned into vector pET-32a, and a recombinant bacterium containing each truncated form was obtained by transforming plasmid DNA into Escherichia coli. After induction with IPTG, seven truncated forms of the pAPN-C gene were expressed (Fig. 1a). Purification of the truncated pAPN-C recombinant proteins was then carried out using the gel-cutting method followed by successive dialysis renaturation. All purified recombinant pAPN-C proteins were of high purity as judged by SDS-PAGE analysis (Fig. 1b).

Expression and purification of the truncated forms of pAPN-C. a Expression of the truncated forms of pAPN-C. Lane M, PageRuler™ Prestained Protein Ladder (10–170 kDa); Lane 1, empty vector control bacteria induced by IPTG; Lanes 2–7, the induced recombinant bacteria of the truncated forms of pAPN-C, rpAPN-C1, rpAPN-C2, rpAPN-C3, rpAPN-C4, rpAPN-C5, rpAPN-C6, and rpAPN-C7, respectively. b Purification of the truncated recombinant proteins of pAPN-C. Lane M, PageRuler™ Prestained Protein Ladder (10–170 kDa); Lanes 1–7, purification of the truncated pAPN-C recombinant proteins, rpAPN-C1, rpAPN-C2, rpAPN-C3, rpAPN-C4, rpAPN-C5, rpAPN-C6, and rpAPN-C7, respectively
Binding competence of the truncated pAPN-C proteins to antibodies against native pAPN
In order to analyze the binding competence of the truncated pAPN-C recombinant proteins to antibodies against native pAPN, an indirect ELISA and western blot were performed using the antiserum against native pAPN. The ELISA showed that all truncated recombinant pAPN-C proteins were recognized by the antiserum against native pAPN (Fig. 2a). In the IRDye 700DX-based western blot, all recombinant pAPN-C proteins showed a strong reaction with the antiserum against native pAPN (Fig. 2b).

Immunogenicity analysis of the truncated pAPN-C recombinant proteins. a ELISA analysis of the truncated pAPN-C recombinant proteins. b Western blot analysis of the truncated pAPN-C recombinant proteins. Lane M, PageRuler™ Prestained Protein Ladder (10–170 kDa); Lanes 1–7, the truncated recombinant proteins of pAPN-C, rpAPN-C1, rpAPN-C2, rpAPN-C3, rpAPN-C4, rpAPN-C5, rpAPN-C6, and rpAPN-C7, respectively; Lane 8, the purified His-tagged protein as negative control
Virus-binding activity of the truncated pAPN-C proteins
The binding activity of the truncated pAPN-C recombinant proteins with TGEV was evaluated using the monoclonal antibody against His-tag by an indirect ELISA. All truncated pAPN-C proteins showed a positive reaction with TGEV particles; the virus-binding activity of the truncated proteins rpAPN-C2 (aa 623–722) and rpAPN-C3 (aa 673–772) were the highest among the truncated pAPN-C recombinant proteins (Fig. 3).

Virus-binding activity of the truncated pAPN-C proteins
Discussion
Aminopeptidase N (APN) is a member of the membrane-bound metallopeptidase family, and has a wide distribution on the surfaces of diverse cell types in humans and most mammals (Lendeckel et al. 2000). APN is a cellular receptor for several coronaviruses (Yeager et al. 1992; Benbacer et al. 1997; Hegyi and Kolb 1998). For swine coronavirus TGEV, the receptor and the catalytic functions, associated with the C and B subunits of pAPN respectively, involve distinct domains of the molecule. Thus pAPN-C holds significant potential for designing an anti-virus strategy against TGEV. In this study, seven overlapping truncated forms of the entire pAPN-C protein were expressed in Escherichia coli. Protein purification, immunogenicity analysis, and tests of the virus-binding activity of these recombinant proteins were carried out in succession.
For protein expression, the vector pET-32a with a medium-sized His-tagged protein was used to generate recombinant pAPN-C proteins. The 18 kDa Trx-His-S Tag protein of the vector pET-32a promoted high-level expression of the target proteins while having little effect on the expressed proteins (Yang et al. 2003; Gasparian et al. 2007). At present, many measures have been reported for the purification of the recombinant proteins expressed in Escherichia coli (Li 2011; Kim et al. 2011; Tzou et al. 2011). In our study, high-purity recombinant pAPN-C proteins were obtained from inclusion bodies by the gel-cutting purification method combined with dialysis refolding, which had been verified as a simple, time-saving, and inexpensive method (Liu et al. 2009; Sun et al. 2011). A polyclonal antibody against native pAPN was used to perform ELISA and western blot analysis of the binding competence of the purified recombinant proteins. The advantage of the polyclonal antibody against the native pAPN was that it recognized any proteins which were consistent with the structure of native pAPN. In our study, all purified recombinant pAPN-C proteins reacted more strongly with antiserum against the native pAPN in both ELISA and western blot. This result provided strong evidence that the purified recombinant proteins possessed similar biological activity to the natural pAPN protein.
Generally, monoclonal antibodies are highly specific, and avoid cross-reaction with non-target proteins. In this study, a monoclonal antibody against His-tag was used to analyze the binding activity of the truncated pAPN-C recombinant proteins with TGEV by an indirect ELISA using the purified TGEV particles as the coating antigen. Of these truncated recombinant proteins, rpAPN-C2 (aa 623–722) and rpAPN-C3 (aa 673–772) showed the highest virus-binding activity. However, rpAPN-C1 (aa 573–672), which overlaps rpAPN-C2, showed low virus-binding activity, while rpAPN-C4 (aa 717–822), overlapping rpAPN-C3, showed medium virus-binding activity. These results suggest that the overlapping region (aa 673–722) between rpAPN-C2 and rpAPN-C3 may be a crucial amino acid region for virus-binding activity of pAPN-C. In addition, the recombinant proteins rpAPN-C4 (aa 717–822) and rpAPN-C5 (aa 773–872) were found to have medium virus-binding activity. The rpAPN-C4 region had been reported previously by Delmas et al. (1994). In this report, the potential virus-binding determinants of pAPN-C are analyzed using a series of truncated recombinant proteins, and the results will provide valuable information for further understanding the function of pAPN-C in infection.
References
Benbacer L, Kut E, Besnardeau L, Laude H, Delmas B (1997) Interspecies aminopeptidase-N chimeras reveal species-specific receptor recognition by canine coronavirus, feline infectious peritonitis virus, and transmissible gastroenteritis virus. J Virol 71:734–737
Delmas B, Rasschaert D, Godet M, Gelfi J, Laude H (1990) Four major antigenic sites of the coronavirus transmissible gastroenteritis virus are located on the amino-terminal half of spike glycoprotein S. J Gen Virol 71:1313–1323
Delmas B, Gelfi J, Haridon LR, Vogel LK, Sjostrom H, Noren O, Laude H (1992) Aminopeptidase N is a major receptor for the enteropathogenic coronavirus TGEV. Nature 357:417–420
Delmas B, Gelfi J, Kut E, Sjöström H, Noren O, Laude H (1994) Determinants essential for the transmissible gastroenteritis virus-receptor interaction reside within a domain of aminopeptidase N that is distinct from the enzymatic site. J Virol 68:5216–5224
Egberink HF, Ederveen J, Callebaut P, Horzinek MC (1988) Characterization of the structural proteins of porcine epizootic diarrhea virus, strain CV777. Am J Vet Res 49:1320–1324
Gasparian ME, Ostapchenko VG, Yagolovich AV, Tsygannik IN, Chernyak BV, Dolgikh DA, Kirpichnikov MP (2007) Overexpression and refolding of thioredoxin/TRAIL fusion from inclusion bodies and further purification of TRAIL after cleavage by enteropeptidase. Biotechnol Lett 29:1567–1573
Gebauer F, Posthumus WP, Correa I, Suñé C, Smerdou C, Sánchez CM, Lenstra JA, Meloen RH, Enjuanes L (1991) Residues involved in the antigenic sites of transmissible gastroenteritis coronavirus S glycoprotein. Virology 183:225–238
Hegyi A, Kolb AF (1998) Characterization of determinants involved in the feline infectious peritonitis virus receptor function of feline aminopeptidase N. J Gen Virol 79:1387–1391
Jongeneel CV, Bouvier J, Bairoch A (1989) A unique signature identifies a family of zinc-dependent metallopeptidases. FEBS Lett 242:211–214
Kenny AJ, Maroux S (1982) Topology of microvillar membrane hydrolases of kidney and intestine. Physiol Rev 62:91–128
Kim YS, Yeom SJ, Oh DK (2011) Production of β-apo-10′-carotenal from β-carotene by human β-carotene-9′,10′-oxygenase expressed in E. coli. Biotechnol Lett 33:1195–1200
Lendeckel U, Kahne T, Riemann D, Neubert K, Arndt M, Reinhold D (2000) Review: the role of membrane peptidases in immune functions. Adv Exp Med Biol 477:1–24
Li Y (2011) The tandem affinity purification technology: an overview. Biotechnol Lett 33:1487–1499
Liu B, Li G, Sui X, Yin J, Wang H, Ren X (2009) Expression and functional analysis of porcine aminopeptidase N produced in prokaryotic expression system. J Biotechnol 141:91–96
Ren X, Glende J, Yin J, Schwegmann-Wessels C, Herrler G (2008) Importance of cholesterol for infection of cells by transmissible gastroenteritis virus. Virus Res 137:220–224
Ren X, Li G, Liu B (2010) Binding characterization of determinants in porcine aminopeptidase N, the cellular receptor for transmissible gastroenteritis virus. J Biotechnol 150:202–206
Reynolds D, Garwes D (1979) Virus isolation and serum antibody-responses after infection of cats with transmissible gastroenteritis virus. Arch Virol 60:161–166
Sánchez CM, Gebauer F, Suñé C, Mendez A, Dopazo J, Enjuanes L (1992) Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 190:92–105
Schwegmann-Wessels C, Bauer S, Winter C, Enjuanes L, Laude H, Herrler G (2011) The sialic acid binding activity of the S protein facilitates infection by porcine transmissible gastroenteritis coronavirus. Virol J 8:435
Semenza G (1986) Anchoring and biosynthesis of stalked brush border membrane proteins: glycosidases and peptidases of enterocytes and renal tubuli. Annu Rev Cell Biol 2:255–313
Sun DB, Feng L, Shi HY, Chen JF, Liu SW, Chen HY, Wang YF (2007) Spike protein region (aa 636–789) of porcine epidemic diarrhea virus is essential for induction of neutralizing antibodies. Acta Virol 51:149–156
Sun DB, Feng L, Shi HY, Chen JF, Cui XC, Chen HY, Liu SW, Tong YE, Wang YF, Tong GZ (2008) Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Vet Microbiol 131:73–81
Sun DB, Wang YQ, Wu GJ, Zhang H, Zhu QH, He XJ, Guo DH, Wu R (2011) A polyclonal antibody against the C subunit of porcine aminopeptidase N expressed in Escherichia coli. Hybridoma 30:457–462
Tzou YM, Huang TS, Huggins KW, Chin BA, Simonne AH, Singh NK (2011) Expression of truncated tobacco osmotin in Escherichia coli: purification and antifungal activity. Biotechnol Lett 33:539–543
Yang Q, Li M, Xu J, Bao Y, Lei X, An L (2003) Expression of gloshedobin, a thrombin-like enzyme from the venom of Gloydius shedaoensis, in Escherichia coli. Biotechnol Lett 25:101–104
Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, Holmes KV (1992) Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357:420–422
Acknowledgments
This work is supported by the State National Key Laboratory of Veterinary Biotechnology (Grant No. SKLVBF201104), the National Natural Science Foundation of China (Grant No. 31001081), and the Technological Innovation Team Building Program of University of Heilongjiang Province (Grant No. 2010td05).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sun, D., Shi, H., Chen, J. et al. Virus-binding activity of the truncated C subunit of porcine aminopeptidase N expressed in Escherichia coli . Biotechnol Lett 34, 533–539 (2012). https://doi.org/10.1007/s10529-011-0795-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-011-0795-1