
Abstract
Lipid A is a major component in the outer membrane of most Gram-negative bacteria. Monophosphoryl lipid A contains no phosphate group at 1-position and can be used as an adjuvant. We constructed an Escherichia coli mutant CW001 by integrating a gene lpxE into the chromosome of E. coli W3110. The gene lpxE encodes an enzyme LpxE which removes the 1-phosphate group of lipid A. CW001 predominantly produces 1-dephosphorylated lipid A in vivo, as adjudged by thin layer chromatography and electro-spray ionization mass spectrometry. This study not only is important for the development of lipid A adjuvants but also provides a novel method for integration of heterologous genes into the chromosome of E. coli.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipopolysaccharide (LPS), known as endotoxin, forms the outer monolayer of the outer membrane in most Gram-negative bacteria. Lipid A is the hydrophobic anchor of LPS and is responsible for the activity of the endotoxin (Wang and Quinn 2010; Raetz et al. 2007; Raetz and Whitfield 2002). Lipid A can be recognized by Toll-Like Receptor 4 (TLR4) existing on the surface of immune cells and trigger various pathophysiological responses, resulting in serious endotoxin shock (Bosshart and Heinzelmann 2007; Munford 2006). The toxicity of lipid A is closely related to its precise chemical structure (Wang et al. 2004, 2006, 2007, 2009).
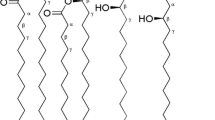
Escherichia coli lipid A contains two phosphate groups and six fatty acid chains (Fig. 1a). The two phosphate groups in lipid A are crucial for its bioactivity; they interact with a cluster of positively charged residues from dimeric TLR4 and MD-2 (Park et al. 2009). Monophosphoryl lipid A (MPLA), a lipid A derivative, which contains only one phosphate group, is much less toxic than the wild type lipid A but still retain many of the immunomodulatory properties (Takayama et al. 1981; Persing et al. 2002). Monophosphoryl lipid A cannot activate caspase-1, one of the key enzymes involved in the synthesis of interleukin-1β (Okemoto et al. 2006). Vaccine development has focused on MPLA analogues which could induce altered cytokine profiles that retain adjuvant activity without toxicity (Baldridge and Crane 1999). Recently, MPLA analogues have been used as vaccine adjuvants and have recently been approved for use in the USA (Mata-Haro et al. 2007; Reed et al. 2009).

Structures of lipid A and MPLA. The blue numbers specify the glucosamine ring positions of lipid A. The black numbers indicate the fatty acid chain lengths of lipid A. a Lipid A purified from E. coli W3110. b MPLA purified from CW001. c Avanti product 699200P, MPLA purified from Salmonella Minnesota R595. d Avanti product 699800P, Synthetic MPLA
Currently, MPLA analogues are prepared either by extracting the lipid A from Salmonella minnesota R595 (Fig. 1c and then removing the 1-phosphate group chemically (Persing et al. 2002), or by chemical synthesis. Salmonella minnesota R595 makes multiple lipid A species which are difficult to separate from each other. Chemically-synthetic lipid A has a slightly different structure from E. coli lipid A (Fig. 1d). Unlike S. minnesota R595, E. coli can only produce one type of lipid A. Here we have constructed an E. coli mutant CW001 which produces MPLA in vivo, by expressing the gene lpxE from Francisella novicida (Wang et al. 2004). The gene lpxE was inserted in the chromosome of CW001. The production of MPLA in CW001 was confirmed by TLC and ESI–MS. This provides a solid foundation for the development of lipid A vaccine adjuvants and the attenuated living vaccine strains. In addition, a rapid and efficient method was also developed for integrating heterologous genes into the E. coli chromosome.
Materials and methods
DNA preparation and PCR techniques
All bacterial strains and plasmids used are listed in Supplementary Table 1. Restrictions enzymes, calf intestine alkaline phosphatase, T4 DNA ligase, and DNA Ladder were from Sangon (Shanghai, China). Plasmid DNA was prepared by using the EZ-10 spin column plasmid mini-preps kit form Bio Basic Inc (Markham, Canada). All the PCR reaction mixtures used in this study are 50 μl containing 10 μl 5× PrimerSTAR buffer, 4 μl dNTP mixture (2.5 mM each), 1 μl plasmid template (100 mg l−1), 1 μl forward primer (20 μM), 1 μl reverse primer (20 μM), 0.5 μl DNA polymerase. The PCR reaction was usually started at 94°C for 5 min, followed by 35 cycles of denaturation (30 s at 94°C), annealing, and extension. At the end, an additional 10 min incubation at 72°C was used. PCR products were purified by using the TIANgel midi purification kit from Tiangen (Beijing, China). Primers synthesis and DNA sequencing were performed by Sangon. The sequences of primers used in this study are listed in Supplementary Table 2.
Chromosomal integration of the lpxE gene in Escherichia coli
The detailed procedure to construct the lpxE mutant strain is shown in Fig. 2. DNA fragment, Fkan, a kanamycin resistance gene flanked by FRT sites, was amplified from the plasmid pKD13 by using primers Fkan-F and Fkan-R. The PCR product was digested with XhoI, and ligated into pWSK29-FnlpxE, which was similarly digested and treated with calf intestine alkaline phosphatase. The resulting plasmid is designated as pWSK29-FnlpxE-Fkan. The lpxE-Fkan cassette was PCR amplified from the plasmid pWSK29-FnlpxE-Fkan by using primers LacZ-F and LacZ-R. During PCR, the annealing was at 55°C for 15 s, and the extension was at 72°C for 150 s. Both ends of the PCR product are the two split fragments from lacZ-α. The PCR products were purified from the gel and transformed into W3110 carrying a plasmid pKD46 (Datsenko and Wanner 2000). The cell suspension was then spread onto agar plates containing 30 μg kanamycin/ml, 24 μg IPTG/ml and 40 μg X-Gal/ml, and the white colonies were selected. The strain containing the correct recombination was designated W3110-lpxE-kan. Next, the antibiotic resistance cassette kan was eliminated from the chromosome using the plasmid pCP20 (Cherepanov and Wackernagel 1995) and the final strain was designated CW001.

The procedure of the chromosomal integration of lpxE gene in E. coli. DNA fragment Fkan, a kanamycin resistance gene flanked by FRT sites, was ligated into pWSK29-FnlpxE to form the plasmid pWSK29-FnlpxE-Fkan. The lpxE-Fkan cassette was PCR amplified and transformed into W3110 carrying a plasmid pKD46. The recombination occurred and the lpxE-Fkan cassette was inserted in the chromosome. At the end, the cassette kan was eliminated from the chromosome using the plasmid pCP20. Rep, replication initiation protein; lacZ-α, a fragment encodes the α subunit of LacZ protein; bla, ampicillin resistance gene
Isolation of lipid A from Escherichia coli
An overnight culture of E. coli was inoculated in 200 ml LB broth, grown to an OD600 of 1, harvested and washed twice with phosphate-buffered saline. Cell pellets were re-suspended in 76 ml chloroform/methanol/water (1:2:0.8, by vol), and held at room temperature for 1 h. Insoluble material was collected by centrifugation at 1000×g for 20 min and washed once with the same solvent. To cleave the glycosidic linkage between polysaccharide and lipid A in LPS, the pellets were re-suspended in 27 ml 12.5 mM sodium acetate (pH 4.5), heated at 100°C for 30 min, followed by sonication. After cooling to room temperature, 30 ml chloroform and 30 ml methanol were added, mixed thoroughly and centrifuged. The lower phase containing lipid A was pooled and dried using a rotary evaporator.
Chromosomal integration of the cat gene in Escherichia coli
Briefly, Fkan was ligated into pBluescript II SK (+), resulting the plasmid pBSK-Fkan. The cat gene was PCR amplified from the plasmid pACYC184, using forward primer Cat-F and reverse primer Cat-R. The primer Cat-F carries a SD sequence and a NotI restriction site at the 5′-end, while the primer Cat-R carries a SalI restriction site at the 5′-end. During PCR, the annealing was at 58°C for 15 s, and the extension was at 72°C for 60 s. The PCR product was digested with NotI and SalI, and ligated into pBSK-Fkan which was similarly digested. The resulting plasmid is designated as pBSK-cat-Fkan, which contains the cat-Fkan cassette under the lac promoter. Next, the cat-Fkan cassette was PCR amplified by using primers lacZ-F and lacZ-R. The PCR products were purified from the gel, and transformed into W3110/pKD46. The cell suspension was spread onto agar plates containing kanamycin, IPTG and X-Gal, and the white colonies were selected. The correct recombination was confirmed by PCR and the strain containing correct recombination is designated W3110-cat-kan. The kan cassette was then eliminated using the plasmid pCP20 and the final strain was designated W3110-cat.
Tachypleus amebocyte lysate assay
Chromogenic end-point Tachypleus amebocyte lysate kit was from Xiamen Houshiji (Xiamen, China). E. coli W3110 and CW001 were grown in LB broth containing IPTG, harvested by centrifugation, and washed three times with phosphate-buffered saline. LPS were prepared using the hot phenol/water method (Westphal and Jann 1965), and quantified using the method described by Lee and Tsai (1999). LPS samples and standard were mixed separately with Tachypleus amebocyte lysate reagent, and incubated at 37°C for 8 min. The activities were determined using p-nitroanilide as chromogenic substrate. The experiment was repeated thrice.
Results
Construction of a MPLA production strain CW001 by chromosomal integration of lpxE gene in Escherichia coli
Monophosphoryl lipid A adjuvants are currently prepared either by chemical synthesis, or by extracting the lipid A from bacteria and then removing the 1-phosphate group chemically (Persing et al. 2002). To construct bacteria that could produce MPLA in vivo, the lpxE gene was integrated in the chromosome of E. coli as shown in Fig. 2. The Fkan DNA fragment was first cloned into the plasmid pWSK29-FnlpxE, to form a plasmid pWSK29-FnlpxE-Fkan which contains a lac promoter and Shine-Dalgarno sequence (SD). The lpxE-Fkan cassette was then amplified by PCR, and transformed into W3110/pKD46. The Red enzymes expressed by pKD46 facilitated the recombination of the lpxE-Fkan cassette at the lacZ locus in the chromosome. The insertion of lpxE-Fkan cassette would break the lacZ gene; therefore E. coli containing correct insertion of the lpxE-Fkan in the chromosome would show white colonies on plates containing IPTG and X-gal. At the end, the plasmid pCP20 which can transiently express FLP recombinase was introduced into the host cell, and the kan was removed. The correct insertion of the lpxE and the later removal of kan were confirmed by the loss of kanamycin resistance and PCR analysis. This lpxE mutant E. coli is designated CW001.
The growth rate of CW001 was found similar to that of wild type W3110. The stability studies showed that the integrated lpxE could stay at the lacZ locus in the chromosome of CW001 even after 50 generations.
Analysis of MPLA produced by CW001
Lipid A were isolated from strains of W3110, CW001 and W3110/pWSK29-FnlpxE, and separated on TLC. A major band, the typical lipid A band, was shown on the TLC in samples isolated from W3110 (Fig. 3, lane 1), but it is significantly weaker in samples isolated from W3110/pWSK29-FnlpxE and CW001 (Fig. 3, lanes 2, 3). However, a more rapidly migrating substance, presumed to be 1-dephospho-lipid A was present in both W3110/pWSK29-FnlpxE and CW001 (Fig. 3, lanes 2, 3). Due to the hydrophilic property of phosphate, lipid A containing two phosphate groups should migrate slower on TLC than 1-dephospho-lipid A. This experiment suggests that LpxE expressed from the chromosome of CW001 functions equally as well as that expressed from the plasmid pWSK29 in the strain W3110/pWSK29-FnlpxE (Fig. 3, lanes 2, 3).

Analysis of lipid A and 1-dephospho-lipid A on TLC. The lipid A samples were dissolved in chloroform/methanol (4:1, by vol), spotted onto a silica gel 60 TLC plate, which was developed in chloroform/methanol/acetic acid/water (40:25:4:2, by vol). After drying, the lipid A bands on the TLC plate were visualized by spraying a 10% H2SO4 (by vol) in ethanol, followed by charring at 180°C. Lane 1, sample from W3110; lane 2, sample from CW001; lane 3, sample from W3110/pWSK29-FnlpxE
Lipid A and its derivatives isolated from W3110, CW001 and W3110/pWSK29-FnlpxE were further analyzed by ESI/MS. The lipid A isolated from W3110 created a major peak at m/z 1797.5 (Fig. 4a), suggesting the peak is created by the molecular ion [M-H]- of lipid A (Fig. 1a). The peak at m/z 1717.5 might arises by loss of a phosphate during the extraction procedure. The peaks at m/z 1819.5 likely arises by the sodium adduct of the molecular ion of lipid A; the peak at m/z 1769.5 could be created by minor lipid A species which differ by 28 Da, that is, two methylene units. The 1-dephospho-lipid A isolated from CW001 and W3110/pWSK29-FnlpxE showed the similar spectra which contain a major peak at m/z 1717.5 as expected (Fig. 4b, c). These results indicate that the lpxE gene in the chromosome of CW001 was expressed and its product LpxE can modify the structure of lipid A in vivo.

ESI/MS analysis of lipid A or 1-dephospho-lipid A isolated from E. coli W3110 (a), CW001 (b) and W3110/pWSK29-FnlpxE (c). Lipid A samples were dissolved in chloroform/methanol (4:1, by vol) and subjected to ESI/MS in the negative ion mode. The mass spectra were acquired on a Waters SYNAPT Q-TOF mass spectrometer equipped with an ESI source. Data acquisition and analysis were performed by using MassLynx V4.1 software
Comparison of the stimulation activities of LPS isolated from W3110 and CW001
LPS was purified from both W3110 and CW001, and their stimulation activities were measured by tachypleus amebocyte lysate. The stimulation activity of W3110 LPS (0.005 μM) was 0.3 EU ml−1, while the activity of the same amount of CW001 LPS was 0.64 EU ml−1. The dephosphorylation of lipid A in LPS produced by CW001 might have higher binding affinity to the Factor C, the LPS-sensitive protein, and facilitate the agglutination reaction. Earlier studies have shown that the molecular interaction between LPS and Sushi 1 domain of Factor C is basically the hydrophobic interaction between the lipid tail of LPS and the Sushi 1 peptide (Tan et al. 2000; Li et al. 2006). Therefore, the enhanced hydrophobicity of MPLA produced in CW001 due to the absence of 1-phosphate might cause the stronger interaction with Sushi 1 peptide of Factor C.
Chromosome integration of the report gene cat in Escherichia coli
To test the generality of the method we used for the integration of lpxE in the chromosome of E. coli, we also integrated the reporter gene cat into the chromosome of E. coli. The integration of the cat gene into the chromosome of E. coli was evaluated by antibiotics. As shown in Fig. 5, wide type W3110 could not grow on plates containing chloramphenicol or kanamycin, but W3110-cat-kan and W3110/pBSK-cat-Fkan grew well on all plates. W3110-cat, however, could grow on the plate containing chloramphenicol and IPTG, but not on the plate containing kanamycin. This indicates that the reporter gene cat has been inserted and the kan gene has been removed in the chromosome of W3110-cat. This experiment proves that the method we used in this study has a generality for the insertion of a heterologous gene in the chromosome of E. coli.

Analysis of different E. coli strains containing the reporter gene cat. (A) The plain LB plate. (B) The LB plate containing 30 μg ml−1 chlorophenicol. (C) The LB plate containing 30 μg ml−1 kanamycin. The plates were divided into four section on which the following four E. coli strains were spread. 1, W3110; 2, W3110/pBSK-cat -Fkan; 3, W3110-cat-kan; 4, W3110-cat
Discussion
MPLA has become an important target molecule for the development of novel vaccine adjuvants. To obtain MPLA, the 1-phosphate group in lipid A is usually removed by chemical method. In this study, we constructed a recombinant, plasmid-free E. coli strain CW001 that predominantly produce 1-dephosphorylated lipid A, by expressing lpxE in the chromosome of W3110. The lpxE gene in the chromosome of CW001 is stable and its expression does not affect the growth of the cell. The stimulation activity of CW001 LPS is higher than W3110. Strains expressing lpxE possess 1-dephospho-lipid A in their LPS and could have many potential applications. Lipid A detoxification by LpxE may be a useful tool for improving live vaccines to prevent infectious disease.
In addition, we also developed a rapid and efficient method to integrate heterologous genes in the chromosome of E. coli by combining the Red recombination system (Sharan et al. 2009; Albermann et al. 2010; De Mey et al. 2010) and the blue-white screen. Heterologous genes are usually expressed in E. coli using plasmids as the vector. But plasmids in bacteria are not stable and usually need antibiotics as selection markers. Therefore, chromosomal integration is the ideal approach to express heterologous genes in E. coli.
Because many E. coli plasmids, such as pMD18-T, pMD19-T, pGEM-T, pBluescript II SK(±), pUC18, pUC19, and pWSK29, contain the lacZ gene or the lacZ-α segment, the lacZ locus in the chromosome of E. coli could be selected at the special location for the integration of heterologous genes. The lacZ segment in plasmids usually contains the lac promoter and an internal multiple cloning site which facilitates the expression and insertion of heterologous genes. After insertion of the heterologous gene, the lacZ segment is separated into two parts which simply become the homology extensions for the integration of the gene at the lacZ locus of chromosome in E. coli. The homology extensions produce by this way are long, which significantly reduces the rate of false-positive clones by the Red recombination system. By combining the Red recombination and the blue-white screen, almost all white colonies showed accurate integration of lpxE or cat at the lacZ locus in the chromosome of E. coli, suggesting that the method has the generality for the insertion of genes into the chromosome of E. coli.
References
Albermann C, Trachtmann N, Sprenger GA (2010) A simple and reliable method to conduct and monitor expression cassette integration into the Escherichia coli chromosome. Biotechnol J 5:32–38
Baldridge JR, Crane RT (1999) Monophosphoryl lipid A (MPL) formulations for the next generation of vaccines. Methods 19:103–107
Bosshart H, Heinzelmann M (2007) Targeting bacterial endotoxin: two sides of a coin. Ann N Y Acad Sci 1096:1–17
Cherepanov PP, Wackernagel W (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645
De Mey M, Maertens J, Boogmans S, Soetaert WK, Vandamme EJ, Cunin R, Foulquie-Moreno MR (2010) Promoter knock-in: a novel rational method for the fine tuning of genes. BMC Biotechnol 10:26
Lee CH, Tsai CM (1999) Quantification of bacterial lipopolysaccharides by the purpald assay: measuring formaldehyde generated from 2-keto-3-deoxyoctonate and heptose at the inner core by periodate oxidation. Anal Biochem 267:161–168
Li P, Sun M, Wohland T, Ho B, Ding JL (2006) The molecular mechanism of interaction between sushi peptide and Pseudomonas endotoxin. Cell Mol Immunol 3:21–28
Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC (2007) The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316:1628–1632
Munford RS (2006) Severe sepsis and septic shock: the role of gram-negative bacteremia. Annu Rev Pathol 1:467–496
Okemoto K, Kawasaki K, Hanada K, Miura M, Nishijima M (2006) A potent adjuvant monophosphoryl lipid A triggers various immune responses, but not secretion of IL-1beta or activation of caspase-1. J Immunol 176:1203–1208
Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458:1191–1195
Persing DH, Coler RN, Lacy MJ, Johnson DA, Baldridge JR, Hershberg RM, Reed SG (2002) Taking toll: lipid A mimetics as adjuvants and immuno modulators. Trends Microbiol 10:S32–S37
Raetz CRH, Whitfield C (2002) Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700
Raetz CRH, Reynolds CM, Trent MS, Bishop RE (2007) Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem 76:295–329
Reed SG, Bertholet S, Coler RN, Friede M (2009) New horizons in adjuvants for vaccine development. Trends Immunol 30:23–32
Sharan SK, Thomason LC, Kuznetsov SG, Court DL (2009) Recombineering: a homologous recombination-based method of genetic engineering. Nat Protoc 4:206–223
Takayama K, Ribi E, Cantrell JL (1981) Isolation of a nontoxic lipid A fraction containing tumor regression activity. Cancer Res 41:2654–2657
Tan NS, Ng ML, Yau YH, Chong PK, Ho B, Ding JL (2000) Definition of endotoxin binding sites in horseshoe crab Factor C recombinant sushi proteins and neutralization of endotoxin by sushi peptides. FASEB J 14:1801–1813
Wang X, Quinn PJ (2010) Lipopolysaccharide: biosynthetic pathway and structure modification. Prog Lipid Res 49:97–107
Wang X, Karbarz MJ, McGrath SC, Cotter RJ, Raetz CRH (2004) MsbA transporter-dependent lipid A 1-dephosphorylation on the periplasmic surface of the inner membrane: topography of Francisella novicida LpxE expressed in Escherichia coli. J Biol Chem 279:49470–49478
Wang X, Ribeiro AA, Guan Z, McGrath SC, Cotter RJ, Raetz CRH (2006) Structure and biosynthesis of free lipid A molecules that replace lipopolysaccharide in Francisella novicida. Biochemistry 45:14427–14440
Wang X, Ribeiro AA, Guan Z, Abraham SN, Raetz CRH (2007) Attenuated virulence of a Francisella mutant lacking the lipid A 4′-phosphatase. Proc Natl Acad Sci USA 104:4136–4141
Wang X, Ribeiro AA, Guan Z, Raetz CRH (2009) Identification of undecaprenyl phosphate-beta-d-galactosamine in Francisella novicida and its function in lipid A modification. Biochemistry 48:1162–1172
Westphal O, Jann K (1965) Bacterial lipopolysaccharides. Extraction with phenol water and further applications of the procedure. In: Whistler RL (ed) Methods in carbohydrate chemistry. Academic Press, New York, pp 83–91
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 30770114 and 30870074), the 111 Project (No. 111-2-06) and Basic Research Priorities Program of Jiangsu Province (BK2009003).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chen, J., Tao, G. & Wang, X. Construction of an Escherichia coli mutant producing monophosphoryl lipid A. Biotechnol Lett 33, 1013–1019 (2011). https://doi.org/10.1007/s10529-011-0521-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-011-0521-z