
Abstract
Colorectal cancer (CRC) is a prevalent cancer with high morbidity and mortality rates worldwide. Late diagnosis is a significant contributor to low survival rates in a minority of cases. The study aimed to perform a robust pipeline using integrated bioinformatics tools that will enable us to identify potential diagnostic and prognostic biomarkers for early detection of CRC by exploring differentially expressed genes (DEGs). In addition to, testing the capability of replacing chemotherapy with plant extract in CRC treatment by validating it using real-time PCR. RNA-seq data from cancerous and adjacent normal tissues were pre-processed and analyzed using various tools such as FastQC, Kallisto, DESeq@ R package, g:Profiler, GNEMANIA-CytoScape and CytoHubba, resulting in the identification of 1641 DEGs enriched in various signaling routes. MMP7, TCF21, and VEGFD were found to be promising diagnostic biomarkers for CRC. An in vitro experiment was conducted to examine the potential anticancer properties of 5-fluorouracile, Withania somnifera extract, and their combination. The extract was found to exhibit a positive trend in gene expression and potential therapeutic value by targeting the three genes; however, further trials are required to regulate the methylation promoter. Molecular docking tests supported the findings by revealing a stable ligand-receptor complex. In conclusion, the study’s analysis workflow is precise and robust in identifying DEGs in CRC that may serve as biomarkers for diagnosis and treatment. Additionally, the identified DEGs can be used in future research with larger sample sizes to analyze CRC survival.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is a prominent global health concern, it is considered the third most commonly diagnosed cancer and the second leading cause of cancer-related mortality worldwide, as reported by the International Agency for Research on Cancer (IARC). The incidence of CRC is raising in many countries, largely attributable to lifestyle and dietary changes. According to the IARC’s 2020 estimates, there were approximately 1.9 million new cases and 935,000 deaths worldwide due to CRC. CRC is often asymptomatic in its early stages, making it a challenging disease to diagnose. However, most CRCs develop from precancerous polyps, which can be detected and removed through screening. The stage of CRC at diagnosis is a major prognostic factor, with patients with localized disease having significantly better survival outcomes than those with regional or distant metastasis. Individuals who are diagnosed with early stage malignancies classified as Dukes A (T1/2N0M0) exhibit a 5-year survival rate of 93.2%. In contrast, individuals diagnosed with advanced-stage cancers categorized as Dukes C (T3/4N1/2M0) experience a significant decrease in the 5-year survival rate, which lowers to 47.7% (Morris et al. 2010). Hence, early recognition of precancerous lesions in the colorectal significantly contributes to enhancing the 5-year rate of survival (Shah et al. 2014). Besides, the improvement of the prognosis as well the increase of survival rates among patients are affected by CRC on timely treatment and early detection (Arhin et al. 2022; Khazaei et al. 2018).
It is difficult to recognize tumors smaller than 1 cm using traditional diagnostic techniques because the sensitivity and specificity of conventional tumor markers are inadequate. Which led to the necessitating identification of corresponding biomarkers (Mármol et al. 2017; Li et al. 2018).
The chemotherapeutic agent known as 5 fluorouracil (5-FU) is extensively utilized in the management of various malignancies, such as colorectal and breast cancers, as well as aerodigestive tract tumors (Sargent et al. 2001). Withania somnifera (L.) Dunal is the scientific name of a plant commonly known as ashwagandha. In Ayurvedic researches, it is sorted as a rejuvenator and it is utilized to enhance mental and physical states, restore the body when incapacitated, and promote longevity. W. somnifera has been found to be beneficial for various neurological conditions, including Alzheimer’s disease, epilepsy, cerebral ischemia, Parkinson’s disorders, and tardive dystonia, as reported by Kulkarni and Dhir (2008); Mukherjee et al. (2021). In addition to its neurological benefits, W. somnifera exhibits a number of other pharmacological qualities, including anti-inflammatory, anti-diabetic, cardioprotective, and anti-tumor capabilities (Behl et al. 2020; Logie and Vanden Berghe 2020).
Bioinformatics analysis has a great role in differential expression analysis to identify DEGs in CRC and other cancer types. Several investigations have been carried out to examine gene expression in CRC through the utilization of microarray and RNA-sequencing technology, alongside publicly available databases such as the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA). Guo et al. (2017) conducted a study with the objective of identifying potential genes and pathways in CRC by utilizing four GEO cohorts where a total of 292 differentially expressed genes (DEGs) were identified from the four datasets, with 165 genes upregulated in addition to 127 genes downregulated. In their study, Wu et al. (2017) conducted an analysis of gene and microRNA expression profiles obtained from GEO datasets. Their findings revealed the identification of seven downregulated differentially expressed miRNAs and 13 upregulated DE miRNAs, as well as approximately 600 upregulated DEGs as well as 283 downregulated genes (Liang et al. 2016; Sun et al. 2019).
Additionally, molecular docking plays a role in validating the anti-tumor effect of W. somnifera extract against different targets, in our case the resulted DEGs, by finding the ligand-receptor interaction that has greater stability by lower binding affinity (Tantawy et al. 2020).
Based on the aforementioned literature, the objective of this study is to identify the most notable and robust DEGs associated with CRC through the utilization of integrated bioinformatics methodologies and test the cytotoxicity effect of 5-FU (as a chemotherapy treatment used for CRC) and W. somnifera extract on HCT-116 cells. In addition, we aim to test the expression of the most three potential DEGS resulted from in-silico analysis by q-PCR to validate them on a molecular level. Finally, we intent to validate the efficacy of W. somnifera extract by knowing its binding affinity to the target genes compared to other ligands using AutoDock tools. Thus, this study will give more details about the ability of our robust bio-informatics pipeline to investigate the most potential differential gene expression in CRC, which may contribute in recognizing new CRC bio-markers, as well as the capability to replace the chemotherapy as 5-FU with W. somnifera extract.
Methodology
Data Retrieval
An investigation for the availability of RNA-Seq transcriptomic data related to CRC from the GEO (Barrett et al. 2012) and Sequence Read Archive (SRA) were done. These databases are managed by the National Center for Biotechnology Information for profiling gene expression and RNA methylation. Microarrays and RNA-Seq techniques provide high-throughput genomic data screening. The accession number GSE156451 was used to retrieve raw RNA sequence paired-end data for 50 samples, 25 tumor samples, and 25 normal tissue samples.
Pre-processing of Raw Data
Raw Data Quality Check
The quality of the reads obtained with FastQ was checked using FASTQC (Andrews et al. 2010) followed using MultiQC (Ewels et al. 2016) for combining FASTQC reports. The adapters in the raw data were trimmed using Cutadapt (version 4.1) (Martin 2011) with normal parameters. Read quality was retested after adapter trimming which were improved using by Trimmomatic and remove contamination that appeared as over-represented sequences. Reads with a Phred quality score of > 25 and minimum length of 36 nt were selected.
Reads Mapping to Reference Genome and Gene Expression Quantification
Good-quality reads were mapped to the latest human genome assembly (GRCh38), which was retrieved from the Ensemble genomic browser with the corresponding annotation GTF file using Kallisto pseudo-aligner (version 0.46.0) (Bray et al. 2016) for quantifying the abundance of RNA-seq data. Kallisto can process both single- and paired-end reads and provides the number of transcripts per million mapped reads (TPM). In this study, the pair-ended default running mode were used, in which, FASTQ files were represented as pairs. Gene expression quantification was performed automatically through Kallisto after the pseudoalignment step and a quantification table was generated as. tsv file format, which was then used for the DeSeq2 R package.
Differential Gene Expression Analysis
Gene expression quantification data were transferred as gene expression data to the Deseq2 package in R software (Love et al. 2014) to investigate significant DEGs in both colorectal tumor samples and nearby non- cancerous samples (control). Relevant parameters ((|log2FC|> 1.2 and padj < 0.05) were set to filter out the DEGs. The DESeq2 package were used to pair the information for all samples by employing a design of the form “Samples + Tissue” for the Sample Table. The “samples” column was used to record the patient’s identification number, while the “tissue” column was used to designate the tissue type, whether normal or cancerous tissue.
Visualization of Differentially Expressed Genes
The data were visualized by RStudio (R version 4.2.0) to obtain the maximum perspective on the resulting data, diverse types of plotting were implemented and maintained. Among these plots is the volcano plot, which is a scatter plot that represents the differential expression of genes in this study. In addition, a heatmap was applied using a heatmap function to plot the value versus frequency, in which annotation colors were established, and data scaling was performed to provide a color scale for each map’s values
Network Construction
The GeneMANIA-Cytoscape app was used to study the interactions and correlations between DEGs in the dataset, in which direct physical interactions, pathway interactions, and co-expression interactions were only considered for the Homo sapiens database. The top ten most closely related genes were included in the network construction. The STRING database was used to visualize gene interactions based on the K-means clustering method, enabling the identification of significant hub gene nodes for each network. Moreover, the CytoHubaa blugin in Cytoscape was used to calculate the Maximal Clique Centrality and other 6-centralities as: closeness, bottleneck, betweenness, eccentricity, radiation, and stress (Chin et al. 2014).
Functional Enrichment Analysis
The g:Profiler enrichment analysis was carried out by analyzing the KEGG and Reactome pathways to explore related pathways, biological processes, and molecular functions (MFs) of the aforementioned DEGs. The g:Profiler cut-off score was used with a detection rate threshold of 0.05 FDR. The parameters utilized to detect significant genes included the number of genes in the dataset (1641), the statistical domain was limited to annotated genes, and a significant threshold of Benjamini–Hochberg FDR was applied, along with a user threshold of 0.05. These parameters were used as the basis for the selection of highly expressed genes.
Cell Viability Assay
Cell Culture
HCT-116 cells derived from CRC tissues were procured from Nawah Scientific Inc. (Mokatam, Cairo, Egypt). These cells were cultured in RPMI media supplemented with 100 mg/mL streptomycin, 100 units/mL penicillin, and 10% heat-inactivated fetal bovine serum under humidified conditions in a 5% (v/v) CO2 atmosphere at 37 °C.
Sulforhodamine B (SRB) Cytotoxicity
Cell viability was assessed using the SRB assay to ascertain the IC50 concentration for each treatment group. A suspension containing 5 × 103 cells was seeded in each well of a 96-well plate and subsequently incubated in complete media for 24 h. Subsequently, the cells were treated with 100 L of media containing various drug concentrations. After 72 h of drug exposure, the cells were fixed by substituting the media with 150 L of 10% trichloroacetic acid (TCA) and incubated at 4 °C for 1 h. The TCA solution was removed, and the cells were washed five times with distilled water.
An amount of 70 L SRB solution containing 0.4% (weight/volume) was introduced and incubated under light-deprived conditions at ambient temperature for 10 min. The plates were washed three times with 1% acetic acid solution, followed by an overnight period of air-drying. Subsequently, 150 L of TRIS (10 mM) was added to facilitate dissolution of the protein-bound SRB stain. The absorbance was then measured at 540 nm with the aid of a BMG LABTECH®- FLUOstar Omega microplate reader (Ortenberg, Germany), according to the sources cited (Skehan et al. 1990; Allam et al. 2018). The HCT-116 cell lines were categorized into four groups, as shown in Table 1, based on drug type and concentration. Each drug was administered at five different concentrations to determine the IC50 values.
RNA Extraction and cDNA Synthesis
Total RNA was extracted and purified from the cell line pellets using the Qiagen RNeasy Mini kit (Cat No.74104) following the manufacturer’s instructions. RNA samples were quantified, and their quality was assessed using a Nanodrop spectrophotometer at A230, A260, and A280. First-strand cDNA synthesis was performed on RNA samples obtained from the four groups using the RevertAid First-Strand cDNA Synthesis Kit (Cat K1622, Thermo ScientificTM) according to the manufacturer’s instructions. The cDNA was synthesized using a Bio-Rad TM 100 Thermal Cycler.
Real-Time PCR to Test Gene Expression
The cDNA was then amplified with the TB Green Permix EXTTaq PCR Master Kit in a 48-well plate using the Stratagene Mx3005P, Agilent Technologies, as follows: 30 s. at 95 °C for enzyme activation, followed by 45 cycles of 5 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C for the amplification step. A quantity of 0.25 uM was used from both primers specific for each target gene. The total volume of the reaction was completed with ddH2O of 20 µL. The relative gene expression foldChange was calculated using the PCR analysis R package according to the equation \(2^{ - \Lambda \Lambda C_{\text{t}} }\).
Molecular Docking and the Prediction of Pharmacological Targets
The X-ray crystallographic structure of each gene was obtained from the RCSB PDB (Protein Data Bank) (RCSB.org) while the 2D structure of each ligand was obtained from PubChem. The Drug Gene Interaction Database (DGIdb) and Drug Interaction Checker were used to identify possible druggable gene interactions of our target genes and drug–drug interactions, respectively. The Withania somnifera extract was searched in the drug interaction checker to identify ligands that may have common active ingredients.
Precise docking was performed using AutoDocvina v.1.2.0. The binding energy (measured in kcal/mol) was used to assess the precision of the docking process with 70 runs taken and a maximum of 2000 interactions, as well a population size of 200, and an energy threshold of 100. Furthermore, during each iteration the minimum torsional, transitional, and rotational values were evaluated. Then, the configuration with the lowest energy was selected. The hydrophobic and electrostatic preferences were set to 1. To identify the binding site of the target, grid box dimensions were adjusted using Mgltools to ensure a distance of 1 Å. Finally, Pymol software was used to visualize ligand-target binding.
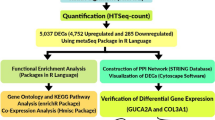
Methodology Diagram
See Fig. 1.

Diagrammatic methodology pipeline
Results and Discussion
Check for Data Quality
The sequence count in the 100 samples ranged from 10 M to 45 M, for all samples, the duplicated read counts accounted for more than 50% of the total read count, which was expected in RNA-seq experiments. As they showed some adapter contamination, we applied cutadapt software to cut this part from the read.
Although the quality Phred score of the samples ranged from 30 to 40, the quality of the reads in terms of adapters was not significant, as the MultiQC report showed remaining adapters in 17 samples (two of them failed and others were not satisfied). The over-represented sequences were analyzed by BLAST and were shown to be bacterial and viral contaminants in the GEO datasets. Consequently, quality trimming was applied to the trimmed reads, which resulted in the removal of approximately 85 percent of the low-quality reads or contaminants, as shown in Fig. 2a–c.

a Mean quality score, b GC content after trimming and c Heat-map status check
Differentially Gene Exression
The Kallisto aligner succeeded in quantifying 204,563 reads in each sample and resulted in an expression matrix containing the ensemble transcript ID for each transcript and its abundance. Screening the control and tumor samples using DESeq2 with the previously mentioned parameters resulted in the identification of 1641 DEGs, including 773 upregulated genes and 869 downregulated genes. The top 20 upregulated and 20 downregulated genes were investigated for use in the wet lab experiment validation using real-time PCR, as shown in Tables 2 and 3. Matrix metallopeptidase 7 (MMP7) has crucial regulatory roles in numerous pathophysiological processes in humans. Since its discovery, MMP-7 has been identified as a regulatory protein in wound healing, bone development, and remodelling. Subsequently, it was shown that MMP-7 regulates the formation and development of malignancies, mediates the proliferation, differentiation, metastasis, and invasion of several types of cancer cells through multiple methods, and is overexpressed in cancer cells. (Liao et al. 2021). In addition, transcription factor-21 (TCF-21), also called epicardin, capsulin, or Pod1, is involved in epithelial-mesenchymal interactions, including epithelial differentiation and branching morphogenesis, during kidney and lung morphogenesis. Possible participant in the specification or differentiation of one or more subsets of epicardial cell types Helix-loop-helical protein structures. TCF-21, which is positioned on 6q23, is known to function as a tumor suppressor and is dysregulated in several malignancies, including breast cancer, gastric cancer, and clear cell renal cell carcinoma (Liao et al. 2021). Furthermore, the vascular endothelial growth factor D (VEGFD) gene, which is stimulated by chronic inflammation, is essential for tumor angiogenesis, tumor growth, and tumor spread (Hu et al. 2015).
Visualization of DEGs
Typically, the most upregulated genes lie towards the right of the zero value of the x-axis (logFoldChange), while the most downregulated genes are towards the left. Additionally, the most statistically significant genes are usually scattered towards the top, as shown in Fig. 3a.

a Volcano plot and b heatmap visualizing the CRC DEGs
Depending on the log2FoldChange value (positive or negative), we determined which of the differentially expressed results were upregulated and which were downregulated. When comparing the number of up-regulated genes with their respective plots, it can be observed that the colored spots in blue on the right side of the volcano plot matched the aforementioned number of up-regulated genes. It can be concluded that the case would be similar to that of the down-regulated genes. Therefore, volcano plots can be a reliable visualization method for identifying significant DE genes.
Based on the RNA-Seq data, only the first 500 DE genes were selected and visualized using a heatmap to demonstrate the relationship between the values and frequencies of these genes, as shown in Fig. 3b. Heatmap clustering analysis was used to cluster the DEGs into two groups. Considering the high number of genes observed in this section. Furthermore, the top bar indicates which of them are normal (orange) patients and which are patients with primary tumors (blue).
Network Analysis
DEGS interactions were investigated using GeneMania-cytoScape and CytoHubba to rank the nodes depending on MCC calculation and other centralities, such as closeness and betweenness. According to CytoHubba, REG1B, TCF-21, MMP7, and VEGF-D were among the top 20 ranking genes, depending on betweenness. This was also validated by STRING, which resulted in a network of 36 nodes and 13 edges representing the interactions between the nodes, which could be physical, pathway, or co-expression interactions. The top five hub genes based on the node ranking scores were MMP7, REG1A, GUCA2A, UGT2B17, and DEFA6, with scores ranges from 0.6:0.4. In addition, STRING applies K-means clustering and provides three clusters of interacting genes, as shown in Figs. 4 and 5.

Network analysis of the top 40 CRC DEGs

Clustering network of the top 40 CRC DEGs after applying K-means clustering method
Enrichment Analysis of CRC DEGS
Enrichment analysis of DEGs for CRC using g:Profiler software, the database identified 1573 out of 1641 genes and converted them to ID Entrez using GO Molecular function, GO Biological Process, GO Cellular Component, KEGG pathways, and Reactome Pathways, as shown in Fig. 6. Gene Ontology (GO) analysis indicated that alterations in the biological process (BP) of the DEGs were considerably enriched in the stimulus response, immune system process, and immune response. This study revealed that alterations in cell composition (CC) were predominantly concentrated in the cell periphery, with 512 genes exhibiting enrichment. Additionally, 476 genes were enriched in the plasma membrane, whereas 368 genes were enriched in the extracellular region. Differential expression analysis revealed that alterations in the MF of DEGs were predominantly enriched in three categories: signaling receptor regulator activity (89 genes), signaling receptor binding (166 genes), and cytokine activity (53 genes). KEGG pathway analysis revealed that DEGs were significantly enriched in cytokine–cytokine receptor interactions, chemokine signaling pathways, and neuroactive ligand–receptor interactions as shown in Fig. 7a–d.

Enriched analysis of CRC DEGs

a Enrichment analysis for molecular function, b enrichment analysis for biological process, c enrichment analysis for cellular component and d enrichment pathways of CRC
The analysis showed that MMP7, AGT, ADORA3, VEGFD, and CCL2 are common genes in many biological processes, MFs, and pathways, in addition to those reported in the literature.
Cytotoxicity Results
To determine the IC50 concentration for each treatment, cell viability was evaluated using the SRB assay and the absorbance was measured at 540 nm using a microplate reader (BMG LABTECH®- FLUOstar Omega model (Ortenberg, Germany) (Skehan et al. 1990; Allam et al. 2018).
The SRB test results in the following IC50 values for each treatment group are shown in Tables 4, 5 and 6 and Figs. 8, 9 and 10.

Concentration against cell viability in extract group

Concentration against cell viability in mixture group

Concentration against cell viability in 5- FU group
Real-Time PCR Results
The resulting data were analyzed using the PCR analysis package in R, which uses \(2^{ - \Lambda \Lambda C_{\text{t}} }\) method according to Hu et al. (2015), Schmittgen and Livak (2008) and is illustrated as bar graphs, as shown in Fig. 11a–c. Relative expression of MMP7 was 1.1, 1.3 and 0.7 in groups II, III, and IV, respectively. In contrast, the relative expression of TCF21 was 0.4, 0.09 and 0.1 for the three groups. In addition, the relative expression of the VEGFD gene was 1.4, 1.9 and 1.3 in the same three groups.

a MMP7 foldChange compared to control group, b TCF21 foldChange compared to control group and c VEGFD foldChange compared to control group
This also was proved by testing the significant difference depending on p-value using ANOVA test, Shapiro test and Tuckey test. For MMP7, The Shapiro test gives a p-value = 0.2818 which means that this data is normalized. Additionally after analyzing the data using both ANOVA and TukeyHSD tests, the results showed significance between group I and group IV with p-value = 0.0417 “*”. In case of VEGF-D gene, the results revealed that the WS extract group (III) gave the most up-regulation by 0.9 foldChange compared to control group (I), with significant p-value = 0.038 (**). However, the combination group (IV) was slightly over-expressed by 0.3 with no significant p-value.
Finally, for TCF-21 the results showed that the group III gave the most significant down-regulation by 0.91 foldChange compared to control group with significant p-value = 0.0000299 “**” by both ANOVA and Tuckey tests. In addition to Shapiro test p-value = 0.1197 which showed that this data is normalized. This can be concluded that there was significant difference between all groups.
Molecular Docking Results
Withanolide A has a low binding affinity for MMP7 in the catalytic part of the protein (−12). Withaferin A has the lowest binding affinity with VEGFD and TCF21 in the catalytic part of the protein (−10.3 and −10.1, respectively) as shown in Figs. 12, 13 and 14 and Tables 7, 8 and 9 and these binding values revealed the strength of the interaction between those ligands and their target.

MMP7 active site docked with Withanolide A with MMP7

VEGFD active site docked with Withanolide A with VEGFD

TCF21 active site docked with Withanolide A with TCF21
Discussion
CRC is a common gastrointestinal neoplasm that is distinguished by high rates of morbidity and mortality. Early diagnosis and corresponding intervention are considered the most effective approaches for increasing survival time and decreasing the mortality caused by CRC (Arhin et al. 2022; Khazaei et al. 2018). There is an unrelenting necessity for potential biomarkers discovery of this type of cancer to be identified, which will promote early detection and hence a higher treatment rate (Huo et al. 2018; Chen and Ke 2021). Several studies involving microarray analysis have documented the recognition of gene expression patterns in adenomas and cancers. To determine potential CRC DEGs, the gene expression profiles of GSE156451 were analyzed of 50 paired-end tumor samples and their adjacent normal tissue samples. We recognized 1641 DEGS (773 upregulated and 896 downregulated genes). To have a better understanding of the interactions between the resulted DEGS, we performed a functional enrichment analysis on GProfiler, The analysis showed that MMP7, AGT, ADORA3, VEGFD, and CCL2 are common genes in many biological process, MFs and pathways, which also reported in Chun et al. (2015); Ding et al. (2021); Ma et al. (2010). Then PPI-network was constructed using GeneMANIA-Cytoscape software and STRING database. About 20 hub genes were screened using CytoHubba MCC. Consequently, we chose three genes (MMP7, VEGFD and TCF-21) from the top 10 hub genes to be validated their relative expression on molecular level by q-PCR. Which resulted to be synchronized with the bio-informatics results. Additionally, it also necessary to know the ability to replace the traditional treatments (chemotherapy) with other treatments to avoid the side-effects of chemotherapy. We tested the extract of Withania somnifera plant in different concentrations by SRB cytotoxicity test, as it is recently known to exhibits anti-tumor capabilities. Finally, to validate the efficacy of W. Withania somnifera, molecular docking was performed by AutoDockvina to test the binding affinity of the target ligand (extract) in the catalytic part of the proteins (MMP7, VEGFD and TCF-21). Withanolides showed the lowest binding affinities, which indicates the effectiveness of them against the target proteins.
Based on functional enrichment analyses, the DEGs were significantly enriched in biological functions including immune response, immune system process, adaptive immune response, stimulus response, leukocyte migration, response to external biotic stimulus and response to stress A prior study identified immunological destruction, which leads to chronic inflammation, as a key cause of CRC; consequently, our transcriptome findings are consistent with earlier studies that found inflammation to be a major characteristic of the tumor microenvironment in CRC (Schottelius and Dinter 2006; Hammad et al. 2021).
Based on the construction of PPI network, MMP7, REG1A, GUCA2A, UGT2B17, and DEFA6, VEGFD, TCF-21, AGT, CCL2 and ADORA3 with a high degree of connectivity were identified as hub genes. Some of them were significantly downregulated in CRC tissues compared with normal tissues, while the others was significantly upregulated. The relative expression for three of them were validated by q-PCR as MMP-7 was upregulated in cancer cells (Chen and Ke 2021; Powell and Matrisian 1996). Furthermore, Fan et al. discovered that MMP7 was extremely important in the chemotherapeutic treatment of colon cancer (Peng et al. 2019). MMP was found to be significant in the epithelial-mesenchymal transition and invasion of colon carcinoma by Kobayashi et al. (Yamada et al. 2013). In addition, the association between MMP7 and invasive development of tumors as well as distant metastasis has been observed in colorectal tumors (CRC) (Sun et al. 2015). In this study, the treatment group IV cause significantly down- regulation by p-value 0.0417 compared to control group (I). This was also mentioned by Kyakulaga et al., which states that WS extract can target many cancer pathways as cytotoxicity, cell apoptosis, angiogenesis, inflammation, and immune modulation, they overlap in many forms of cancer (Kyakulaga et al. 2018).
Concerning VEGF-D, Vascular endothelial growth factors-D play crucial role as an angiogenic factor that control angiogensos process (Hanrahan et al. 2003). The angiogenic effects of VEGF-D have been demonstrated on endothelial cells in both in vitro and in vivo applications and its regulation is influenced by proinflammatory cytokines. Moreover, VEGF-C and VEGF-D are known to exert significant influence on the process of lymphangiogenesis (George et al. 2001). VEGFD was relatively up-regulated in the treated groups compared to the control group, which validated the in-silico results as the VEGFD showed to be down-regulated in cancer cells, as agreed with (Hanrahan et al. 2003; George et al. 2001). Moehler et al., recognized downregulation of VEGF-D after using cetuximab treatment (Moehler et al. 2008) and other study provides evidence that solid tumors can develop lymphatic vessels and suggests that VEGF family members play a crucial role in determining the pattern of metastasis by Stacker et al. (2001). However, our data didn’t come in line with other studies, there are many factors affecting VEGF-A and VEGF-D mRNA expression as age and VEGF-receptors (Hanrahan et al. 2003; Mazeda et al. 2020). The reduction of VEGF-D levels observed in polyps and carcinomas may facilitate the enhanced binding of VEGF-A and VEGF-C to the VEGF receptors (George et al. 2001). Moreover, all vascular endothelial growth factors A, B, and C showed low levels of expression in elderly patients, except for VEGF-D, which was slightly down-regulated in younger ages than old ages with no significant results (Mazeda et al. 2020; Ito et al. 2005), given that according to American Type Culture Collection (ATCC), our used HCT-116 cell line were collected from adult patients. In addition, Wang et al. said that the formation and development of tumors are influenced by inflammation, which operates through several mechanisms such as the release of inflammatory mediators, vascular abnormalities, and epigenetic changes that modify gene expression (Mazeda et al. 2020). This was proved by our previously mentioned enrichment analysis as VEGF-D was significantly enriched in many BP concerning the immune response and immune system process.
Finally, TCF-21 plays a crucial role an anti-angiogenic factor and recognized as a tumour suppressor in various types of cancers, as demonstrated by Arab et al. (2011). The up-regulation of TCF21 has been found to impede the motion of melanoma cells. Additionally, it has been observed to diminish the proliferation of cancer cells and the formation of colonies in lung and cervical cancers. In contrast to literature, the current work showed significant downregulation of TCF-21 among the treatment groups compared to control group. This was preceded by in-silico studies with the same conclusion that TCF-21 is a gene that is up-regulated in cancer cells. This may be explained by Dai et al. (2016), where they stated that some treatments activate the hyper-methylation promoter which cause dysregulation of TCF-21. Dia et al., has shown in their work that the di-activation of methylation promotor will regulate the expression of TCF-21 to do its anti-angiogenesis role normally. This also was proved by Jones (2002), the tumor suppressor genes are largely silenced as a result of alterations in DNA methylation. From other perspective, TCF-21 may be dysregulated by the action of VEGF group, it was mentioned by Chen et al. in 2019 that VEGF could boost cancer cells’ production of miR-205 which was found to inhibit the action of TCF-21 (Chen et al. 2019).
In conclusion, this analysis pipeline is robust and accurate in identifying the most notable CRC DEGs consistent with the literature. The identified genes can be used as potential biomarkers for CRC diagnosis and treatment as mentioned before early recognition of precancerous lesions in the colorectal is significantly important to enhance the survival rate and also help in timely treatment. Additionally, our research may offer novel approaches for analyzing the most significant biological functions in CRC focusing on the immune response. Furthermore, those biomarkers can be used later on studies with an increased sample size for survival analysis of CRC and cross-analysis using different types of Geo datasets. Nevertheless, this study does possess several drawbacks. First, there is further trials needed to test the regulation of the methylation promoters and also overcome many clinical challenges to clinically translate the in vitro results as the clinical information on the databases is not enough due to the absence of comprehensive grade data pertaining to patients diagnosed with CRC, as well as the limited availability of follow-up information subsequent to surgical interventions, presents a significant gap in current academic research. In addition, additional investigation needed to explore the therapeutic potential of biomarkers that have been found which will be also the focal point of our subsequent research endeavors. Finally, it is anticipated that this study’s results will contribute to the identification of novel diagnostic and prognostic biomarkers, as well as potential therapeutic targets, for CRC.
Data Availability
The data are available under accession number in NCBI, Gene Expression Omnibus (GEO): GSE156451.
References
Allam RM, Al-Abd AM, Khedr A, Sharaf OA, Nofal SM, Khalifa AE, Mosli HA, Abdel-Naim AB (2018) Fingolimod interrupts the cross talk between estrogen metabolism and sphingolipid metabolism within prostate cancer cells. Toxicol Lett 291:77–85
Andrews S et al (2010) FastQC: a quality control tool for high throughput sequence data
Arab K, Smith LT, Gast A, Weichenhan D, Huang JP-H, Claus R, Hielscher T, Espinosa AV, Ringel MD, Morrison CD et al (2011) Epigenetic deregulation of TCF21 inhibits metastasis suppressor KISS1 in metastatic melanoma. Carcinogenesis 32(10):1467–1473
Arhin N, Ssentongo P, Taylor M, Olecki EJ, Pameijer C, Shen C, Oh J, Eng C (2022) Age-standardised incidence rate and epidemiology of colorectal cancer in Africa: a systematic review and meta-analysis. BMJ Open 12(1):e052376
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M et al (2012) Ncbi geo: archive for functional genomics data sets—update. Nucleic Acids Res 41(D1):D991–D995
Behl T, Sharma A, Sharma L, Sehgal A, Zengin G, Brata R, Fratila O, Bungau S (2020) Exploring the multifaceted therapeutic potential of withaferin a and its derivatives. Biomedicines 8(12):571
Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34(5):525–527
Chen L, Ke X (2021) MMP7 as a potential biomarker of colon cancer and its prognostic value by bioinformatics analysis. Medicine 100(9):e24953
Chen S-N, Chang R, Lin L-T, Chern C-U, Tsai H-W, Wen Z-H, Li Y-H, Li C-J, Tsui K-H (2019) MicroRNA in ovarian cancer: biology, pathogenesis, and therapeutic opportunities. Int J Environ Res Public Health 16(9):1510
Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y (2014) cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 8(4):1–7
Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN, Garrett WS (2015) CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep 12(2):244–257
Dai Y, Duan H, Duan C, Zhou R, He Y, Tu Q, Shen L (2016) Down-regulation of TCF21 by hypermethylation induces cell proliferation, migration and invasion in colorectal cancer. Biochem Biophys Res Commun 469(3):430–436
Ding Q, Wu Y, Liu W (2021) Molecular mechanism of reproductive toxicity induced by Tripterygium Wilfordii based on network pharmacology. Medicine 100(27):e26197
Ewels P, Magnusson M, Lundin S, Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32(19):3047–3048
George ML, Tutton MG, Janssen F, Arnaout A, Abulafi AM, Eccles SA, Swift RI (2001) “VEGF-A, VEGF-C, and VEGF-D in colorectal cancer progression. Neoplasia 3(5):420–427
Guo Y, Bao Y, Ma M, Yang W (2017) Identification of key candidate genes and pathways in colorectal cancer by integrated bioinformatical analysis. Int J Mol Sci 18(4):722
Hammad A, Elshaer M, Tang X (2021) Identification of potential biomarkers with colorectal cancer based on bioinformatics analysis and machine learning. Math Biosci Eng 18(6):8997–9015
Hanrahan V, Currie MJ, Gunningham SP, Morrin HR, Scott PA, Robinson BA, Fox SB (2003) The angiogenic switch for vascular endothelial growth factor (VEGF)-A, VEGF-B, VEGF-C, and VEGF-D in the adenoma–carcinoma sequence during colorectal cancer progression. J Pathol 200(2):183–194
Hu T, Fei Li L, Shen J, Zhang L, Hin Cho C (2015) Chronic inflammation and colorectal cancer: the role of vascular endothelial growth factor. Curr Pharm Des 21(21):2960–2967
Huo X, Zhang L, Li T (2018) Analysis of the association of the expression of KiSS-1 in colorectal cancer tissues with the pathology and prognosis. Oncol Lett 15(3):3056–3060
Ito Y, Betsuyaku T, Nagai K, Nasuhara Y, Nishimura M (2005) Expression of pulmonary VEGF family declines with age and is further down-regulated in lipopolysaccharide (LPS)-induced lung injury. Exp Gerontol 40(4):315–323
Jones PA (2002) DNA methylation and cancer. Oncogene 21(35):5358–5360
Khazaei Z, Jarrahi AM, Momenabadi V, Ghorat F, Adineh H, Sohrabivafa M, Goodarzi E (2019) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide stomach cancers and their relationship with the human development index (HDI). World Cancer Res J 6(9):e1257
Kulkarni SK, Dhir A (2008) Withania somnifera: an Indian ginseng. Prog Neuropsychopharmacol Biol Psychiatry 32(5):1093–1105
Kyakulaga AH, Aqil F, Munagala R, Gupta RC (2018) Withaferin a inhibits epithelial to mesenchymal transition in non-small cell lung cancer cells. Sci Rep 8(1):1–14
Li M, Zhao L-M, Li S-L, Li J, Gao B, Wang F-F, Wang S-P, Hu X-H, Cao J, Wang G-Y (2018) Differentially expressed lncRNAs and mRNAs identified by NGS analysis in colorectal cancer patients. Cancer Med 7(9):4650–4664
Liang B, Li C, Zhao J (2016) Identification of key pathways and genes in colorectal cancer using bioinformatics analysis. Med Oncol 33(10):1–8
Liao H-Y, Da C-M, Liao B, Zhang H-H (2021) Roles of matrix metalloproteinase-7 (MMP-7) in cancer. Clin Biochem 92:9–18
Logie E, Vanden Berghe W (2020) Tackling chronic inflammation with withanolide phytochemicals—a withaferin a perspective. Antioxidants 9(11):1107
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):1–21
Ma D-F, Kondo T, Nakazawa T, Niu D-F, Mochizuki K, Kawasaki T, Yamane T, Katoh R (2010) Hypoxia-inducible adenosine A2B receptor modulates proliferation of colon carcinoma cells. Hum Pathol 41(11):1550–1557
Mármol I, Sánchez-de Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ (2017) Colorectal carcinoma: a general overview and future perspectives in colorectal cancer. Int J Mol Sci 18(1):197
Martin M (2011) CUTADAPT removes adapter sequences from high-throughput sequencing reads. Embnet J 17(1):10–12
Mazeda I, Martins SF, Garcia EA, Rodrigues M, Longatto A (2020) VEGF expression in colorectal cancer metastatic lymph nodes: clinicopathological correlation and prognostic significance. Gastrointest Disord 2(3):25
Moehler M, Frings C, Mueller A, Gockel I, Schimanski CC, Biesterfeld S, Galle PR, Holtmann MH (2008) VEGF-D expression correlates with colorectal cancer aggressiveness and is downregulated by cetuximab. World J Gastroenterol 14(26):4156–4167
Morris E, Forman D, Thomas J, Quirke P, Taylor E, Fairley L, Cottier B, Poston G (2010) Surgical management and outcomes of colorectal cancer liver metastases. J Br Surg 97(7):1110–1118
Mukherjee PK, Banerjee S, Biswas S, Das B, Kar A, Katiyar C (2021) Withania somnifera (l.) dunal-modern perspectives of an ancient rasayana from ayurveda. J Ethnopharmacol 264:113157
Peng Z, Chen J, Drachenberg CB, Raufman J-P, Xie G (2019) Farnesoid X receptor represses matrix metalloproteinase 7 expression, revealing this regulatory axis as a promising therapeutic target in colon cancer. J Biol Chem 294(21):8529–8542
Powell W, Matrisian L (1996) Complex roles of matrix metalloproteinases in tumor progression. In: Günthert U, Birchmeier W (eds) Attempts to understand metastasis formation I: metastasis-related molecules. Springer, Berlin, pp 1–21
Sargent DJ, Goldberg RM, Jacobson SD, Macdonald JS, Labianca R, Haller DG, Shepherd LE, Seitz JF, Francini G (2001) A pooled analysis of adjuvant chemotherapy for resected colon cancer in elderly patients. N Engl J Med 345(15):1091–1097
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3(6):1101–1108
Schottelius AJ, Dinter H (2006) Cytokines, nf-κb, microenvironment, intestinal inflammation and cancer. In: Dalgleish AG, Haefner B (eds) The link between inflammation and cancer: wounds that do not heal. Springer, Boston, MA, pp 67–87
Shah R, Jones E, Vidart V, Kuppen PJ, Conti JA, Francis NK (2014) Biomarkers for early detection of colorectal cancer and polyps: systematic review. Cancer Epidemiol Biomark Prev 23(9):1712–1728
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82(13):1107–1112
Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S-I, Kubo H, Achen MG (2001) VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 7(2):186–191
Sun D-W, Zhang Y-Y, Qi Y, Zhou X-T, Lv G-Y (2015) Prognostic significance of mmp-7 expression in colorectal cancer: a meta-analysis. Cancer Epidemiol 39(2):135–142
Sun G, Li Y, Peng Y, Lu D, Zhang F, Cui X, Zhang Q, Li Z (2019) Identification of differentially expressed genes and biological characteristics of colorectal cancer by integrated bioinformatics analysis. J Cell Physiol 234(9):15215–15224
Tantawy MA, El-Sherbeeny NA, Helmi N, Alazragi R, Salem N, Elaidy SM (2020) Synthetic antiprotozoal thiazolide drug induced apoptosis in colorectal cancer cells: implications of IL-6/JAK2/STAT3 and p53/caspases-dependent signaling pathways based on molecular docking and in vitro study. Mol Cell Biochem 469(1):143–157
Yamada D, Kobayashi S, Wada H, Kawamoto K, Marubashi S, Eguchi H, Ishii H, Nagano H, Doki Y, Mori M (2013) Role of crosstalk between interleukin-6 and transforming growth factor-beta 1 in epithelial–mesenchymal transition and chemoresistance in biliary tract cancer. Eur J Cancer 49(7):1725–1740
Acknowledgements
The authors express their gratitude to Prof. Abdelrazeik A. B. (Head of the genetics department, Faculty of Agriculture at Ain Shams University in Cairo, Egypt), for his valuable support. Additionally, the authors extend their appreciation to NanoFab Lab for their assistance with the plant extract. We also want to sincerely thank Eng. Ahmad Mohamed Sebaq (Associate R&D Engineer at the Applied Innovation Center) for his appreciated assistance and stimulating bash discussion.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
Collection and assembly of data: AA. Analysis and interpretation of the bioinformatics data: ME, WA-A, AA and MO. Supply of extract: AA. Design of wet lab experiments: MS, AA and AA. Measurement of the fold changes of the target genes by qPCR: MT, SR and AA. Evaluation of wet lab experiments’ results (Cell Culturing, qPCR and Study gene expression ): AA, MS, AA. Statistical Analysis of the obtained results: AA, ME, WA-A, AA and MO. Writing of the article and master thesis : AA. Scientific review of the research article: ME, MS, AA and WA-A.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declared no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elsayed, A.M.A., Oweda, M., Abushady, A.M. et al. Identification of Differentially Expressed Genes in Human Colorectal Cancer Using RNASeq Data Validated on the Molecular Level with Real-Time PCR. Biochem Genet 62, 3260–3284 (2024). https://doi.org/10.1007/s10528-023-10593-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-023-10593-5