
Abstract
The loss of muscle mass and function with age, termed sarcopenia, is an inevitable process, which has a significant impact on quality of life. During ageing we observe a progressive loss of total muscle fibres and a reduction in cross-sectional area of the remaining fibres, resulting in a significant reduction in force output. The mechanisms which underpin sarcopenia are complex and poorly understood, ranging from inflammation, dysregulation of protein metabolism and denervation. However, there is significant evidence to demonstrate that modified ROS generation, redox dis-homeostasis and mitochondrial dysfunction may have an important role to play. Based on this, significant interest and research has interrogated potential ROS-targeted therapies, ranging from nutritional-based interventions such as vitamin E/C, polyphenols (resveratrol) and targeted pharmacological compounds, using molecules such as SS-31 and MitoQ. In this review we evaluate these approaches to target aberrant age-related ROS generation and the impact on muscle mass and function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The age-related decline in muscle mass, strength and function, termed sarcopenia, is a significant contributor to frailty (Lexell 1993). Approximately 600 million people over the age of 60 years old suffering from sarcopenia in 2000 and by 2050 this population is estimated to grow to 2 billion people (Dhillon and Hasni 2017). Sarcopenia contributes considerably to the frailty seen in the elderly population, with sarcopenia being associated with generalised weakness, impaired mobility, poor balance and stamina (Walston 2012). Consequently, this can lead to an increasing number of falls, disability and mortality (Cruz-Jentoft et al. 2014).
Loss of muscle mass typically begins from around 50 years of age, occurring at a rate of around 1–2% per year, with around a 50% reduction by 80 years of age (Lexell 1993). Ageing muscle displays a reduction (atrophy) in overall fibre cross-sectional area, with a predominant loss of type II muscle fibres (Lexell 1993). Consequently, this results in an overall slow twitch phenotype, characterised by a reduction in force generation and activation velocity (Nilwik et al. 2013). The impact of sarcopenia is compounded via unloading and disuse of muscle as a consequence of a progressively sedentary lifestyle (Breen et al. 2013).
The mechanisms that underpin sarcopenia are not fully understood, however, it is a complex multifactorial network of interacting dysfunctional systems. Specifically, modified protein synthesis (Breen and Phillips 2013) and anabolic blunting (Cuthbertson et al. 2005), alongside chronic low-grade inflammation (Roubenoff 2003), mitochondrial dysfunction (Sakellariou et al. 2017), aberrant ROS generation (Vasilaki and Jackson 2013), denervation (Pollock et al. 2017; Scalabrin et al. 2019) and a reduced regenerative capacity (Almada andWagers 2016) are some of the key processes implicated in sarcopenia. In this review we will evaluate the ROS generation in skeletal muscle and the role these processes play in sarcopenia and current avenues for therapeutic intervention.
Reactive oxygen and nitrogen species (RONS) generation and regulation in skeletal muscle
Reactive oxygen and nitrogen species are radical and non-radical bi products of cellular respiration, formed via the molecular reduction of atmospheric oxygen. Skeletal muscle as an excitable tissue with a high energetic demand is widely described as a significant generator of RONS. In the context of ageing, seminal studies in the 1950’s developed the field and investigation of RONS, formulating the “free radical theory of ageing” (Gerschmann 1954; Haraan 1955). An increased activity of reactive oxygen species (ROS) has been implicated in the processes underlying ageing and, in all species, tissues (including skeletal muscle) of aged organisms contain increased amounts of oxidative damage to lipids, DNA and proteins (Vasilaki et al. 2006).
There are a diverse family of RONS in skeletal muscle, with superoxide (O2.−) and nitric oxide (NO−) are the primary species generated at rest and in response to contractile activity (Sakellariou et al. 2014). Moreover, there is a complex network of enzymatic and non-enzymatic antioxidant defence mechanisms which regulate RONS generation (Halliwell and Guttridge 2015).
Superoxide is generated from complex I, II and III of the electron transport chain and the nicotinamide adenine dinucleotide phosphate NAD(P)H oxidases, xanthine oxidase and lipoxygenase enzymes (Hellsten et al. 1997; Zuo et al. 2004; Muller et al. 2004; Goncalves et al. 2015). Superoxide has a relatively long biological half-life of ~ 10−6 s, is membrane impermeable, bar movement via specific membrane channels (TOM40/VDAC/BAX). Superoxide has a low oxidising capacity towards cellular macromolecules; however, it may react with other species such as NO−, forming the highly reactive peroxynitrite (ONOO−) (Halliwell and Guttridge 2015). Typically, superoxide is dismutated to hydrogen peroxide (H2O2) by the superoxide dismutase (SOD) family of enzymes (Fukai and Ushio-Fukai 2011). The SOD family of enzymes are characterised by their ability to convert O2.− to H2O2 are localised to different cellular compartments and differentiated by the transition metal bound to the active site. SOD1 termed CuZnSOD is localised to both the cytosol and mitochondrial intermembrane space; SOD2 termed MnSOD is enriched within the mitochondrial matrix and SOD3 (extracellular SOD) has a CuZn cofactor and is found in the interstitial spaces (Halliwell and Guttridge 2015). Approximately 35% of all cellular SOD activity is localised to the mitochondria to skeletal muscle (Ji et al. 1998). Modification of SOD activity and function has been shown to induce dysfunction in skeletal muscle. Specifically, SOD1 gene knockout mice display elevated markers of oxidative damage and an overall accelerated sarcopenic phenotype and is a model for amyotrophic lateral sclerosis (Deepa et al. 2019). In response to contractile activity, skeletal muscle upregulates the expression of both SOD1 and 2, with a higher activity in type I slow oxidative muscle fibres (Kojda and Hambrecht 2005).
Nitric oxide (NO−) is generated by the nitric oxide synthase (NOS) enzymes, catalysing the conversion of L-arginine to citrulline (Korhonen et al. 2005). NO− has a biological half-life of ~ 1–10−1 s and functions via s-nitrosylation of cysteine residues (Stamler and Meissner 2001). There are three key members of the NOS enzyme family: inducible (iNOS), neuronal (nNOS) and endothelial (eNOS). NO− has a well-established function in vasodilation and the immune response and has an important role in muscle physiology. A recent study using the nNOS−/− mouse, demonstrated NO− helps to regulate muscle fibre type, fatiguability and post-exercise recovery (Moon et al. 2017). NO− levels are elevated in the muscle fibres of old mice following contractile activity, associated in with elevated marked of nitrosylation (3-nitrotryrosine) and eNOS activity (Pearson et al. 2015).
Hydrogen peroxide is a non-radical species, generated from dismutation of superoxide by the SODs, and as a bi product of protein folding in the endoplasmic reticulum via the thiol-disulphide exchange mechanism (Hudson et al. 2015). Comparative to other radical RONS species, H2O2 has a relatively long half-life 10−5 s and is membrane permeable. H2O2 levels are regulated by the enzymes catalase, glutathione peroxidase (Gpx) and the peroxiredoxin (PRXs) family, converting it to water and oxygen. Elevated H2O2 generation has been observed in the muscle of old mice and is reported to be derived from increased mitochondria superoxide generation (Mansouri et al. 2006). Moreover, H2O2 has been heavily implicated as an important mediator of muscle ageing, based on several studies of the transgenic mCAT model, which overexpress the human form of catalase in the mitochondria (Basisty et al. 2016). mCAT mice have been shown to have increased longevity (Schriner et al. 2005) and improved muscle function—the latter due to decreased ca2+ leakage from the sarcoplasmic reticulum (Umanskaya et al. 2014).
Peroxynitrite (ONOO−) is a highly reactive radical species, formed via the reaction between superoxide and nitric oxide, with a half-life of 10−2 s. Thiol (-SH) groups are highly sensitive to peroxynitrite (resulting in disulphide formation), which can rapidly deplete the overall thiol pools within the cell. Peroxynitrite induces the nitration of tyrosine and S-nitrosylation of cysteine residues and can induce significant damage to DNA and in some instances, affect enzyme activity and supress signalling cascades. Studies in isolated muscle fibres have demonstrated that peroxynitrite directly suppresses ca2+ stimulated force generation (Dutka et al. 2011). Studies of old mice (26–28 month) and in the SOD1−/− model have shown a three-fold increase in 3-nitrotyrosine (3-NT) residues compared with young/WT controls (Sakellariou et al. 2011; Pearson et al. 2015).
Hydroxyl radical (OH−) is a highly reactive species, with a very short half-life ~ 10−9 s. The hydroxyl radical is formed via the fenton reaction:
Transition metals such as iron (Fe+) or copper (Cu2+) can reduce H2O2 to the hydroxyl radical. Contracting skeletal muscle has been shown to generate the hydroxyl radical (O’Neill et al. 1996) and it has been reported to modify calcium sensitivity and force generation in rodent muscle fibres (Murphy et al. 2008). However, there are some elements of controversy in terms of the fenton reaction, given the relatively low concentrations of transition metals detected in vivo (Halliwell and Guttridge 2015).
Mitochondrial dysfunction in skeletal muscle ageing
Skeletal muscle is richly abundant in mitochondria, due to the high energetic demand of contractile activity. Thus, given the role of mitochondria in ROS generation, there has been significant interest in understanding what if any part mitochondria may play in muscle wasting and dysfunction. Aberrant ROS generation and oxidative damage have been associated with many aspects of mitochondrial dysfunction in skeletal muscle ageing. Firstly, accumulation of mitochondrial DNA mutations (mtDNA) in human aged skeletal muscle is evident (Bua et al. 2006; Herbst et al. 2007; Melov et al. 1995). Various in vitro studies have shown association with mtDNA mutations and with reduced content of the etc. complexes, oxidative phosphorylation, and membrane potential (Hiona et al. 2010; Sahu et al. 2018), as well as increased expression of mitochondrial markers of oxidative damage, decreased antioxidant defense system and mitochondrial biogenesis, indicative to a defective adaptive response to mitochondrial dysfunction in mtDNA mutator mice (Kolesar et al. 2014). These studies are consistent with results in aged rats that showed reduced levels of mitochondrial biogenesis and function markers, accompanying with swollen mitochondria (Zhu et al. 2019; Sahu et al. 2018). Furthermore, there is an age-related decline in mitofusin 2 (Mfn2) a key regulator of mitochondrial fusion (Sebastian et al. 2016), which an important process which helps regulate and accommodate the energetic demands of the cell. Moreover, ablation of Mfn2 in mice induced an ageing phenotype, associated with impaired mitophagy (Sebastian et al. 2016). The latter observation may be related the accumulation of damaged mitochondria during ageing. Changes have been observed in both intermyofibrillar (IMF) and subsarcolemmal (SS) mitochondria in aged mice. IMF mitochondria appeared longitudinal and branched in shape, whereas SS mitochondria had a larger and less circular appearance—in, contrast they did not observe changes in absolute Mfn2 levels. However, there was alteration in the ratio of genes which regulate mitochondrial fusion, fission and biogenesis—suggesting dysregulation of the overall system (Leduc-Gaudet et al. 2015). Further rodent studies have also demonstrated ageing and the loss of muscle mass and function to be associated with a downregulation of genes encoding key mitochondrial function (Ibebunjo et al. 2013).
Targeting RONS to combat sarcopenia
Given the ever-ageing population and the socioeconomic impact of sarcopenia, there is an urgent need to develop effective targeted therapies. Moreover, the significant evidence of aberrant RONS generation and mitochondrial dysfunction in muscle ageing suggest that therapies to target these processes are worthy of investigation.
Vitamins
Nutritional antioxidants are considered as a potential approach in an effort to combat the age-related loss of muscle mass and function, due to their antioxidant and anti-inflammatory activity. A study showed that both Vitamin E and C supplementation could improve muscle function by increasing antioxidant activity and preventing oxidative stress in aged rats (Ryan et al. 2010). A further insight into the effects of the two subgroups of vitamin E, tocopherol and tocotrienols, suggested that both α-tocopherol and tocotrienol-rich fraction (TRF) can encourage myogenic differentiation during replicative senescence, with TRF having superior effects (Khor et al. 2016). Data demonstrated that TRF can promote muscle regeneration during oxidative stress-induced premature senescence, by both increasing the proliferation capacity of myoblasts and maintaining the renewal of satellite cells (Lim et al. 2019). A further study using a vitamin E analog, TROLOX, showed that it could prevent oxidative stress and maintain skeletal muscle mass in an animal ageing model with muscle specific Opa1 deletion (Tezze et al. 2017). A clinical trial on female athletes has shown that supplementation of Vitamin E and C together or Vitamin C alone could reduce muscle damage, alongside with decreased levels of oxidative stress markers, induced by aerobic exercise (Taghiyar et al. 2013). Further enhancing previous evidence of beneficial effects of vitamins supplementation in muscle ageing, a recent study showed a positive association between high dietary intake of vitamin E, C, and carotenoids with improved sarcopenic indices (muscle mass and power); however, this association was only noticeable in women younger than 65 years (Welch et al. 2019).
However, it is important to note that there is compelling evidence demonstrating the use of broad-spectrum antioxidants such as vitamin C and E, can in fact suppress the beneficial adaptations in response to exercise training (Ristow et al. 2009). In contrast, mitoQ supplementation during endurance training, displayed no tangible beneficial or negative effects in terms of adaptations (Shill et al. 2016). Moreover n-acetylecysteine supplementation has been reported to suppress fatigue following repeated bouts of intermittent exercise (Cobley et al. 2011). However, RONS are no longer considered to be purely toxic/damaging molecules and instead are important mediators and regulators of an array of signalling cascades and pathways (Cobley et al. 2015).
Resveratrol
Resveratol has been widely studying in muscle ageing due to its antioxidant activity, as well as its effects on peroxisome proliferator-activated receptor gamma coactivator-1a, an important regulator of mitochondrial biogenesis (Gülçin 2010). The anti-ageing effects of resveratrol supplementation for 4 weeks, have been recently reported, highlighting its capability of inhibiting lipid peroxidation and increasing catalase and superoxide dismutase activity, by targeting mitochondrial mass and function, associated with improved physical endurance in aged mice (Muhammad and Allam 2018). In an in vitro study, resveratrol has shown dose-dependent effects on muscle cell plasticity, with low doses preventing ROS generation and inducing muscle regeneration, effects not observed in high doses (Bosutti and Degens 2015). However, various studies have obtained controversial results on the effect of resveratrol on ageing-associated muscle dysfunction. A 6–7-week low-to-moderate daily resveratrol intake has produced no effects in muscular strength and function in aged mice (Baumann et al. 2014; Zhou et al. 2019). Similarly, long-term (10 months) low-to-moderate daily resveratrol intake, even though it could mitigate ageing-induced oxidative stress, no beneficial effects were observed on muscle mass and function in aged mice, while mitochondrial biogenesis was also unaffected by resveratrol (Park et al. 2018). Overall, it seems that resveratrol has protective effects against ageing-induced abnormalities, even the incongruent findings, as they can likely be explained by variations in the age of experimental species, as well as the dosage and duration of supplementation; those variations highlight the need for further investigation for acute and chronic resveratrol intake both in vitro and in vivo with consistent experimental approach.
Targeted antioxidant therapies
When evaluating studies of antioxidants, there is a large degree of heterogeneity in terms of efficacy and outcomes. This is likely due in part to a poor distribution and uptake of such compounds into cells and tissues, resulting in the need to administer such antioxidants in high concentrations. In the case of the latter, this may be a reason for the observed pro-oxidant effect of some purported antioxidants (Pearson et al. 2006). Thus, development of targeted antioxidant therapies has provided a novel and promising avenue of research, in an effort to combat the pitfalls of broad-spectrum approaches.
Given the aforementioned potential role of mitochondrial dysfunction in sarcopenia, the use of novel antioxidant compounds to this organelle have been investigated in sarcopenia and a range of myopathies and neuromuscular disease (Sakellariou et al. 2017). The Szeto-Schiller (SS-) peptide family are small molecules, with potent antioxidant capacity towards a range of radical species (Lightfoot et al. 2015). The Dmt-D-Arg-Phe-atnDAP-NH2 structure of the SS- peptides results in their accumulation at the inner mitochondrial membrane (IMM). The accumulation of SS- peptides at the IMM does not rely on the mitochondrial membrane potential, which offers a unique opportunity to target mitochondria which may have underlying damage (Zhao et al. 2004).
SS-31 has been explored as a potential therapy against the age-related loss of muscle mass and function (sarcopenia). Modified ROS generation and oxidative damage has been widely characterised as an important mediator of muscle wasting and dysfunction (Lightfoot et al. 2014). In this study, mice were administered sub-cutaneous SS-31 peptide (1.5 mg/kg), over a four-month period from 24 to 28 months of age. Data showed significant reductions in markers of oxidative damage in the muscles of old mice, however, this did prevent the age-related loss of muscle mass and function (Sakellariou et al. 2016a). This clearly supports the potent ability of SS peptides as antioxidants, however, the lack of change in underlying pathology in this instance may be due to the complex and multi-factorial nature of sarcopenia. In contrast, 8-weeks of SS-31 (3 mg/kg) treatment in 26-month old mice, showed improvements in mitochondrial structure, function and homeostasisalongside an increased in exercise tolerance (Campbell et al. 2019). Moreover, short-term (1-h) SS-31 treatment (3 mg/kg) of 27-month-old mice, improved mitochondrial function and muscle performance (Siegel et al. 2013). Thus, differences in dose and duration of treatment with SS-31 in these models may be significant factors in the observed differences in outcome.
More recently, SS-31 has been explored in the context of a murine model of doxorubicin-induced toxicity (Montalvo et al. 2019). Doxorubicin is a highly potent chemotherapy agent, which has significant side-effects on muscle. In this study, SS-31 (3 mg/kg) attenuated mitochondrial ROS generation and proteolytic pathway activity in the muscles of doxorubicin treated mice (Montalvo et al. 2019). Furthermore, studies of leukocytes taken from patients with type 2 diabetes, demonstrated a reduction in inflammation, via down regulation of TNF-alpha and NF-kB and upregulation of SIRT1, in response to treatment with SS-31 peptide (Escribano-Lopez et al. 2018). Thus, there are clear and compelling evidence demonstrating the potent antioxidant ability of SS family of peptides in a range of disorders, which can have direct functional effects.
Mitoquinone is a compound derived from ubiquinone, which has a lipophilic cation (triphenylphosphonium) conjugated to it, which drives accumulation of the molecule within mitochondria (Murphy and Smith 2007). Ubiquione is a crucial component of the electron transport chain as a two-electron carrier, as well as having antioxidant capacity (Nicholls and Ferguson 2002). MitoQ readily accumulates within the cytosol of the cell driven by the plasma membrane potential, however, is found in > 100-fold greater concentration within the mitochondria at the matrix face of the inner membrane, driven by the mitochondrial membrane potential (Murphy and Smith 2007). Upon accumulation in the mitochondria, MitoQ is converted to its active antioxidant form (ubiquinol) via reduction at complex II (Murphy and Smith 2007). When acting as an antioxidant, MitoQ is oxidised to ubiquinone and then rapidly reduced back to ubiquinol, rejuvenating its antioxidant capacity and thus, is a marker of the efficacy of MitoQ. MitoQ has an affinity for an targets the hydroxyl (.OH) radical, thus preventing lipid peroxidation (Tauskela 2007), alongside quenching superoxide generation (James et al. 2005). In the context of ageing-induced mitochondrial dysfunction, a recent study has clearly found that MitoQ was able to alleviate ageing-induced changes in respiratory chain, ATP production, and membrane potential, highlighting the role of peroxynitrite in ageing deficits (Maiti et al. 2018). However, its effects on ageing-induced muscle atrophy seem to be less clear. A study of ageing mice (24–26 months) reported failure of long-term MitoQ supplementation to protect against age-related loss of muscle mass function and oxidative damage (Sakellariou et al. 2016b). A further novel mitochondria-targeted antioxidant is XJB-5-131, which is TEMPO derivative that has a gramicidin S moiety conjugated to it, which encourages accumulation in the mitochondria (Robinson et al. 2018). XJB-5-131 has been found to decrease mitochondrial ROS and membrane depolarization (Escobales et al. 2014), increase the activity of the electron transport chain complexes, and improve the single fibre contractile properties in aged rats (Javadov et al. 2015).
Conclusions
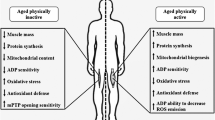
Aberrant ROS generation and redox dis-homeostasis have clear and important role to play in the age-related loss of muscle mass and function (Fig. 1). However, there is no clear consensus on intervention to target ROS. There remains significant discordance in findings, likely due to differences in model systems (species) in tandem with dosing regimens. Moreover, this raises questions on the ability to translate these mechanistic findings to humans—where there is a need to pursue this line of research further.

A schematic summary of the role of ROS and putative interventions in the age-related loss of muscle mass and function
References
Almada AE, Wagers AJ (2016) Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat Rev Mol Cell Biol 17(5):267
Basisty N, Dai DF, Gagnidze A, Gitari L, Fredrickson J, Maina Y, Beyer RP, Emond MJ, Hsieh EJ, MacCoss MJ, Martin GM (2016) Mitochondrial-targeted catalase is good for the old mouse proteome, but not for the young:‘reverse’antagonistic pleiotropy? Aging Cell 15(4):634–645
Baumann CW, Rogers RG, Lees SJ, Otis JS (2014) Muscular strength is unaffected by shortterm resveratrol supplementation in aged mouse muscle. Int J Clin Exp Physiol 1(4):253–257
Bosutti A, Degens H (2015) The impact of resveratrol and hydrogen peroxide on muscle cell plasticity shows a dose-dependent interaction. Sci Rep 5:8093
Breen L, Phillips SM (2013) Interactions between exercise and nutrition to prevent muscle waste during ageing. Br J Clin Pharmacol 75(3):708–715
Breen L, Stokes KA, Churchward-Venne TA, Moore DR, Baker SK, Smith K, Atherton PJ, Phillips SM (2013) Two weeks of reduced activity decreases leg lean mass and induces “anabolic resistance” of myofibrillar protein synthesis in healthy elderly. J Clin Endocrinol Metabol 98(6):2604–2612
Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM (2006) Mitochondrial DNA–deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet 79(3):469–480
Campbell MD, Duan J, Samuelson AT, Gaffrey MJ, Merrihew GE, Egertson JD, Wang L, Bammler TK, Moore RJ, White CC, Kavanagh TJ (2019) Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic Biol Med 134:268–281
Cobley JN, McGlory C, Morton JP, Close GL (2011) N-Acetylcysteine’s attenuation of fatigue after repeated bouts of intermittent exercise: practical implications for tournament situations. Int J Sport Nutr Exerc Metab 21(6):451–461
Cobley JN, Moult PR, Burniston JG, Morton JP, Close GL (2015) Exercise improves mitochondrial and redox-regulated stress responses in the elderly: better late than never! Biogerontology 16(2):249–264
Cruz-Jentoft AJ, Landi F, Schneider SM, Zúñiga C, Arai H, Boirie Y, Chen LK, Fielding RA, Martin FC, Michel JP, Sieber C (2014) Prevalence of and interventions for sarcopenia in ageing adults: a systematic review. Report of the International Sarcopenia Initiative (EWGSOP and IWGS). Age Ageing 43(6):748–759
Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, Wackerhage H, Taylor PM, Rennie MJ (2005) Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J 19(3):422–424
Deepa SS, Van Remmen H, Brooks SV, Faulkner JA, Larkin L, McArdle A, Jackson MJ, Vasilaki A, Richardson A (2019) Accelerated sarcopenia in Cu/Zn superoxide dismutase knockout mice. Free Radic Biol Med 132:19–23
Dhillon RJ, Hasni S (2017) Pathogenesis and management of sarcopenia. Clin Geriatr Med 33(1):17–26
Dutka TL, Mollica JP, Lamb GD (2011) Differential effects of peroxynitrite on contractile protein properties in fast-and slow-twitch skeletal muscle fibers of rat. J Appl Physiol 110(3):705–716
Escobales N, Nuñez RE, Jang S, Parodi-Rullan R, Ayala-Peña S, Sacher JR, Skoda EM, Wipf P, Frontera W, Javadov S (2014) Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J Mol Cell Cardiol 77:136–146
Escribano-Lopez I, Diaz-Morales N, Iannantuoni F, Lopez-Domenech S, de Marañon AM, Abad-Jimenez Z, Bañuls C, Rovira-Llopis S, Herance JR, Rocha M, Victor VM (2018) The mitochondrial antioxidant SS-31 increases SIRT1 levels and ameliorates inflammation, oxidative stress and leukocyte-endothelium interactions in type 2 diabetes. Sci Rep 8(1):15862
Fukai T, Ushio-Fukai M (2011) Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 15(6):1583–1606
Gerschman R (1954) Oxygen poisoning and x-irradiation: a mechanism in common In Glutathione. Academic Press, Cambridge, pp 288–291
Goncalves RL, Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Brand MD (2015) Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J Biol Chem 290(1):209–227
Gülçin İ (2010) Antioxidant properties of resveratrol: a structure–activity insight. Innov Food Sci Emerg Technol 11(1):210–218
Halliwell B, Gutteridge JM (2015) Free radicals in biology and medicine. Oxford University Press, Oxford
Harraan D (1955) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11:298–300
Hellsten Y, Frandsen U, Orthenblad N, Sjødin B, Richter EA (1997) Xanthine oxidase in human skeletal muscle following eccentric exercise: a role in inflammation. J Physiol 498(1):239–248
Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM (2007) Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J Gerontol Ser A 62(3):235–245
Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, Someya S, Miyakawa T, Nakayama C, Samhan-Arias AK, Servais S (2010) Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 5(7):e11468
Hudson DA, Gannon SA, Thorpe C (2015) Oxidative protein folding: from thiol–disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic Biol Med 80:171–182
Ibebunjo C, Chick JM, Kendall T, Eash JK, Li C, Zhang Y, Vickers C, Wu Z, Clarke BA, Shi J, Cruz J (2013) Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol Cell Biol 33(2):194–212
James AM, Cochemé HM, Smith RA, Murphy MP (2005) Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem 280(22):21295–21312
Javadov S, Jang S, Rodriguez-Reyes N, Rodriguez-Zayas AE, Hernandez JS, Krainz T, Wipf P, Frontera W (2015) Mitochondria-targeted antioxidant preserves contractile properties and mitochondrial function of skeletal muscle in aged rats. Oncotarget 6(37):39469
Khor SC, Razak AM, Ngah WZW, Yusof YAM, Karim NA, Makpol S (2016) The tocotrienol-rich fraction is superior to tocopherol in promoting myogenic differentiation in the prevention of replicative senescence of myoblasts. PLoS ONE 11(2):e0149265
Kojda G, Hambrecht R (2005) Molecular mechanisms of vascular adaptations to exercise. Physical activity as an effective antioxidant therapy? Cardiovasc Res 67(2):187–197
Kolesar JE, Safdar A, Abadi A, MacNeil LG, Crane JD, Tarnopolsky MA, Kaufman BA (2014) Defects in mitochondrial DNA replication and oxidative damage in muscle of mtDNA mutator mice. Free Radic Biol Med 75:241–251
Korhonen R, Lahti A, Kankaanranta H, Moilanen E (2005) Nitric oxide production and signaling in inflammation. Curr Drug Targets-Inflamm Allergy 4(4):471–479
Leduc-Gaudet JP, Picard M, Pelletier FSJ, Sgarioto N, Auger MJ, Vallée J, Robitaille R, St-Pierre DH, Gouspillou G (2015) Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6(20):17923
Lexell J (1993) Ageing and human muscle: observations from Sweden. Can J Appl Physiol 18(1):2–18
Lightfoot AP, McCormick R, Nye GA, McArdle A (2014) Mechanisms of skeletal muscle ageing; avenues for therapeutic intervention. Curr Opin Pharmacol 16:116–121
Lightfoot AP, Sakellariou GK, Nye GA, McArdle F, Jackson MJ, Griffiths RD, McArdle A (2015) SS-31 attenuates TNF-α induced cytokine release from C2C12 myotubes. Redox Biol 6:253–259
Lim JJ, Zurinah W, Ngah W, Mouly V, Norwahidah AK (2019) Tocotrienol-rich fraction (TRF) treatment promotes proliferation capacity of stress-induced premature senescence myoblasts and modulates the renewal of satellite cells: microarray analysis. Oxid Med Cell Longev 2019:9141343
Maiti AK, Spoorthi BC, Saha NC, Panigrahi AK (2018) Mitigating peroxynitrite mediated mitochondrial dysfunction in aged rat brain by mitochondria-targeted antioxidant MitoQ. Biogerontology 19(3–4):271–286
Mansouri A, Muller FL, Liu Y, Ng R, Faulkner J, Hamilton M, Richardson A, Huang TT, Epstein CJ, Van Remmen H (2006) Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech Ageing Dev 127(3):298–306
Melov S, Shoffner JM, Kaufman A, Wallace DC (1995) Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res 23(20):4122–4126
Montalvo RN, Doerr V, Min K, Szeto HH, Smuder AJ (2019) Doxorubicin-induced oxidative stress differentially regulates proteolytic signaling in cardiac and skeletal muscle. Am J Physiol-Regulat Integr Comp Physiol. https://doi.org/10.1152/ajpregu.00299.2019
Moon Y, Balke JE, Madorma D, Siegel MP, Knowels G, Brouckaert P, Buys ES, Marcinek DJ, Percival JM (2017) Nitric oxide regulates skeletal muscle fatigue, fiber type, microtubule organization, and mitochondrial ATP synthesis efficiency through cGMP-dependent mechanisms. Antioxid Redox Signal 26(17):966–985
Muhammad MH, Allam MM (2018) Resveratrol and/or exercise training counteract aging-associated decline of physical endurance in aged mice; targeting mitochondrial biogenesis and function. J Physiol Sci 68(5):681–688
Muller FL, Liu Y, Van Remmen H (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279(47):49064–49073
Murphy MP, Smith RA (2007) Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 47:629–656
Murphy RM, Dutka TL, Lamb GD (2008) Hydroxyl radical and glutathione interactions alter calcium sensitivity and maximum force of the contractile apparatus in rat skeletal muscle fibres. J Physiol 586(8):2203–2216
Nicholls DG, Ferguson SJ (2002) Bioenergetics. Elsevier, Amsterdam
Nilwik R, Snijders T, Leenders M, Groen BB, van Kranenburg J, Verdijk LB, van Loon LJ (2013) The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Exp Gerontol 48(5):492–498
O’Neill CA, Stebbins CL, Bonigut S, Halliwell B, Longhurst JC (1996) Production of hydroxyl radicals in contracting skeletal muscle of cats. J Appl Physiol 81(3):1197–1206
Park SY, Kwon OS, Andtbacka RHI, Hyngstrom JR, Reese V, Murphy MP, Richardson RS (2018) Age-related endothelial dysfunction in human skeletal muscle feed arteries: the role of free radicals derived from mitochondria in the vasculature. Acta Physiol 222(1):e12893
Pearson P, Lewis SA, Britton J, Young IS, Fogarty A (2006) The pro-oxidant activity of high-dose vitamin E supplements in vivo. BioDrugs 20(5):271–273
Pearson T, McArdle A, Jackson MJ (2015) Nitric oxide availability is increased in contracting skeletal muscle from aged mice, but does not differentially decrease muscle superoxide. Free Radic Biol Med 78:82–88
Pollock N, Staunton CA, Vasilaki A, McArdle A, Jackson MJ (2017) Denervated muscle fibers induce mitochondrial peroxide generation in neighboring innervated fibers: role in muscle aging. Free Radic Biol Med 112:84–92
Ristow M, Zarse K, Oberbach A, Klöting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Blüher M (2009) Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl AcadSci 106(21):8665–8670
Robinson AR, Yousefzadeh MJ, Rozgaja TA, Wang J, Li X, Tilstra JS, Feldman CH, Gregg SQ, Johnson CH, Skoda EM, Frantz MC (2018) Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol 17:259–273
Roubenoff R (2003) Catabolism of aging: is it an inflammatory process? Curr Opin Clin Nutr Metabol Care 6(3):295–299
Ryan MJ, Dudash HJ, Docherty M, Geronilla KB, Baker BA, Haff GG, Cutlip RG, Alway SE (2010) Vitamin E and C supplementation reduces oxidative stress, improves antioxidant enzymes and positive muscle work in chronically loaded muscles of aged rats. Exp Gerontol 45(11):882–895
Sahu A, Mamiya H, Shinde SN, Cheikhi A, Winter LL, Vo NV, Stolz D, Roginskaya V, Tang WY, Croix CS, Sanders LH (2018) Age-related declines in α-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nat Commun 9(1):1–14
Sakellariou GK, Pye D, Vasilaki A, Zibrik L, Palomero J, Kabayo T, McArdle F, Van Remmen H, Richardson A, Tidball JG, McArdle A (2011) Role of superoxide–nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu, Zn superoxide dismutase. Aging Cell 10(5):749–760
Sakellariou GK, Jackson MJ, Vasilaki A (2014) Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD (P) H oxidases. Free Radic Res 48(1):12–29
Sakellariou GK, Pearson T, Lightfoot AP, Nye GA, Wells N, Giakoumaki II, Vasilaki A, Griffiths RD, Jackson MJ, McArdle A (2016) Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci Rep 6:33944
Sakellariou GK, Pearson T, Lightfoot AP, Nye GA, Wells N, Giakoumaki II, Griffiths RD, McArdle A, Jackson MJ (2016) Long-term administration of the mitochondria-targeted antioxidant mitoquinone mesylate fails to attenuate age-related oxidative damage or rescue the loss of muscle mass and function associated with aging of skeletal muscle. FASEB J 30(11):3771–3785
Sakellariou GK, Lightfoot AP, Earl KE, Stofanko M, McDonagh B (2017) Redox homeostasis and age-related deficits in neuromuscular integrity and function. J Cachexia Sarcopenia Muscle 8(6):881–906
Scalabrin M, Pollock N, Staunton CA, Brooks SV, McArdle A, Jackson MJ, Vasilaki A (2019) Redox responses in skeletal muscle following denervation. Redox Biol 26:101294
Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308(5730):1909–1911
Sebastián D, Sorianello E, Segalés J, Irazoki A, Ruiz-Bonilla V, Sala D, Planet E, Berenguer‐Llergo A, Muñoz JP, Sánchez‐Feutrie M, Plana N (2016) Mfn2 deficiency links age‐related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J 35(15):1677–1693
Shill DD, Southern WM, Willingham TB, Lansford KA, McCully KK, Jenkins NT (2016) Mitochondria-specific antioxidant supplementation does not influence endurance exercise training‐induced adaptations in circulating angiogenic cells, skeletal muscle oxidative capacity or maximal oxygen uptake. J Physiol 594(23):7005–7014
Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS, Marcinek DJ (2013) Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 12(5):763–771
Stamler JS, Meissner G (2001) Physiology of nitric oxide in skeletal muscle. Physiol Rev 81(1):209–237
Taghiyar M, Darvishi L, Askari G, Feizi A, Hariri M, Mashhadi NS, Ghiasvand R (2013) The effect of vitamin C and e supplementation on muscle damage and oxidative stress in female athletes: a clinical trial. Int J Prevent Med 4(Suppl 1):S16
Tauskela JS (2007) MitoQ–a mitochondria-targeted antioxidant. IDrugs 10(6):399–412
Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, Ciciliot S, Soriano ME, Morbidoni V, Cerqua C, Loefler S (2017) Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metabol 25(6):1374–1389
Umanskaya A, Santulli G, Xie W, Andersson DC, Reiken SR, Marks AR (2014) Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci 111(42):15250–15255
Vasilaki A, Mansouri A, Van Remmen H, Van Der Meulen JH, Larkin L, Richardson AG, McArdle A, Faulkner JA, Jackson MJ (2006) Free radical generation by skeletal muscle of adult and old mice: effect of contractile activity. Aging Cell 5(2):109–117
Vasilaki A, Jackson MJ (2013) Role of reactive oxygen species in the defective regeneration seen in aging muscle. Free Radic Biol Med 65:317–323
Walston JD (2012) Sarcopenia in older adults. Curr Opin Rheumatol 24(6):623
Welch AA, Jennings A, Kelaiditi E, Skinner J, Steves CJ (2019) Cross-sectional associations between dietary antioxidant vitamins C, E and carotenoid intakes and sarcopenic indices in women aged 18–79 years. Calcif Tissue Int 106:1–12
Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH (2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279(33):34682–34690
Zhou J, Liao Z, Jia J, Chen JL, Xiao Q (2019) The effects of resveratrol feeding and exercise training on the skeletal muscle function and transcriptome of aged rats. PeerJ 7:.e7199
Zhu Y, Liu X, Ding X, Wang F, Geng X (2019) Telomere and its role in the aging pathways: telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 20(1):1–16
Zuo L, Christofi FL, Wright VP, Bao S, Clanton TL (2004) Lipoxygenase-dependent superoxide release in skeletal muscle. J Appl Physiol 97(2):661–668
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thoma, A., Akter-Miah, T., Reade, R.L. et al. Targeting reactive oxygen species (ROS) to combat the age-related loss of muscle mass and function. Biogerontology 21, 475–484 (2020). https://doi.org/10.1007/s10522-020-09883-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-020-09883-x