
Abstract
Naturally occurring goethites often show Al for Fe substitution approaching 33 mol% Al. This substitution has potential to influence the rate of goethite dissolution and therefore the supply of bioavailable Fe. Siderophores such as ferrichrome and enterobactin have considerable potential to dissolve Fe from Fe3+ rich minerals, including Al-substituted goethites. Here, we show that Al substitution in synthetic goethites (0.021 ≥ x ≥ 0.098) gives rise to a significant increase in both ferrichrome- and enterobactin-mediated dissolution rates. In the presence of ferrichrome, Al-goethite (x = 0.033) yields a dissolution rate of 19.0 × 10−3 µmol m−2 h−1, nearly twice that of pure goethite, whereas dissolution of the most highly substituted Al-goethite (x = 0.098) is 36.9 × 10−3 µmol m−2 h−1, more than threefold greater than the pure mineral. Similarly, in the presence of enterobactin, the dissolution rate of Al-goethite increases with increasing Al substitution. Ferrichrome is a less effective ligand than enterobactin in its dissolution of both pure goethite and the range of Al-goethites, an observation we ascribe to the lower affinity of the hydroxamate functional groups of ferrichrome for both Fe3+ and Al3+. Despite greater affinity of both ferrichrome and enterobactin for Fe3+ over Al3+, we observe a broadly congruent dissolution of all our Al-goethites.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Soil goethites often show Al substitution for Fe, [Fe1 − x Al x OOH], where Al content can reach 33 mol% [i.e. Al/(Al + Fe) = 0.33] (Alvarez et al. 2007; Cornell and Schwertmann 2003; Schwertmann and Taylor 1989). This substitution changes unit-cell dimensions, crystal size, surface area, surface chemistry, domain morphology, and other structural properties that influence the mechanisms and rates of goethite dissolution (Ainsworth et al. 1989; Cornell and Schwertmann 2003; Schulze and Schwertmann 1984). Given the potential for Al substitution to influence the dissolution of goethite and other Fe(III) oxides, and thus the supply of bioavailable Fe(III), a growing body of work has explored the rates and mechanisms of dissolution for both natural and synthetic Al-substituted goethites (Cervini-Silva and Sposito 2002; Dominik et al. 2002; Ekstrom et al. 2010; Kukkadapu et al. 2001; Maurice et al. 2000).
The reductive dissolution of synthetic Al-goethites (where x = 0.051 or 0.33) by the Fe-reducing bacterium Clostridium butyricum revealed incongruent dissolution of Fe and Al and a rate of Fe release which decreased with increasing Al substitution (Bousserrhine et al. 1998). Accumulation of Al at the goethite surface during dissolution was thought to block the reactive surface sites, thus decreasing the rate of Fe release. In contrast, the reductive dissolution of a natural Al-goethite (x = 0.17) by Shewanella putrefaciens showed that dissolution was unaffected by Al sorption (Kukkadapu et al. 2001). In this latter study, reductive dissolution was thought to occur at sites distant from those of Al precipitation or adsorption. More recently, Ekstrom et al. (2010) have shown that the effect of Al substitution on reductive dissolution varies mainly with Fe(III) oxide type. For example, these workers observed no significant difference in Fe(III) reduction for either lepidocrocite or goethite over a range of Al substitutions. Rather, Fe(III) reduction rates were shown to vary only for ferrihydrite, with rates for pure ferrihydrite more than twice that for Al-ferrihydrite where x = 0.13. These conflicting studies demonstrate the considerable and ongoing uncertainty surrounding the effect of Al substitution on the reductive dissolution of Fe(III) oxides.
The dissolution of Al-substituted goethite has also been investigated for oxic environments. For example, when synthetic Al-goethite (0 ≤ x ≤ 0.088) was incubated under batch conditions (Maurice et al. 2000) with siderophore-producing bacteria (Pseudomonas mendocina), Fe release increased with increasing Al content, in contrast to the trends observed for reductive dissolution. Substitution of Al3+ for Fe3+ evidently increases the population of reactive surface sites which, under oxic conditions, enhances microbially mediated dissolution. Furthermore, a small number of studies have explored the release of Fe from Al-goethite in the presence of individual ligands such as oxalate and siderophores (Bousserrhine et al. 1999; Cervini-Silva and Sposito 2002). These latter ligands comprise a structurally diverse group of Fe(III) chelators secreted by microbes in response to Fe stress (Haselwandter 2008; Watteau and Berthelin 1994). The steady-state dissolution of synthetic Al-goethite (x < 0.10) in the presence of the trihydroxamate siderophore, desferrioxamine B (DFOB), has been examined by Cervini-Silva and Sposito (2002). These workers observed that the rate of DFOB-mediated Fe release increased with Al substitution and also with DFOB concentration. This latter effect was apparent only to 100 µM DFOB, above which a plateau in Fe release was observed.
Although DFOB is the siderophore most commonly used in batch dissolution experiments with Fe(III) oxides, including Al-substituted goethite, more than 500 other siderophore types have been identified, of which approximately half have been structurally characterised (Boukhalfa and Crumbliss 2002; Boukhalfa et al. 2003). Among the more notable of these are ferrichrome, a trihydroxamate fungal siderophore with a 1:1 stability constant, K Fe(III), of 1029.07 (Wawrousek and McArdle 1982) and enterobactin, a bacterial catecholate siderophore whose 1:1 stability constant with Fe(III) = 1049 (Loomis and Raymond 1991). Generally, both hydroxamate and catecholate functional groups show a lower affinity for Al3+ than for Fe3+. For example, acetohydroxamate forms a weaker 1:1 association with Al3+ (log K = 8.0) than with Fe3+ (log K = 11.4) (Martell et al. 2004), while catechol forms a weaker 1:1 complex with Al3+ (log K = 16.22; Sikora and McBride 1989) than with Fe3+ (log K = 20.0; Martell et al. 2004). Importantly, major divalent cations such as Ca2+ complex only weakly with ferrichrome (log K = 4.3) and enterobactin (log K = 4.6) (Hider et al. 1982).
Given the wide occurrence of ferrichrome and enterobactin in soils and other Earth surface environments, and their considerable affinity for Fe(III) as indicated by the high 1:1 stability constants, these two siderophores have potential to influence the release of Fe from Al-substituted goethites. Catecholate siderophores such as enterobactin are of particular interest due to their exceedingly high affinity for Fe(III) and the limited studies examining their interaction with Fe(III) oxides. The objective of our experiments was therefore twofold: to measure, for the first time, the dissolution kinetics of synthetic Al-goethite at pH 6.5 (1) as a function of Al substitution (x < 0.1) and (2) in the presence of either ferrichrome or enterobactin under steady-state conditions.
2 Methodology
Pure goethite (α-FeOOH) and a series of four Al-substituted goethites were prepared as described in Schwertmann and Cornell (1991). Briefly, for the Al-goethite, we made fresh solutions of 0.5 M Al(NO3)3, 1 M Fe(NO3)3, and 5 M KOH. An aluminate solution was then prepared by combining 300 mL of KOH and 500 mL of Al(NO3)3 solutions while stirring constantly. Aliquots of the aluminate solution (20, 40, 80, and 120 mL) were then added to 2 L high density polyethylene (HDPE) bottles followed by addition of, respectively, 178, 176, 170, and 165 mL KOH stock solution. Immediately following addition of the KOH, 100 mL of 1 M Fe(NO3)3 was introduced to each bottle, which were subsequently brought to 2 L with deionised water. The suspensions were mixed for 10 min then incubated at 70 °C for 14 days and shaken daily. At the end of the 14 day reaction, the suspensions were centrifuged then washed three times with 1 M KOH to remove excess Al. The solids were washed with deionised water, freeze-dried, and stored in a dessicator at 23 °C for later use. As the goethites were dried and stored at temperatures well below the temperature at which dehydroxylation occurs (~110 °C), we avoid the formation of intracrystal micro- and mesopores observed by others (Naono et al. 1987; Ruan and Gilkes 1996).
The solids were confirmed as goethite and Al-substituted goethite by X-ray diffraction with an Enraf–Nonius PSD 120 diffractometer using Cu Kα1 radiation (45 mV, 45 kV) and fitted with an INEL 120° position sensitive detector. All samples appeared monomineralic (i.e. only goethite) at the detection limit of the XRD (~5 %). Ruan and Gilkes (1995) and Schwertmann and Cornell (1991) report smaller unit-cell dimensions for goethite following Al for Fe isomorphous substitution, shifting several peak positions to higher angles. We therefore interpret the progressive shift of the 111, 511, and 610 reflections to higher angles within the series of Al-substituted goethites as confirmation of increasing Al for Fe substitution in our goethites (Fig. 1).

X-ray diffraction patterns for the four synthetic Al-goethites. Bragg reflections for the Al-goethites are indicated by the hkl indices, with progressive shifting of the 111 reflection to higher angles indicating increasing Al for Fe substitution. Samples AG1 through AG4 represent Al-goethites with, respectively, 2.1, 3.3, 4.6, and 9.8 mol% Al
The Al and Fe content of the goethites were determined by first digesting 100 mg samples of each oxide in 20 mL of 12 M HCl (80 °C) for 30 min or until dissolution was complete. The solutions were then diluted 100-fold with 10 % HCl prior to analysis for Al and Fe by ICP-OES (Varian Vista Pro, ICP Expert version 4.1.0). Surface area of the goethites was determined by N2-BET analysis following degassing of each sample with N2 for 24 h at 100 °C (Micrometrics Gemini III 2375). To ensure accuracy of surface area measurements, we analysed a reference kaolinite (15.9 ± 0.8 m2 g−1) alongside the goethite samples.
Acid dissolution experiments were conducted on the series of goethites to determine their dissolution kinetics and also to assess their Al substitution (Schwertmann and Carlson 1994). Briefly, 250 mg of each oxide were suspended in 250 mL of 6 M HCl (25 °C) with continuous stirring. Five mL subsamples of the suspension were removed periodically then passed through 25 mm Millipore® nitrocellulose membrane filters (pore size 0.025 µm) into clear polythene tubes prior to Al and Fe analysis by ICP-OES. The reaction was allowed to proceed for 45 h.
The siderophore-promoted dissolution kinetics of pure goethite and the series of Al-goethites were measured in batch reactors at pH 6.5 and 25 °C. We chose pH 6.5 to ensure minimal proton-promoted dissolution during the 335 h reaction period and because it is representative of many Earth surface environments (e.g. soils) in which siderophores are abundant. The goethite concentration was 0.5 g L−1, and the background electrolyte was 10-mM NaNO3 (AnalaR, BDH) mixed with 1 mM MOPS [3-(N-morpholino) propanesulfonic acid, VWR] a noncomplexing pH buffer. We then added either iron-free ferrichrome [C27H45N9O12] or enterobactin [C30H27N3O15] (EMC Microcollection, Tübingen, Germany) (Fig. 2) to achieve an initial siderophore concentration of 240 µM, and pH was adjusted to 6.5 with 0.1 M NaOH or HClO4. We chose 240 µM as the initial siderophore concentration as it falls within a range of concentrations (20–1000 µM) commonly used in similar model systems, thus allowing comparison with results from other studies (e.g. Cervini-Silva and Sposito 2002; Cocozza et al. 2002; Wolff-Boenisch and Traina 2007; Stewart et al. 2013). Total volume of each batch reactor, prepared in triplicate, was brought to 200 mL by addition of background electrolyte. All samples were placed in a water bath at 25 °C for 335 h with constant stirring. Aliquots were periodically removed from each suspension then filtered through 25 mm Millipore® membrane filters of pore size 0.025 µm. Supernatant solutions were acidified to pH 1.5 with HCl prior to analyses for Al and Fe by ICP-OES. Siderophore concentrations in the filtrate solutions were measured by the chelometric method as described in detail elsewhere (Cheah et al. 2003; Stewart et al. 2016). Briefly, concentrations of the Fe(III)–siderophore complex for each filtrate were measured spectrophotometrically by absorption at 467 nm and compared against those for a series of Fe(III)-siderophore standards, the latter containing predetermined quantities of enterobactin or ferrichrome to construct the calibration curve. Samples were then placed in 1-mL UV microcuvettes (10-mm path length) and absorbance at 467 nm measured on a Shimadzu UV-1800 spectrophotometer. The surface excess for each siderophore was then derived by dividing the enterobactin or ferrichrome lost from solution by the goethite surface area. On the basis of previous studies that show there is an optimal reaction period during which ligand adsorption is achieved, but where dissolution is minimal (Cocozza et al. 2002; Simanova et al. 2010), we choose 30 min reaction time to determine maximum siderophore surface excess.

Structural representations of a ferrichrome and b enterobactin showing, respectively, their hydroxamate and catecholate functional groups. The proton dissociation constants for ferrichrome are: pK a1 = 8.11; pK a2 = 9.00; pK a3 = 9.83 (Anderegg et al. 1963), while those for enterobactin are: pK a1 = 6.0 ± 0.5; pK a2 = 7.5 ± 0.2; pK a3 = 8.6 ± 0.1 (Loomis and Raymond 1991)
The initial dissolution rates were derived by performing a linear least-square regression analysis. We chose the first five data points of the dissolution curve for the regression because they most closely approach linearity. The same number of points was used for each dissolution, giving regression coefficients (R 2) greater than 0.93 for all but one of the least-square fits.
3 Results and Discussion
3.1 Acid Dissolution of Synthetic Goethites
The Al content of the substituted goethites varied from 2.1 to 9.8 mol% Al (Table 1), while their surface areas decreased with increasing Al substitution, from 41.0 m2 g−1 for the least substituted Al-goethite to 29.7 m2 g−1 for the most highly substituted. Dissolution of these synthetic Al-substituted goethites alongside pure goethite in 6 M HCl yields S-shaped dissolution curves (Fig. 3). Generally, the shape of the curve became more sigmoidal, and the dissolution rate decreased with increasing Al substitution, a trend also observed by others (Cervini-Silva and Sposito 2002; Cornell and Schwertmann 2003; Schwertmann 1991). The shapes of our dissolution curves indicate that interdomain boundaries and surface defects may be created during the early stages of dissolution (Ruan and Gilkes, 1995), thereby increasing surface area and the rate of proton-promoted dissolution.

Dissolution-time curves of synthetic goethite and four synthetic Al-substituted goethites in 6 M HCl at 25 °C. Samples AG1 through AG4 represent Al-goethites with, respectively, 2.1, 3.3, 4.6, and 9.8 mol% Al
The time required to give 50 % dissolution of the solid, which we refer to as t 50, is given for Fe and Al for each oxide and provides a measure of the rate of goethite dissolution (Table 1). Our data show that the rate of dissolution decreases with increasing Al substitution, an observation consistent with previous studies indicating that Al substitution in goethite reduces proton-promoted dissolution (Cervini-Silva and Sposito 2002; Cornell and Schwertmann 2003). Moreover, the observation that dissolution rate decreases also with decreasing surface area (Table 1) indicates that proton-promoted dissolution of our goethites is surface-controlled. Given the similarity of Fe t50 and Al t50 for each oxide, we infer that there is no significant difference in the rate of dissolution of these two cations from the solids. This observation also indicates that Al is broadly randomly distributed within the oxides.
3.2 Siderophore-Promoted Dissolution
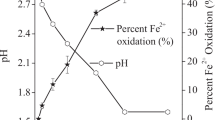
The Fe release kinetics for each oxide in the presence of ferrichrome or enterobactin at pH 6.5 and 240 µM initial siderophore concentration are shown in Figs. 4a, b, respectively. For both siderophores, the rate of Fe release is rapid initially then decreases at t > 20 h. Aluminium substitution in the oxides increases Fe release, with sample AG4 yielding the greatest soluble Fe at all reaction times for both ferrichrome and enterobactin. These observations are consistent with results from the dissolution of Al-goethite in the presence of DFOB (Cervini-Silva and Sposito 2002) and in the presence of siderophore-producing bacteria (Maurice et al. 2000). However, our results contrast with those for the dissolution of Al-goethite by Fe(III)-reducing bacteria (Bousserrhine et al. 1998; Ekstrom et al. 2010).

Iron release from synthetic goethite and four synthetic Al-substituted goethites in the presence of a ferrichrome and b enterobactin. Samples AG1 through AG4 represent Al-goethites with, respectively, 2.1, 3.3, 4.6, and 9.8 mol% Al. Initial siderophore concentration: 240 μM; solid concentration: 0.5 g L−1; pH 6.5; 25 °C
The dissolution kinetics at t < 20 h (Figs. 5a, b) show that dissolved Fe concentration depends approximately linearly on time, which is typical when dissolution reactions are far from equilibrium (Lasaga 1998; Sposito 1994; Stumm and Furrer 1987). The slope of the regression line equation is therefore equal to the rate coefficient for Fe release (Table 2, column 3). The initial dissolution rates of pure goethite by ferrichrome and enterobactin are 9.91 × 10−3 and 35.9 × 10−3 µmol m−2 h−1, respectively. This compares with dissolution rates of 3.86 × 10−3 µmol m−2 h−1 (Cocozza et al. 2002) and 5.98 × 10−3 µmol m−2 h−1 (Stewart et al. 2013) for goethite in the presence of 240 µM DFOB. It is notable that enterobactin induces significantly greater initial dissolution rates for goethite than either ferrichrome (Table 2, column 4) or desferrioxamine B at equivalent siderophore concentrations.

Initial iron release kinetics from synthetic goethite and four synthetic Al-substituted goethites in the presence of a ferrichrome and b enterobactin. Samples AG1 through AG4 represent Al-goethites with, respectively, 2.1, 3.3, 4.6, and 9.8 mol% Al. The lines through the data are linear least-square regressions (Table 2). Initial siderophore concentration: 240 μM; solid concentration: 0.5 g L−1; pH 6.5; 25 °C
Aluminium substitution gives rise to a significant increase in both the ferrichrome- and enterobactin-mediated initial dissolution rates. For example, in the presence of ferrichrome, sample AG1 (x = 0.021) yields a dissolution rate of 11.1 × 10−3 µmol m−2 h−1 whereas that for AG4 (x = 0.098) is more than threefold greater at 36.9 × 10−3 µmol m−2 h−1 (Table 2, column 4). Similarly, in the presence of enterobactin, the dissolution rate of the Al-goethites increases with Al substitution such that the rate for sample AG4 (170 × 10−3 µmol m−2 h−1) is approximately three times greater than that for sample AG1 (46.8 × 10−3 µmol m−2 h−1). Aluminium(III) for Fe3+ substitution evidently increases the population of reactive surface sites available for complexation by ferrichrome or enterobactin.
It is notable that there is little apparent difference in ferrichrome-mediated initial dissolution rates for goethite and sample AG1 (Fig. 5a), despite AG1 containing 2.1 mol% Al. However, when one considers the siderophore surface excess values for all samples, it is evident that the ferrichrome surface excess for goethite is about 10 % greater than that for AG1 (Table 2, column 5). When the dissolution rates are normalised for siderophore surface excess through a calculation of the pseudo-first-order rate coefficient, we observe an increase in dissolution kinetics in line with Al substitution. The pseudo-first-order rate coefficient is commonly used to characterise ligand-promoted dissolution kinetics far from equilibrium (Cocozza et al. 2002) and is obtained as a ratio by dividing the surface normalised initial dissolution rate by the surface excess of the ligand promoting the dissolution.
Steady-state Al release rates for each dissolution experiment are given in Table 3. If the Al-goethites dissolve congruently, the rate of Al release would vary with the Fe release rate according to the ratio x/(1 − x), where x is the Al mole fraction. This ratio for the Al-goethite samples AG1 through AG4 is 0.021, 0.034, 0.048, and 0.109, respectively. As shown in Table 3, the Al release rates derived experimentally for the Al-goethite samples in the presence of either ferrichrome or enterobactin approximate that predicted for the congruent dissolution of the oxide. Furthermore, the congruent dissolution we observe in the presence of these two siderophores corroborates our observation of congruence during proton-promoted dissolution in 6 M HCl (Table 1, columns 4, 5). These data together indicate that the Al in the Al-goethites is in solid solution with Fe and that there are no significant occurrences of gibbsite-like coatings or inclusions, even where x = 0.098 (i.e. for sample AG4), as these would give rise to differing responses to ligands and protons. Furthermore, given these data, we can infer that there is no significant secondary precipitation of Al or Fe released during dissolution of the solids.
3.3 The Efficacy of Enterobactin Versus Ferrichrome
Enterobactin is a more effective ligand than ferrichrome both in the magnitude of its effect (e.g. compare Fe release in Fig. 4a with that in Fig. 4b) as well as the consistency of its dissolution enhancement across diverse samples, from pure goethites to Al-goethites where x ~ 0.1 (i.e. sample AG4). The greater dissolution of goethite that we observe with enterobactin can not be ascribed to a greater initial ligand concentration, as both enterobactin and ferrichrome were present at starting concentrations of 240 µM. Likewise, the surface excess of the siderophores are broadly similar, varying from 0.558 to 0.871 µmol m−2 for enterobactin and 0.631 to 0.827 µmol m−2 for ferrichrome (Table 2). Differences in pK a values for the two siderophores (Fig. 2) appear not to have significantly influenced adsorption. It is also notable that neither siderophore reaches surface saturation, with less than 10 % of the total initial ligand load sorbed to the oxide surface, similar to surface excess values reported by Wolff-Boenisch and Traina (2007) for enterobactin and DFOB. Furthermore, both siderophores are hexadentate, forming 1:1 octahedral complexes with Fe3+ where each organic molecule contributes three, bidentate functional groups that together satisfy the sixfold coordination of Fe3+.
In addition to each siderophore’s initial concentration, surface excess, and number of functional groups, another important property involved in the dissolution of oxides is the nature of the functional groups. Comparing data in Fig. 4a with that in Fig. 4b illustrates that the catechol functional groups of enterobactin are more effective than the hydroxamate groups of ferrichrome in dissolving both pure goethite and Al-goethite. The greater efficacy of enterobactin that we report here has been observed previously by others during the dissolution of metal oxides. For example, Brainard et al. (1992) observed during the dissolution of PuO2 that the rate constant in the presence of enterobactin was at least an order of magnitude greater than that for ferrichrome. Similarly, Wolff-Boenisch and Traina (2007) reported that enterobactin-promoted goethite dissolution rates at pH 6 are approximately five times greater than comparable DFOB-mediated dissolution rates. These results indicate that catechol containing siderophores are significantly more effective than their hydroxamate counterparts in the dissolution of various metal oxides.
Catechol siderophores, with pK a1 values of 6–8 for the dissociation of the first proton, are generally more acidic than hydroxamate siderophores, which have pK a1 values of 8–9 (Raymond et al. 1987). As protonation of the functional groups competes with metal chelation, the pK a values of the donor groups must be considered when comparing the effectiveness of Fe(III) complexation by various ligands. Thus, given the lower pK a1 value for enterobactin (6.0 ± 0.5; Loomis and Raymond 1991) as compared to that for ferrichrome (8.11; Anderegg et al. 1963), one would predict greater affinity of enterobactin for Fe(III) at the pH of our systems (i.e. 6.5). Furthermore, as well as more favourable proton dissociation characteristics, enterobactin is uniquely predisposed to metal binding owing to the dynamic conformation of the free enterobactin molecule as described by Raymond et al. (2003). Specifically, the conformation of the uncomplexed ligand favours rapid initial binding of an Fe(III) atom, whereas the subsequent conformation change arising from proton loss and Fe(III) chelation promotes full encapsulation of the Fe(III) atom.
Thermodynamic parameters for Fe(III) complexes with enterobactin and ferrichrome are given in Table 4. For comparison, we also show data for the well studied trihydroxamate siderophore, DFOB. The significantly higher 1:1 stability constant for the enterobactin–Fe3+ complex (K = 1049), contrasts sharply with that for the two hydroxamate containing siderophores, namely the ferrichrome–Fe3+ complex (K = 1029.07) and the DFOB-Fe3+ complex (K = 1030.60). The E° values further demonstrate the greater thermodynamic influence conferred by enterobactin on the dissolution of Fe3+ from goethite, although it is apparent that all three siderophores induce considerable deviation from equilibrium.
Given the very low, and broadly similar, surface excess values for both enterobactin and ferrichrome, it is plausible that the steep thermodynamic gradients created by these siderophores are rate controlling. The presence of powerful chelators such as enterobactin and ferrichrome creates a thermodynamic sink, with enterobactin in particular demonstrating a greater pull for iron release from goethite. Further, the apparent contradiction between the relatively high oxide dissolution rates combined with low surface excess values has been considered previously by Kraemer (2004) who concluded that the oxide dissolution reaction is influenced by both metal affinity phenomena as well as ligand adsorption factors. Our data are consistent with this view. However, given the magnitude of the stability constants and E° values, we attribute greater relevance to chemical affinity than to ligand adsorption.
Both the catecholate and hydroxamate functional groups show a lower affinity for Al3+ than for Fe3+. This is illustrated most clearly for catecholate by considering the model ligand, catechol, where the 1:1 Al3+-catechol complex gives a log K of 16.22 (Sikora and McBride 1989), and the 1:1 Fe3+-catechol complex has a log K of 20.0 (Martell et al. 2004). Likewise, the monohydroxamate analogue, acetohydroxamate, forms a weaker 1:1 association with Al3+ (log K = 8.0) than with Fe3+ (log K = 11.4) (Martell et al. 2004). These trends occur also for DFOB, the linear trihydroxamate siderophore, such that the log K for the 1:1 complex of Al3+ with deprotonated DFOB is significantly smaller than that of Fe3+ (Albrecht-Gary and Crumbliss 1998; Desroches et al. 1999):
We attribute these trends in log K to Al3+ being a softer, less electronegative cation (χ = 1.61) than Fe3+ (χ = 1.96) and, therefore, less likely to form stable complexes with the hard-O donor atoms (OD) of either catcholate or hydroxamate. Also, because Al3+ has a lower electronegativity, this cation promotes less electron delocalisation along OD–Fe–O–Al linkages and, therefore, a lower bond energy for the neighbouring Fe–O groups. Despite these differences in Al3+ and Fe3+ affinity arising from the different functional groups of enterobactin and ferrichrome, we observe in this study a broadly congruent dissolution of Al-goethites at pH 6.5. Also, we note that both enterobactin and ferrichrome induce greater goethite dissolution as Al substitution increases over the range of Al contents examined in this study (i.e. 0.021 ≤ x ≤ 0.098). Clearly, further work is needed to elucidate the mechanisms underlying these various phenomena.
References
Ainsworth CC, Girvin DC, Zachara JM, Smith SC (1989) Chromate adsorption on goethite: effects of aluminum substitution. Soil Sci Soc Am 53:411–418
Albrecht-Gary AM, Crumbliss AL (1998) Iron transport and storage in microorganisms, plants, and animals. In: Sigel A, Sigel H (eds) Metal ions in biological systems, vol 35. Marcel Dekker, New York, pp 239–327
Alvarez M, Rueda EH, Sileo EE (2007) Simultaneous incorporation of Mn and Al in the goethite structure. Geoderma 140:8–16
Anderegg G, L’Eplattenier F, Schwarzenbach G (1963) Hydroxamatkomplexe III: eisen(III)-austausch zwischen sideraminen und komplexonen. Helv Chim Acta 46:1409–1422
Boukhalfa H, Crumbliss AL (2002) Chemical aspects of siderophores mediated iron transport. Biometals 15:325–339
Boukhalfa H, Lack J, Reilly SD, Hersman L, Neu MP (2003) Siderophore production and facilitated uptake of iron and plutonium in P. putida. AIP Conf Proc 673:343–344
Bousserrhine N, Gasser U, Jeanroy E, Berthelin J (1998) Effect of aluminium substitution on ferri-reducing bacterial activity and dissolution of goethites. Comptes Rendus Acad Sci Paris Earth Planet Sci 326:617
Bousserrhine N, Gasser UG, Jeanroy E, Berthelin J (1999) Bacterial and chemical reductive dissolution of Mn-, Co-, Cr-, and Al-substituted goethites. Geomicrobiol J 16:245–258
Brainard JR, Strietelmeier BA, Smith PH, Langston-Unkefer PJ, Barr ME, Ryan RR (1992) Actinide binding and solubilization by microbial siderophores. Radiochim Acta 58(59):357–363
Cervini-Silva J, Sposito G (2002) Steady-state dissolution kinetics of aluminum-goethite in the presence of desferrioxamine-B and oxalate ligands. Environ Sci Technol 36:337–342
Cheah SF, Kraemer SM, Cervini-Silva J, Sposito G (2003) Steady-state dissolution kinetics of goethite in the presence of desferrioxamine B and oxalate ligands: implications for the microbial acquisition of iron. Chem Geol 198:63–75
Cocozza C, Tsao CCG, Cheah SF, Kraemer SM, Raymond KN, Miano TM, Sposito G (2002) Temperature dependence of goethite dissolution promoted by trihydroxamate siderophores. Geochim Cosmochim Acta 66:431–438
Cooper SR, McArdle JV, Raymond KN (1978) Siderophore electrochemistry: relation to intracellular iron release mechanism. Proc Natl Acad Sci 75:3551–3554
Cornell RM, Schwertmann U (2003) the iron oxides—structure, properties, reactions, occurrences, and uses, 2nd edn. Wiley, Weinheim
Desroches S, Biron F, Berthon G (1999) Aluminum speciation studies in biological fluids. Part 5. A quantitative investigation of Al(III) complex equilibria with desferrioxamine, 2,3-dihydroxybenzoic acid, Tiron, CP20 (L1), and CP94 under physiological conditions, and computer-aided assessment of the aluminum-mobilizing capacities of these ligands in vivo. J Inorg Biochem 75:27–35
Dominik P, Pohl HN, Bousserrhine N, Berthelin J, Kaupenjohann M (2002) Limitations to the reductive dissolution of Al-substituted goethites by Clostridium butyricum. Soil Biol Biochem 34:1147–1155
Ekstrom EB, Learman DR, Madden AS, Hansel CM (2010) Contrasting effects of Al substitution on microbial reduction of Fe(III) (hydr)oxides. Geochim Cosmochim Acta 74:7086–7099
Harris WR, Carrano CJ, Raymond KN (1979) Spectrophotometric determination of the proton-dependent stability constant of ferric enterobactin. J Am Chem Soc 101:2213–2214
Haselwandter K (2008) Structure and function of siderophores produced by mycorrhizal fungi. Mineral Mag 72:61–64
Hider RC, Rahim Mohd-Nor A, Silver J, Neilands JB (1982) Coordination of calcium by iron enterobactin. J Inorg Chem 17:205–213
Kraemer SM (2004) Iron oxide dissolution and solubility in the presence of siderophores. Aquat Sci 66:3–18
Kukkadapu RK, Zachara JM, Smith SC, Fredrickson JK, Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments. Geochim Cosmochim Acta 65:2913–2924
Lasaga AC (1998) Kinetic theory in the earth sciences. Princeton University Press, Princeton
Loomis LD, Raymond KN (1991) Solution equilibria of enterobactin and metal–enterobactin complexes. Inorg Chem 30:906–911
Martell AE, Smith RM, Motekaitis RJ (2004) NIST critically selected stability constants of metal complexes. National Institute of Standards and Technology, Gaithersburg
Maurice PA, Lee YJ, Hersman LE (2000) Dissolution of Al-substituted goethites by an aerobic Pseudomonas mendocina var. bacteria. Geochim Cosmochim Acta 64:1363–1374
Naono H, Nakai K, Sueyoshi T, Yagi H (1987) Porous texture in hematite derived from goethite: mechanism of thermal decomposition of goethite. J Colloid lnterface Sci 120:439–450
Raymond KN, Cass ME, Evans SL (1987) Metal sequestering agents in bioinorganic chemistry: enterobactin mediated iron transport in E. coli and biomimetic applications. Pure Appl Chem 59:771–778
Raymond KN, Dertz EA, Kim SS (2003) Enterobactin: an archetype for microbial iron transport. Proc Natl Acad Sci USA 100:3584–3588
Ruan HD, Gilkes RJ (1995) Dehydroxylation of aluminous goethite—unit cell dimentions, crystal size and surface area. Clays Clay Miner 43:196–212
Ruan HD, Gilkes RJ (1996) Micropore formation in heated synthetic Al-goethites from a ferrous system. Clay Miner 31:75–79
Schulze DG, Schwertmann U (1984) The influence of aluminium on iron oxides. X: properties of Al-substituted goethites. Clay Miner 19:521–539
Schwertmann U (1991) Solubility and dissolution of iron oxides. Plant Soil 130:1–25
Schwertmann U, Carlson L (1994) Aluminium influence on iron oxides: XVII. Unit-cell parameters and aluminium substitution of natural goethites. Soil Sci Soc Am J 58:256–261
Schwertmann U, Cornell RM (1991) Iron oxides in the laboratory: preparation and characterization. VCH Publishers, New York
Schwertmann U, Taylor RM (1989) Iron oxides. In: Dixon JB, Weed SR (eds) Minerals in soils environments. Soil Science Society of America Inc, Madison, pp 379–439
Sikora FJ, McBride MB (1989) Aluminum complexation by catechol as determined by ultraviolet spectrophotometry. Environ Sci Technol 23:349–356
Simanova AA, Persson P, Loring JS (2010) Evidence for hydrolysis and Fe(III) reduction in the dissolution of goethite by desferrioxamine B. Geochim Cosmochim Acta 74:6706–6720
Spasojevic I, Armstrong SK, Brickman TJ, Crumbliss AL (1999) Electrochemical behavior of the Fe(III) complexes of the cyclic hydroxamate siderophores alcaligin and desferrioxamine E. Inorg Chem 38:449–454
Sposito G (1994) Chemical equilibria and kinetics in soils. Oxford University Press, Oxford
Stewart AG, Hudson-Edwards KA, Dubbin WE (2013) Mechanisms of goethite dissolution in the presence of desferrioxamine B and Suwannee River fulvic acid at pH 6.5. Geochim Cosmochim Acta 115:1–14
Stewart AG, Hudson-Edwards KA, Dubbin WE (2016) Effect of desferrioxamine B and Suwannee River fulvic acid on Fe(III) release and Cr(III) desorption from goethite. Geochim Cosmochim Acta 178:62–75
Stumm W, Furrer G (1987) The dissolution of oxides and aluminum silicates; examples of surface-controlled kinetics. In: Stumm W (ed) Aquatic surface chemistry. Wiley Interscience, New Jersey, pp 197–219
Watteau F, Berthelin J (1994) Microbial dissolution of iron and aluminium from soil minerals: efficiency and specificity of hydroxamate siderophores compared to aliphatic acids. Eur J Soil Biol 30:1–9
Wawrousek EF, McArdle JV (1982) Spectroelectrochemistry of ferrioxamine B, ferrichrome, and ferrichrome A. J Inorg Biochem 17:169–183
Wolff-Boenisch D, Traina S (2007) The effect of desferrioxamine B, enterobactin, oxalic acid, and Na-alginate on the dissolution of uranyl-treatedgoethite at pH 6 and 25 & #xB0;C. Chem Geol 243:357–368
Acknowledgments
We thank Emma Humphreys-Williams and Stanislav Strekopytov for assistance with chemical analysis and Jens Najorka for XRD analysis. Funding for this study was provided through a grant from the Natural Environment Resesarch Council (UK).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dubbin, W.E., Bullough, F. Dissolution of Al-Substituted Goethite in the Presence of Ferrichrome and Enterobactin at pH 6.5. Aquat Geochem 23, 61–74 (2017). https://doi.org/10.1007/s10498-016-9304-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10498-016-9304-4