
Abstract
Lung cancer is one of the most common malignant tumors. Despite decades of research, the treatment of lung cancer remains challenging. Non-small cell lung cancer (NSCLC) is the primary type of lung cancer and is a significant focus of research in lung cancer treatment. The deubiquitinase ubiquitin-specific protease 28 (USP28) plays a role in the progression of various tumors and serves as a potential therapeutic target. This study aims to determine the role of USP28 in the progression of NSCLC. We examined the impact of the USP28 inhibitor AZ1 on the cell cycle, apoptosis, DNA damage response, and cellular immunogenicity in non-small cell lung cancer. We observed that AZ1 and siUSP28 induce DNA damage, leading to the activation of Noxa-mediated mitochondrial apoptosis. The dsDNA and mtDNA released from DNA damage and mitochondrial apoptosis activate tumor cell immunogenicity through the cGAS-STING signaling pathway. Simultaneously, targeting USP28 promotes the degradation of c-MYC, resulting in cell cycle arrest and inhibition of DNA repair. This further promotes DNA damage-induced cell apoptosis mediated by the Noxa protein, thereby enhancing tumor cell immunogenicity mediated by dsDNA and mtDNA. Moreover, we found that the combination of AZ1 and cisplatin (DDP) can enhance therapeutic efficacy, thereby providing a new strategy to overcome cisplatin resistance in NSCLC. These findings suggest that targeting USP28 and combining it with cisplatin are feasible strategies for treating NSCLC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
USP28 is a member of the deubiquitinase (DUBs) family of ubiquitin-specific proteases. It is typically considered an oncoprotein that can stabilize many potent oncogenic proteins commonly found in cancer cells, such as c-MYC, c-JUN, NOTCH1, STAT3, and LIN28, thereby promoting cancer cell vitality and growth. After stabilizing FOXC1, USP28 enhances aerobic glycolysis and promotes cell proliferation. USP28 can also stabilize ZNF304, leading to hypermethylation and transcriptional silencing of tumor suppressor genes. Additionally, USP28 regulates processes such as angiogenesis, metastasis, and cellular adaptation to hypoxia by stabilizing HIF-1α [1]. However, some studies have indicated that USP28 also exhibits tumor-suppressive effects. USP28 can induce H2A-mediated activation of tumor suppressor gene transcription [2]. Additionally, USP28 has been shown to reduce FN-1 and increase E-cadherin levels, which can inhibit tumor invasion and migration [3]. In certain types of tumors, the overexpression of USP28 is primarily associated with tumor-promoting effects, as seen in gliomas, hepatocellular carcinoma, bladder cancer, and colorectal cancer [4,5,6,7,8,9]. However, in some tumors, overexpression of USP28 demonstrates tumor-suppressive effects, as observed in breast cancer [10, 11]. These findings suggest that the role of USP28 in tumors is highly complex and can vary among different types of cancer. Therefore, further research is needed to explore the potential role of USP28 in cancer therapy.
Cisplatin (DDP) is one of the commonly used chemotherapeutic drugs in cancer treatment. Platinum compounds cause DNA damage by forming covalent adducts with cellular DNA, leading to the induction of cell apoptosis and exerting their anti-tumor activity [12]. However, clinical application has revealed that cisplatin treatment can lead to nephrotoxicity, neurotoxicity, bone marrow suppression, and gastrointestinal toxicity. The intrinsic DNA repair mechanisms of cells can make tumor cells resistant to DNA-damaging chemotherapeutic drugs, which is a significant factor contributing to platinum resistance in cancer patients. Toxic reactions and drug resistance are the primary factors that restrict the clinical application of platinum-based chemotherapeutic drugs such as cisplatin. Combination therapy is a common strategy used to reduce toxicity and enhance the efficacy of chemotherapy drugs. Combination with drugs that inhibit DNA repair is also one of the emerging application strategies for platinum-based chemotherapy drugs. The combination therapy of the PARP-1 inhibitor niraparib with cisplatin has been clinically utilized to overcome acquired platinum resistance in cancer cells [13]. USP28 is closely related to DNA repair. Studies have shown that USP28 can mediate cellular adaptive changes after DNA damage [14], stabilize the expression of the RecQ family of deubiquitinating enzymes [15], and participate in mitotic checkpoint pathways [16]. Therefore, targeting USP28 in combination with cisplatin may be an effective strategy for tumor chemotherapy.
AZ1 is a USP28 inhibitor that has been less well studied and is more focused on neurological disorders. In this study, we observed the phenomenon of apoptosis induced by AZ1 alone and in combination with cisplatin in non-small cell lung cancer through in vitro studies and explored the underlying mechanisms. And we further explored the effect of AZ1 in combination with cisplatin in the treatment of non-small cell lung cancer through in vivo experiments.
Materials and methods
Cell culture and regents
NSCLC cell lines (A549, H460, Calu-3, H3122) were purchased from the Shanghai Cell Resource Centre Academy of Sciences. The cell lines underwent testing by STR (QuiCell Biological, Shanghai, China) which confirmed that no cross-contamination between the cell lines was present. Cells were cultured in complete culture medium containing 90% DMEM (No. 10–013-CVRV, CORNING, China) and 4 mM glutamine (no. 2323081, Glibco) and 1% penicillin–streptomycin (no.C0224, Beyotime, China) and 10% fetal bovine serum (No. FSP500, ExCell Bio) in a cell culture vessel at 37 °C, 5% CO2 in a cell culture incubator. Cells were passaged every 1–2 d. Cells in the logarithmic growth phase were selected for the experiment. The USP28 inhibitor AZ1 was purchased from Good Laboratory Practice bioscience.AZ1 was stored in dimethyl sulfoxide (DMSO) solution. Cisplatin injection purchased from Hansoh Pharma (China).
Cell viability and colony formation
Human NSCLC cell lines A549, H460, H3122 and Clu-3 were inoculated into 96-well plates and treated with AZ1 or DMSO (0.1%) after 16–20 h. Cell viability was determined after 48 h using Cell Counting Kit-8 (CCK-8 Kit) (Beyotime Institute of Biotechnology, China).
Cell growth was also assessed by a colony formation assay. Suspension cells were aliquoted into 6-well plates, with 1000–2000 cells per well, treated with DMSO (0.1%) or AZ1 and then incubated for 7–10 days. Cell colonies were fixed with 4% paraformaldehyde (Solarbio, China) for 30 min and then stained with crystal violet (Beyotime, China) for 30 min. Enumeration of the cell colonies was performed.
Cell cycle
A549 and H460 cells were treated with AZ1 or DMSO (0.1%) for 24 h.Following treatment, cells were collected and fixed with 70% ethanol at 4 °C overnight. After centrifugation, the supernatant was discarded and the cells were resuspended in PBS containing 10 μg/ml PI and 50 μg/ml RNaseA and incubated for 30 min at 37 °C in the dark. The cell sorting instrument, Becton Dickinson FACScan (Becton–Dickinson, San Jose, CA, USA), was utilized for the detection of fluorescently activated cells. The obtained results were processed using MODFit LT 3.1 software.
Western blotting assay
AZ1 or DMSO (0.1%) was used to treat A549 and H460 cells for 48 h. RIPA lysis buffer (Beyotime) was used to lyse the cells on ice for 30 min to obtain samples for Western blotting assay. Antibodies against p27(#3686), CyclinD1(#55,506), CyclinE1(#4129),CDK2(#18,048), CDK4(#23,972) cleaved CASP9(#7237), cleaved CASP3(#9664),cleaved-PARP(#5625), Bax(#5023), Bak(#12,105), Bcl-xl(#2764), Mcl-1(#5453), XIAP(#2045), CIAP1(#7065), Bcl-2(#15,071), Y-H2AX(#80,312) Noxa(#14,766), and c-MYC(#18,583)were obtained from CST (Cell Signaling Technology, Danvers, MA, USA). Antibodies against ATM(AF1399), ATR(AF6267), CHK1(AF1849), CHK2(AF2020), BRCA1(AF6339), RAD51(AF7860)from Beyotime Biotechnology(Shanghai, China) Antibodies against GAPDH (TA-08, ZGSB-Bio, China) were used as controls.
Apoptosis detection
Following treatment with AZ1 or DMSO (0.1%) for 48 h, A549 and H460 cells were subjected to apoptosis detection using the AnnexinV EGFP/PI dual staining cell apoptosis detection kit (KeyGEN BioTECH, Nanjing, China) according to the manufacturer’s instructions.
Detection of mitochondrial membrane potential
After AZ1 or DMSO (0.1%) treatment of A549 and H460 cells for 24 h, the JC-1 mitochondrial membrane potential assay kit (Yeasen, Shanghai, China) was used according to the instructions.
Immunofluorescence
A549 and H460 cells were inoculated into 96-well plates and treated with AZ1 or DMSO (0. 1%) for 48 h.The cells were then fixed with ice-cold anhydrous methanol at − 20 °C for 30 min. Subsequently, the cells were incubated at room temperature for 2 h with 5% bovine serum albumin (BSA). The cells were further incubated overnight with γ-H2AX antibody (AF5836, Beyotime, China) diluted in 5% BSA (1:300). Following this, the cells were incubated at room temperature for 2 h with Cy3-conjugated goat anti-rabbit secondary antibody (Beyotime, China). Finally, DAPI (Beyotime, China) was incubated for 20 min.
The comet experiment
A549 and H460 cells were treated with AZ1 or DMSO (0.1%) for 24 h, followed by treatment according to the instructions of the comet assay kit (KeyGEN BioTECH). The cells were collected and mixed with low melting point agarose and added dropwise to a well solidified normal melting point agarose gel. When the upper layer of gel was well solidified, it was placed in lysis solution and lysed at − 4 °C for 2 h. It was placed in pre-cooled electrophoresis solution, deconvoluted for 20 min, followed by electrophoresis for 20–30 min at 25 V. The samples were neutralized at 4 °C and subsequently stained by PI. Keep out of the light. Subsequently, they were observed and photographed under a fluorescence microscope.
Establishment and treatment of mouse lung cancer models.
Five-week-old female BALB/c mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Each mouse was injected subcutaneously with Lewis lung cancer cell suspension (5 × 105 cells/100ul PBS), 100 μl, to establish a mouse subcutaneous lung cancer model. Four days post-injection, the tumour-bearing mice were randomly divided into four groups for treatment (AZ1: 20 mg/kg [17] and DDP: 20 mg/kg), and treatment was performed every three days. Mice were euthanized six days after the first treatment and tumors were surgically removed. Tumor tissue was fixed in liquid nitrogen or 4% paraformaldehyde for preservation. The mouse experiments were conducted in a specific pathogen-free (SPF) animal facility. Animal experiments were carried out in accordance with the guidelines approved by the Institutional Animal Care and Use Committee of Zhengzhou University.
Flow cytometry
Cells extracted frm tumor tissue and spleen were incubated in the dark with fluorescent-labeled surface antibodies for 30 min. Subsequently, the cells were fixed with 4% paraformaldehyde for 20–30 min, followed by incubation with a membrane permeabilization agent for 30 min. The cells were then incubated with corresponding intracellular factor antibodies for 30 min. Finally, flow cytometry analysis was performed.
RNA extraction and quantitative real-time fluorescence quantitative PCR
A549 and H460 cells were treated with drugs or siRNA for 48 h. RNA was then extracted and reverse transcribed using the PrimeScript™ RT reagent kit (Cat. No. RR047A, Takara, Dalian, China) with a gDNA eraser. The cDNA was amplified and analyzed using the QuantiNova™ SYBR Green PCR reagent kit (Cat. No. 208054, QIAGEN, Germany) and a q-PCR system (Applied Biosystems, Foster City, California) following the instructions provided. For each sample, the mRNA abundance was normalized to the quantity of β-actin. The relative expression was calculated as fold change = 2−∆∆Ct.
The primer sequences are as follows:
β-actin F:CATGTACGTTGCTATCCAGGC, R:CTCCTTAATGTCACGCACGAT; USP28 F:CACTGTTGCTACAGAACCATC, R:TGGGAGACTCCAGTAGACTCA; CGAS F:TAACCCTGGCTTTGGAATCAAAA, R:TGGGTACAAGGTAAAATGGCTTT; STING F:CACTTGGATGCTTGCCCTC, R:GCCACGTTGAAATTCCCTTTTT; CCL5 F:CCAGCAGTCGTCTTTGTCAC, R:CTCTGGGTTGGCACACACTT; CXCL10 F:GTGGCATTCAAGGAGTACCTC, R:TGATGGCCTTCGATTCTGGATT
Silencing of genes by siRNA
Transfection of siRNA into A549 and H460 with jetPRIME Transfection Reagent (No. 6).The siRNA sequences are as follows: siNoxa:GUAAUUAUUGACACAUUUC[18]; siControl: GUUCUCCGAACGUGUCACGU; siUSP25-1: GCCAGUGCAUACUGUUUAATT; siUSP25-2: CCCACCAGAAACCGAUUAUT; siUSP28-1GCUGCCAAAUGCUGUUAAATT; siUSP28-2: GGGCCUAUAUCUAUAAUCATT。All siRNAs were synthesised by GenePharma Ltd (Shanghai, China).
Statistical analysis
The experimental data were statistically analyzed using SPSS 21.0 software and GraphPad Prism 9 software. One-way analysis of variance (ANOVA) and t-tests were used for normally distributed data. The t-tests were used to compare parameters between different groups. ns indicates non-significant results; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Results
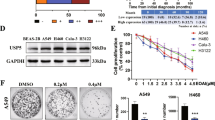
AZ1 inhibited the proliferation of NSCLC cells by inducing G0/G1 phase arrest. We first detected the USP28 protein expression level after 48 h of AZ1 treatment by Western blot. The results showed that AZ1 could effectively inhibit USP28 in non-small cell lung cancer (Fig.S1A). To determine whether USP28 could be a new target for NSCLC therapy, we exposed four NSCLC cell lines to varying concentrations of AZ1 and evaluated cell viability. The results showed that AZ1 (0–20 μM) inhibited the growth of the four NSCLC cell lines in a dose-dependent manner (Fig. 1A), as well as colony formation (Fig. 1B, C). The results suggest that targeting USP28 is a promising approach for the treatment of NSCLC. We analyzed the effect of AZ1 on the NSCLC cell cycle by flow cytometry and observed that AZ1 treatment of both A549 and H460 cells led to an increase in the number of cells in the G0/G1 phase (Fig. 1D, E). To further validate the cell cycle block and understand its underlying mechanism, we detected the expression of some cell cycle-related proteins by Western blotting. The results showed that AZ1 treatment increased the expression of P27 and decreased the expression of c-MYC, CyclinD1, CyclinE1, CDK4, CDK2, and P21 (Fig. 1F). These results suggest that AZ1 treatment induces G1 phase blockade in NSCLC.

AZ1 restrained NSCLC cell growth, colony formation and induced cell cycle arrest A AZ1 affects NSCLC cell proliferation. The cells were treated with either AZ1 or DMSO (0.1%), and cell viability was assessed after 48 h using the CCK-8 kit. The EC50 was calculated using SPSS software. B AZ1 impacts NSCLC cell colony formation. NSCLC cells were subjected to varying concentrations of AZ1 for a period of 10 days, fixed, stained and subsequently photographed. C The intergroup differences in the number of colonies were analyzed using GraphPad Prism 9.0. D AZ1 induces G0/G1 phase arrest in NSCLC cells. A549 and H460 cells were treated with varying concentrations of AZ1 for 24 h, stained with PI, and analyzed using flow cytometry and ModFit software. Representative images are shown. F AZ1 affects the expression levels of cell cycle-related proteins. Following a 48 h treatment of AZ1 on A549 and H460 cells, a Western blotting assay was conducted. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, compared with the control group. Data are shown as the mean ± standard deviation
AZ1 induced mitochondria-dependent apoptosis in NSCLC cells mediated by Noxa
After 48 h of treatment with AZ1, we observed dose-dependent cell death in A549 and H460 cells, which was visible to the naked eye. Flow cytometry was then employed to detect the changes, and it was found that AZ1 treatment significantly increased the expression of membrane-associated protein V (Fig. 2A) and prominently altered the mitochondrial membrane potential (Fig. 2B). In addition, cleavage of PARP, caspase-3 (CASP3) and caspase-9 (CASP9), classical markers of apoptosis activation, was also increased in AZ1-treated cells (Fig. 2C). The expression of apoptotic proteins (Noxa, Bak, Bax) increased in AZ1-treated A549 and H460 cells, while the expression of anti-apoptotic proteins (Mcl-1, XIAP, cIAP1, and cIAP2) decreased (Fig. 2C). Western blot results showed that our selected siRNA could effectively knock down the protein expression level of Noxa gene(Fig.S2). Silencing of Noxa through siRNA inhibited AZ1-induced apoptosis in NSCLC cells (Fig. 2D) and reduced PARP cleavage (Fig. 2E).

Noxa mediated mitochondrial apoptosis in NSCLC cells Cells were processed according to the Annexin V-FITC/PI double staining kit and analyzed by flow cytometry for cell fractionation. B Flow cytometry analysis of the effect of AZ1 on mitochondrial membrane potential in NSCLC. C Western blot analysis of the effect of AZ1 on the expression of apoptosis-related proteins. D After introducing Noxa siRNA or control siRNA into A549 and H460 cells, AZ1 treatment was performed for 48 h. Annexin V-FITC/PI double staining was performed. E Western blotting was performed to analyze the efficiency of Noxa knockdown and its effect on cleaved PARP expression. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, compared with the control group. Data are shown as the mean ± standard deviation
AZ1 induced DNA damage and inhibits DNA repair in NSCLC cells
We speculated that AZ1 itself could induce DNA damage, and this speculation was confirmed by immunofluorescence (Fig. 3A, B) and comet assays (Fig. 3C, D). We investigated the protein expression levels of DNA damage response-related proteins and observed that treatment with AZ1 led to a significant decrease in classic DNA repair-related proteins such as ATM, ATR, CHK1, CHK2, BRCA1, and RAD51. Additionally, there was a significant increase in the expression of the DNA damage marker γ-H2AX (Fig. 3E).

Treatment with AZ1 induced DNA damage and inhibited DNA repair in NSCLC cells A Following treatment with AZ1 for 48 h, the cells underwent staining with a primary antibody against γ-H2AX (1:200) overnight at 4 °C. Representative images were captured using a fluorescence microscope. B GraphPad Prism 9.0 was used to statistically analyze the immunofluorescence intergroup differences. C Immunoblot analysis of DNA damage- and repair-related proteins after 48 h of AZ1 treatment. D GraphPad Prism 9.0 was used to analyze the positive rate of the comet assay. E Western blot was used to detect the levels of γ-H2AX and DNA repair-related proteins. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, compared with the control group. Data are shown as the mean ± standard deviation
AZ1 enhanced the immunogenicity of NSCLC cells
Immunotherapy is a new strategy for cancer treatment. dsDNA and mtDNA can activate the cGAS-STING pathway, leading to increased secretion of chemokines, enhancing cellular immunogenicity and promotion of immune-mediated tumor killing. Therefore, we examined the expression of cGAS, STING, CCL5, and CXCL10 genes. We observed through Western blot that AZ1 treatment led to an increase in cGAS and STING protein expression levels (Fig. 4A, B). Due to the low protein levels, CCL5 and CXCL10 proteins could not be validated by Western blot. We further observed with q-PCR that AZ1 upregulated cGAS (Fig. 4C), STING (Fig. 4D), CCL5 (Fig. 4E) and CXCL10 (Fig. 4F) mRNA levels.

AZ1 activated the cGAS-STING signaling pathway A Western blot was used to detect the protein levels of cGAS and STING after treatment with AZ1 for 48 h. B GraphPad Prism 9.0 was used for statistical analysis of the protein abundance of cGAS and STING. Then q-PCR was used to detect the mRNA expression fold changes of cGASC, STINGD, CCL5E, and CXCL10F after treatment with AZ1 for 48 h. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, compared with the control group. Data are shown as the mean ± standard deviation
Silencing the USP28 gene had a similar effect to AZ1 treatment in NSCLC.
To further investigate the role of USP28 in NSCLC, we silenced USP28 using siRNA. The expression of apoptotic marker proteins increased (Fig. 5A), and the levels of cell cycle proteins were also altered. Silencing of USP28 increased the expression of P27 and significantly decreased the expression of Cyclin D1, Cyclin E1, CDK4, and CDK2, consistent with the effect of AZ1 treatment (Fig. 5B). The alterations in DNA damage (Fig. 5C) and inhibition of DNA repair (Fig. 5D) induced by USP28 silencing were also consistent with AZ1 treatment. Silencing USP28 also activates the cGAS-STING signaling pathway (Fig. 5E, F). The gene sequences of USP28 and USP25 are similar, with the overall identity and similarity between the sequences of USP25 and USP28 being 48% and 76%, respectively, including similar insertions. Therefore, USP28 and USP25 have very similar structures, and inhibitors of USP28 typically also inhibit USP25. There is evidence that AZ1 can also inhibit USP25, we also knocked down USP25 with siRNA to observe the effects on apoptosis, cell cycle, DNA damage, and DNA repair in non-small cell lung cancer. The results showed that silencing USP25 alone led to a similar increase in γ-H2AX and activation of apoptosis as AZ1 treatment. However, it did not inhibit DNA repair, and the changes in cycle proteins also significantly differed from those observed with AZ1 treatment (Fig. S3).

USP28 knockdown replicated the effect of AZ1 on NSCLC cells Western blot was used to detect the levels of apoptosis A, cell cycle B, DNA damage D, and DNA repair-related proteins 48 h after siUSP28 treatment. C Immunofluorescence was used to detect the formation of γ-H2AX foci 48 h after siUSP28 treatment. E Western blot was used to detect the levels of cGAS and STING proteins 48 h after siUSP28 treatment. F q-PCR was used to detect the mRNA fold changes of the cGAS-STING signaling pathway. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, compared with the control group. Data are shown as the mean ± standard deviation
AZ1 was used in combination with cisplatin for the treatment of non-small cell lung cancer
In previous experiments, we found that targeting USP28 with AZ1 induces DNA damage in non-small cell lung cancer while also inhibiting DNA repair. Therefore, we considered whether AZ1 could enhance the therapeutic effects of traditional platinum-based chemotherapy drugs and provide a new approach for the application of platinum-based chemotherapy drugs. Consequently, we further explored whether AZ1 could synergize with cisplatin in treating non-small cell lung cancer. As expected, the combination of AZ1 and DDP significantly inhibited the growth of NSCLC (Fig. 6A), colony formation (Fig. 6B), enhanced the cytotoxic effect (Fig. 6C), increased and DNA damage (Fig. 6D) of DDP on NSCLC. Western blotting revealed a significant increase in the cleaved forms of the classical apoptotic proteins PARP and Casp3, as well as Noxa, in the combination group. Additionally, the expression of DNA repair-related proteins was lower in the combination group compared to the DDP monotherapy group (Fig. 6E).

AZ1 can synergistically enhance the treatment of NSCLC with DDP A A Cell Counting Kit-8 (CCK-8) assay was conducted to determine the viability of NSCLC cells after treatment with DDP alone or in combination with DMSO or AZ1 for 48 h. The results of the assay were recorded. Calculate the combination index (CI) of two drugs used in combination using compusyn 1.0, and plot the CI/Fa curve using Origin 2021. B Observation of cell colony formation using the flat cloning method. Cells were treated with DDP alone or in combination with DMSO or AZ1 for 10 days, and photographs were taken for observation. C Apoptosis analysis of NSCLC cells treated with DDP alone or in combination with DMSO or AZ1 using the Annexin V-FITC/PI dual staining kit for flow cytometry. D Immunofluorescence staining of γ-H2AX in NSCLC cells treated with DDP alone or in combination with DMSO or AZ1, showing DNA damage. E Western blot analysis of apoptosis-, DNA damage-, and repair-related protein expression in NSCLC cells treated with DDP alone or in combination with DMSO or AZ1 for 48 h. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, compared with the control group. Data are shown as the mean ± standard deviation
AZ1 combined with cisplatin inhibited the progression of non-small cell lung cancer in mice
To further investigate the efficacy of the combination of AZ1 and cisplatin in treating non-small cell lung cancer in in vivo models, we established a subcutaneous tumor model using LLC cells in mice. After administering drugs to the tumor-bearing mice for 6 days, we euthanized the mice by dislocating the cervical spine, rapidly removed the implanted tumors, took photographs, and weighed them (Fig. 7A). The results showed that the tumor volume and weight in the AZ1 and cisplatin monotherapy groups were significantly smaller than those in the control group, while the combination group had significantly smaller tumor volume and weight than the monotherapy groups (Fig. 7B). This suggests that AZ1 in combination with cisplatin can synergistically inhibit the progression of non-small cell lung cancer in in vivo models. To assess the impact of AZ1 and cisplatin treatment on the anti-tumor immune response in non-small cell lung cancer, we used flow cytometry to detect the positivity rates of CD8 + T cells for IFN-γ and Granzyme B in mouse tumors (Fig. 7C) and spleens (Fig. 7D). The results showed that in both tumor tissues and spleens, the positivity rates of CD8 + T cells for IFN-γ and Granzyme B were higher in the AZ1 and cisplatin monotherapy groups compared to the control group, and even higher in the combination group compared to the monotherapy groups.

AZ1 suppresses tumor growth and improves the effectiveness of DDP in mouse models of lung cancer A Mice were subcutaneously implanted with lung cancer tumors and grouped for drug administration starting from the fourth day. Mice were euthanized on the tenth day. Group a was treated with PBS, Group b with AZ1 alone, Group c with DDP alone, and Group d with AZ1 and DDP. Please refer to the “Materials and Methods” section for details on the drug administration methods B Photographs were taken post-execution to record the tumor volume of mice. The tumors were weighed and subjected to statistical analysis. C Flow cytometry analysis of CD8+ T-cell IFN-γ and GranB positivity rates in mouse tumors under four treatment groups, with intergroup differences analyzed using GraphPad Prism 9.0. D Flow cytometry analysis of CD8+ T-cell IFN-γ and GranB positivity rates in mouse spleens under four treatment groups, with intergroup differences analyzed using GraphPad Prism 9.0.E Immunohistochemical staining of tumor tissue. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, compared with the control group. Data are shown as the mean ± standard deviation
Discussion
Research on lung cancer has been ongoing for many years, but it still remains one of the leading causes of cancer-related deaths globally. The study of treatment strategies for lung cancer is a daunting task. USP28 plays a crucial role in tumor progression, and inhibiting USP28 can suppress the growth of various tumors [1]. Therefore, inhibiting USP28 is a promising strategy for the treatment of lung cancer. AZ1 is a USP28 inhibitor that has demonstrated significant anti-tumor effects in some studies. However, the role and mechanism of action of AZ1 and USP28 in lung cancer are not yet clear. The combination of targeting USP28 therapy with traditional chemotherapy also requires further exploration.
USP28 is involved in various cellular physiological activities, including cell cycle regulation. In previous studies on breast cancer, depletion of USP28 was found to result in an increase in the G2/M fraction in TNBC cells. However, in MDA-MB-231 and HCC1937 cells, it led to an increase in the S phase fraction, while no cell cycle disruption was observed in MCF7 cells [15]. In addition, in hepatocellular carcinoma, MiR-216b was found to impact the cell cycle by inhibiting USP28/c-MYC, downregulating Cyclin E, and upregulating P27 [19]. It has also been found that USP28 depletion did not cause cell cycle arrest in HCT116, HMEC, HEK-293 T, and U2OS cells, but instead increased the cell proliferation rate by reducing P53 [2, 20, 21]. Therefore, USP28 has varying effects on the cell cycle of different types of tumor cells, and further investigation is needed to understand its underlying mechanisms. In our study, inhibiting USP28 led to an increase in the G1/G0 fraction in A549 and H460 cells. CyclinD-CDK4 and CyclinE-CDK2 are G1 → S driving signals [22]. In our research, after inhibiting USP28, the levels of Cyclin D1, CDK4, Cyclin E1, and CDK2 all decreased. P27 is a widely expressed cell cycle inhibitory factor, and a 2–threefold increase in P27 can completely inhibit the G1 → S transition [23]. In our study, the USP28 inhibitor upregulated P27. The changes in cyclin proteins were consistent with the results of cell cycle flow cytometry analysis. MYC is an important transcriptional regulatory factor, overexpressed in approximately 70% of malignant tumors in humans, with c-MYC being the most common form of MYC dysregulation in cancer [24, 25]. Research has shown that c-MYC drives cell proliferation by inducing the activation of CyclinE-CDK2 kinase activity. This induction leads to the release of P27Kip1 from the CDK2 complex, forming a CyclinD-CDK4-P27Kip1 complex [26]. Additionally, it has been reported that MYC can reduce the inactivation of the CyclinD-CDK4 complex by inhibiting the expression of the p15NK4b gene [27]. Furthermore, MYC can directly transactivate CyclinD and CDK4 genes [28]. Therefore, inhibiting c-MYC is a strategy to prevent the G1 → S transition. Additionally, USP28 has been identified as an important deubiquitinase that stabilizes c-MYC [29]. These studies suggest that the potential mechanism of this experiment involves AZ1 targeting USP28, which leads to increased degradation of c-MYC in non-small cell lung cancer. This results in a decrease in cyclin proteins (CyclinD1, CDK4, CyclinE1, CDK2), an increase in P27, and subsequently, cell cycle arrest at the G0/G1 phase.
Multiple studies indicate that P53 is a downstream target protein of USP28, as USP28 can stabilize and activate P53 [2, 16, 20, 30,31,32,33,34]. Additionally, P53 is an important apoptosis-inducing protein [35,36,37,38,39,40,41]. Therefore, it is deduced that the inhibition of USP28 should lead to a reduction in P53-mediated apoptosis. However, some studies have indicated that USP28 deficiency leads to apoptosis [42]. The specific mechanism by which USP28 deficiency induces apoptosis is still unclear. In our study, treatment with AZ1 also significantly induced cell apoptosis. A study in 2021 demonstrated that shUSP28 leads to an increase in cleaved Caspase-3 and cleaved PARP, while Caspase-3 and PARP levels decrease. This suggests that USP28 can affect the caspase cascade reaction [42,43,44]. Reports indicate that the overexpression of Noxa in tumors can significantly activate cell apoptosis, initiating the apoptosis process centered around the caspase cascade reaction[18]. In our study, the USP28 inhibitor AZ1 can upregulate Noxa, thereby activating Noxa-mediated mitochondrial apoptosis. Noxa binds to MCL-1 and Bcl-2, releasing BAX and BAK1, causing pores to form in the outer mitochondrial membrane. This process leads to the release of mitochondrial intermembrane proteins, primarily cytochrome C, into the cytosol [45, 46]. After the release of cytochrome C from the mitochondria, Apaf-1 (apoptotic protease activating factor) polymerizes, recruits, activates, and cleaves Caspase 9, forming the apoptosome [47]. The apoptosome further promotes the activation of initiator caspases, triggering apoptosome assembly. This creates a positive feedback cascade reaction that ultimately leads to cell death [48]. The IAP (inhibitor of apoptosis) family, including main members XIAP, cIAP1, and cIAP2, can regulate cellular adaptability to the environment, block the caspase cascade reaction, and inhibit cell apoptosis[49,50,51,52,53].In our study, knocking down Noxa inhibited the apoptosis induced by AZ1. These findings, combined with our research results, lead to the following conclusion: The USP28 inhibitor AZ1 upregulates Noxa and downregulates IAP, activating mitochondrial apoptosis in non-small cell lung cancer. The increased Noxa protein is a key factor in AZ1-induced apoptosis of NSCLC cells.
DNA damage is an important strategy in cancer therapy. Research has shown that USP28 deubiquitinates histones [2], stabilizes c-MYC [29], promotes the ATM-CHK2/ATR-CHK1 signaling pathway [33, 54], and plays a crucial role in the DNA damage response (DDR). ATM (ataxia-telangiectasia mutated) and ATR (ataxia-telangiectasia and Rad3 related) are responsible for coordinating the response to DNA double-strand breaks (DSBs) and replication stress, respectively. CHK2 is a critical downstream target of ATM, primarily mediating cell cycle arrest. Activated ATR and its major downstream target CHK1 mediate transient cell cycle arrest, facilitating DNA repair, stability, and reinitiation of replication. The ATM-CHK2 and ATR-CHK1 pathways can mutually activate each other [55]. BRCA1 has ATM and CHK2 phosphorylation sites on its surface, which can regulate BRCA1 function. BRCA1 is involved in the initial nucleotide excision of DNA damage, mediating the recruitment of RAD51 to single-stranded DNA (ssDNA) [56]. The RAD51 recombinase mediates the insertion of single DNA strands into homologous double strands, forming a D-loop. Within the D-loop, DNA polymerase synthesizes DNA using the homologous complementary DNA strand as a template, which is a crucial step in homologous recombination repair (HR) [21, 57]. When DNA strand breaks occur, the upregulation of c-MYC can lead to an increased generation of reactive oxygen species (ROS) and oxidative DNA damage. Additionally, c-MYC also promotes DNA repair reactions mediated by ATM and ATR, regulates the expression of RAD51 and BRCA1, and plays a dual regulatory role in DNA replication stress [58, 59]. Furthermore, research suggests that BRCA1 may constitute mediate the ATR-CHK1 signaling pathway[56]. Therefore, BRCA1 and the ATM-CHK2/ATR-CHK1 signaling pathways may form a cascade reaction. Based on these reports, we can infer that AZ1 induces DNA damage, downregulates c-MYC, inhibits the ATM-CHK2 and ATR-CHK1 signaling pathways, thereby blocking RAD51-mediated homologous recombination repair.
After breakthroughs in the clinical translation of immunoregulatory antibodies and immune checkpoint blockade therapies (especially targeting PD-1 and PD-L1), immunotherapy has gradually become an important strategy in cancer treatment [60]. Stimulator of Interferon Genes (STING) is an endoplasmic reticulum membrane protein that regulates innate immune signal transduction. Cyclic GMP-AMP synthase (cGAS) is a nucleotidyltransferase. Both dsDNA and mtDNA can activate the cGAS-STING pathway, leading to the release of chemokines by cells [61]. The chemokines further recruit and activate CD8+ T cells to eliminate tumor cells. In our study, the USP28 inhibitor AZ1 significantly induced DNA damage and activated mitochondrial apoptosis, inevitably causing the release of dsDNA and mtDNA into the cytoplasm. We observed the mRNA levels of A549 and H460 cells treated with AZ1 and siUSP28 and found that targeting USP28 upregulates the gene expression levels of signaling proteins cGAS, STING, and chemokines CCL5 and CXCL10. Therefore, enhancing the immunogenicity of NSCLC cells is one of the intrinsic mechanisms of AZ1 targeting USP28 in the treatment of non-small cell lung cancer.
In addition, Noxa-mediated cell apoptosis is an important genotoxic stress pathway [62,63,64,65]. Summarizing the research findings above, we can link USP28 inhibition and AZ1-induced events such as cell cycle alterations, apoptosis, immune activation, and DNA damage: Targeting USP28 induces DNA damage, activating Noxa-mediated mitochondrial apoptosis; DNA damage and mitochondrial apoptosis release dsDNA and mtDNA which activate tumor cell immunogenicity via the cGAS-STING signaling pathway; simultaneously targeting USP28 leads to c-MYC degradation, causing cell cycle arrest and inhibition of DNA repair, further promoting DNA damage-induced cell apoptosis mediated by Noxa protein, thereby enhancing tumor cell immunogenicity mediated by dsDNA and mtDNA.
Platinum-based drugs are commonly used DNA-damaging agents widely employed in the treatment of malignant tumors. High expression of BRCA1[66, 67] and RAD51 [21, 57] serves as important markers for platinum drug resistance. In our study, we found that AZ1 treatment and siUSP28 can inhibit the DNA damage response (DDR) and downregulate BRCA1 and RAD51, potentially enhancing the efficacy of platinum-based therapy. We conducted in vitro and in vivo experiments to assess the impact of combining AZ1 with cisplatin on non-small cell lung cancer. Compared to the monotherapy groups, the combination group demonstrated stronger tumor suppression and cytotoxic effects both in vitro and in vivo. In in vitro experiments, the two drugs showed significant synergistic effects in inhibiting the proliferation of tumor cells at low and medium concentrations. In Western blot experiments, the combination group exhibited a significant decrease in the expression of DNA repair-related proteins compared to the cisplatin monotherapy group. Therefore, the combination of USP28 inhibitors with platinum-based DNA-damaging agents represents an effective strategy for treating NSCLC and offers a novel approach to addressing platinum drug resistance issues.
Conclusion
Overall, our research provides new insights into the DDR blocking effect, cellular toxicity mechanism, and proliferation inhibition mechanism of USP28 inhibition. Targeting USP28 and combining it with DNA-damaging agents is an effective strategy for cancer treatment.
Data availability
No datasets were generated or analysed during the current study.
References
Prieto-Garcia C, Tomašković I, Shah VJ, Dikic I, Diefenbacher M (2021) USP28: oncogene or tumor suppressor? a unifying paradigm for squamous cell carcinoma. Cells 10(10):2652. https://doi.org/10.3390/cells10102652
Li F, Han H, Sun Q, Liu K, Lin N, Xu C, Zhao Z, Zhao W (2019) USP28 regulates deubiquitination of histone H2A and cell proliferation. Exp Cell Res 2019:11–18. https://doi.org/10.1016/j.yexcr.2019.03.026
Cao C, Vasilatos SN, Bhargava R, Fine JL, Oesterreich S, Davidson NE, Huang Y (2017) Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene 2017:133–145. https://doi.org/10.1038/onc.2016.186
Yang Y, Gao X, Zhang M, Yan S, Sun C, Xiao F, Huang N, Yang X, Zhao K, Zhou H, Huang S, Xie B, Zhang N (2018) Novel role of FBXW7 circular RNA in repressing glioma tumorigenesis. J Natl Cancer Inst 2018:304–315. https://doi.org/10.1093/jnci/djx166
Sun X, Cai M, Wu L, Zhen X, Chen Y, Peng J, Han S, Zhang P (2022) Ubiquitin-specific protease 28 deubiquitinates TCF7L2 to govern the action of the Wnt signaling pathway in hepatic carcinoma. Cancer Sci 2022:3463–3475. https://doi.org/10.1111/cas.15509
Zhou W, Chen J, Wang J (2023) Comprehensive prognostic and immunological analysis of ubiquitin specific Peptidase 28 in pan-cancers and identification of its role in hepatocellular carcinoma cell lines. Aging (Albany NY) 2023:6545–6576. https://doi.org/10.18632/aging.204869
Devrim T, Atac F, Devrim AK, Balci M (2020) The concomitant use of USP28 and p53 to predict the progression of urothelial carcinoma of the bladder. Pathol Res Pract 2020:152774. https://doi.org/10.1016/j.prp.2019.152774
Diefenbacher ME, Popov N, Blake SM, Schulein-Volk C, Nye E, Spencer-Dene B, Jaenicke LA, Eilers M, Behrens A (2014) The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J Clin Invest 2014:3407–3418. https://doi.org/10.1172/JCI73733
Liu Z, Chen M, Xu X, Zhang L, Pan Y, Chen D (2021) USP28 promotes aerobic glycolysis of colorectal cancer by increasing stability of FOXC1. Acta Biochim Pol. https://doi.org/10.18388/abp.2020_5504
Chen B, Sang Y, Song X, Zhang D, Wang L, Zhao W, Liang Y, Zhang N, Yang Q (2021) Exosomal miR-500a-5p derived from cancer-associated fibroblasts promotes breast cancer cell proliferation and metastasis through targeting USP28. Theranostics. https://doi.org/10.7150/thno.53412
Richter K, Paakkola T, Mennerich D, Kubaichuk K, Konzack A, Ali-Kippari H, Kozlova N, Koivunen P, Haapasaari KM, Jukkola-Vuorinen A, Teppo HR, Dimova EY, Bloigu R, Szabo Z, Kerkela R, Kietzmann T (2018) USP28 Deficiency Promotes Breast and Liver Carcinogenesis as well as Tumor Angiogenesis in a HIF-independent Manner. Mol Cancer Res. https://doi.org/10.1158/1541-7786.MCR-17-0452
Hager S, Ackermann CJ, Joerger M, Gillessen S, Omlin A (2016) Anti-tumour activity of platinum compounds in advanced prostate cancer-a systematic literature review. Ann Oncol. https://doi.org/10.1093/annonc/mdw156
Carusillo A, Mussolino C (2020) DNA damage from threat to treatment. Cells. https://doi.org/10.3390/cells9071665
Cuella-Martin R, Hayward SB, Fan X, Chen X, Huang JW, Taglialatela A, Leuzzi G, Zhao J, Rabadan R, Lu C, Shen Y, Ciccia A (2021) Functional interrogation of DNA damage response variants with base editing screens. Cell. https://doi.org/10.1016/j.cell.2021.01.041
Wang J, Dong Y, Ma H, Wu L, Zhen X, Tang L, Jin J, Han S, Zhang P, Peng J (2022) The deubiquitinase USP28 stabilizes the expression of RecQ family helicases and maintains the viability of triple negative breast cancer cells. J Biol Chem. https://doi.org/10.1016/j.jbc.2021.101443
Lambrus BG, Holland AJ (2017) A new mode of mitotic surveillance. Trends Cell Biol. https://doi.org/10.1016/j.tcb.2017.01.004
Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, Moll R, Elledge SJ, Eilers M (2007) The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol. https://doi.org/10.1038/ncb1601
Doffo J, Bamopoulos SA, Köse H, Orben F, Zang C, Pons M, den Dekker AT, Brouwer RW, Baluapuri A, Habringer S, Reichert M (2022) NOXA expression drives synthetic lethality to RUNX1 inhibition in pancreatic cancer. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.2105691119
Zhang JF (2020) MicroRNA-216b suppresses the cell growth of hepatocellular carcinoma by inhibiting Ubiquitin-specific peptidase 28 expression. Kaohsiung J Med Sci. https://doi.org/10.1002/kjm2.12193
Mazzucco AE, Smogorzewska A, Kang C, Luo J, Schlabach MR, Xu Q, Patel R, Elledge SJ (2017) Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program. Genes Dev. https://doi.org/10.1101/gad.304857.117
Feng Y, Wang D, Xiong L, Zhen G, Tan J (2021) Predictive value of RAD51 on the survival and drug responsiveness of ovarian cancer. Cancer Cell Int. https://doi.org/10.1186/s12935-021-01953-5
Coffman JA (2004) Cell cycle development. Dev Cell. https://doi.org/10.1016/s1534-5807(04)00067-x
Razavipour SF, Harikumar KB, Slingerland JM (2020) p27 as a transcriptional regulator: new roles in development and cancer. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-19-3663
Beaulieu ME, Castillo F, Soucek L (2020) structural and biophysical insights into the function of the intrinsically disordered myc oncoprotein. Cells. https://doi.org/10.3390/cells9041038
Duffy MJ, O’Grady S, Tang M, Crown J (2021) MYC as a target for cancer treatment. Cancer Treat Rev. https://doi.org/10.1016/j.ctrv.2021.102154
Zajac-Kaye M (2001) Myc oncogene: a key component in cell cycle regulation and its implication for lung cancer. Lung Cancer. https://doi.org/10.1016/s0169-5002(01)00343-9
Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, Eilers M (2001) Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. https://doi.org/10.1038/35070076
Ruiz EJ, Pinto-Fernandez A, Turnbull AP, Lan L, Charlton TM, Scott HC, Damianou A, Vere G, Riising EM, Da CC, Krajewski WW, Guerin D, Kearns JD, Ioannidis S, Katz M, McKinnon C, O’Connell J, Moncaut N, Rosewell I, Nye E, Jones N, Heride C, Gersch M, Wu M, Dinsmore CJ, Hammonds TR, Kim S, Komander D, Urbe S, Clague MJ, Kessler BM, Behrens A (2021) USP28 deletion and small-molecule inhibition destabilizes c-MYC and elicits regression of squamous cell lung carcinoma. Elife. https://doi.org/10.7554/eLife.71596
Popov N, Herold S, Llamazares M, Schulein C, Eilers M (2007) Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle. https://doi.org/10.4161/cc.6.19.4804
Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK, Oegema K (2016) 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol. https://doi.org/10.1083/jcb.201604081
Rovsing AB, Thomsen EA, Nielsen I, Skov TW, Luo Y, Dybkaer K, Mikkelsen JG (2023) Resistance to vincristine in DLBCL by disruption of p53-induced cell cycle arrest and apoptosis mediated by KIF18B and USP28. Br J Haematol. https://doi.org/10.1111/bjh.18872
Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ (2016) A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol. https://doi.org/10.1083/jcb.201604054
Zhang D, Zaugg K, Mak TW, Elledge SJ (2006) A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. https://doi.org/10.1016/j.cell.2006.06.039
V. Gerakopoulos, P. Ngo, and L. Tsiokas. 2020. Loss of polycystins suppresses deciliation via the activation of the centrosomal integrity pathway. Life Sci Alliance. https://doi.org/10.26508/lsa.202000750
Gottlieb TM, Oren M (1998) p53 and apoptosis. Semin Cancer Biol. https://doi.org/10.1006/scbi.1998.0098
Arakawa H (2005) p53, apoptosis and axon-guidance molecules. Cell Death Differ. https://doi.org/10.1038/sj.cdd.4401601
Sheikh MS, Fornace AJ (2000) Death and decoy receptors and p53-mediated apoptosis. Leukemia. https://doi.org/10.1038/sj.leu.2401865
Neubauer A, Thiede C, Huhn D, Wittig B (1996) P53 and induction of apoptosis as a target for anticancer therapy. Leukemia 1996:S2–S4
Choisy-Rossi C, Yonish-Rouach E (1998) Apoptosis and the cell cycle: the p53 connection. Cell Death Differ. https://doi.org/10.1038/sj.cdd.4400339
Yu Q (2006) Restoring p53-mediated apoptosis in cancer cells: new opportunities for cancer therapy. Drug Resist Update. https://doi.org/10.1016/j.drup.2006.03.001
Bennett MR (1999) Mechanisms of p53-induced apoptosis. Biochem Pharmacol. https://doi.org/10.1016/s0006-2952(99)00153-7
Chen L, Xu Z, Li Q, Feng Q, Zheng C, Du Y, Yuan R, Peng X (2021) USP28 facilitates pancreatic cancer progression through activation of Wnt/beta-catenin pathway via stabilising FOXM1. Cell Death Dis. https://doi.org/10.1038/s41419-021-04163-z
Zhang L, Xu B, Qiang Y, Huang H, Wang C, Li D, Qian J (2015) Overexpression of deubiquitinating enzyme USP28 promoted non-small cell lung cancer growth. J Cell Mol Med. https://doi.org/10.1111/jcmm.12426
Ren K, Li Y, Lu H, Li Z, Han X (2017) miR-3940-5p functions as a tumor suppressor in non-small cell lung cancer cells by targeting cyclin D1 and ubiquitin specific peptidase-28. Transl Oncol. https://doi.org/10.1016/j.tranon.2016.11.004
Guikema JE, Amiot M, Eldering E (2017) Exploiting the pro-apoptotic function of NOXA as a therapeutic modality in cancer. Expert Opin Ther Targets. https://doi.org/10.1080/14728222.2017.1349754
Wang J, Thomas HR, Li Z, Yeo NCF, Scott HE, Dang N, Hossain MI, Andrabi SA, Parant JM (2021) Puma, noxa, p53, and p63 differentially mediate stress pathway induced apoptosis. Cell Death Dis. https://doi.org/10.1038/s41419-021-03902-6
Ramirez MLG, Salvesen GS (2018) A primer on caspase mechanisms. Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2018.01.002
Bao Q, Shi Y (2007) Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. https://doi.org/10.1038/sj.cdd.4402028
Zadoroznyj A, Dubrez L (2022) Cytoplasmic and Nuclear Functions of cIAP1. Biomolecules. https://doi.org/10.3390/biom12020322
Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y (2001) Structural basis of caspase-7 inhibition by XIAP. Cell. https://doi.org/10.1016/s0092-8674(01)00272-0
Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature. https://doi.org/10.1038/40901
Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC (1998) A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem. https://doi.org/10.1074/jbc.273.14.7787
Zhang J, Webster JD, Dugger DL, Goncharov T, Roose-Girma M, Hung J, Kwon YC, Vucic D, Newton K, Dixit VM (2019) Ubiquitin ligases cIAP1 and cIAP2 limit cell death to prevent inflammation. Cell Rep. https://doi.org/10.1016/j.celrep.2019.04.111
Kim H, Choi H, Im JS, Park SY, Shin G, Yoo JH, Kim G, Lee JK (2021) Stable maintenance of the Mre11-Rad50-Nbs1 complex is sufficient to restore the DNA double-strand break response in cells lacking RecQL4 helicase activity. J Biol Chem. https://doi.org/10.1016/j.jbc.2021.101148
Weber AM, Ryan AJ (2015) ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. https://doi.org/10.1016/j.pharmthera.2014.12.001
Roy R, Chun J, Powell SN (2011) BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. https://doi.org/10.1038/nrc3181
Lee JO, Kang MJ, Byun WS, Kim SA, Seo IH, Han JA, Moon JW, Kim JH, Kim SJ, Lee EJ, In PS, Park SH, Kim HS (2019) Metformin overcomes resistance to cisplatin in triple-negative breast cancer (TNBC) cells by targeting RAD51. Breast Cancer. https://doi.org/10.1186/s13058-019-1204-2
Shortt J, Martin BP, Newbold A, Hannan KM, Devlin JR, Baker AJ, Ralli R, Cullinane C, Schmitt CA, Reimann M, Hall MN, Wall M, Hannan RD, Pearson RB, McArthur GA, Johnstone RW (2013) Combined inhibition of PI3K-related DNA damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood. https://doi.org/10.1182/blood-2012-08-446096
Al-Ejeh F, Kumar R, Wiegmans A, Lakhani SR, Brown MP, Khanna KK (2010) Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene. https://doi.org/10.1038/onc.2010.407
Cheng M, Shao Y, Li L, Jiang M, Song Z (2024) Cost-effectiveness of immunotherapies for advanced squamous non-small cell lung cancer: a systematic review. BMC Cancer. https://doi.org/10.1186/s12885-024-12043-w
Wang X, Wang Y, Zhang Y, Shi H, Liu K, Wang F, Wang Y, Chen H, Shi Y, Wang R (2024) Immune modulatory roles of radioimmunotherapy: biological principles and clinical prospects. Front Immunol. https://doi.org/10.3389/fimmu.2024.1357101
Xu T, Ma Q, Li Y, Yu Q, Pan P, Zheng Y, Li Z, Xiong X, Hou T, Yu B, Liu H, Sun Y (2022) A small molecule inhibitor of the UBE2F-CRL5 axis induces apoptosis and radiosensitization in lung cancer. Signal Transduct Target Ther. https://doi.org/10.1038/s41392-022-01182-w
Albert MC, Brinkmann K, Pokrzywa W, Gunther SD, Kronke M, Hoppe T, Kashkar H (2020) CHIP ubiquitylates NOXA and induces its lysosomal degradation in response to DNA damage. Cell Death Dis. https://doi.org/10.1038/s41419-020-02923-x
Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, Dewson G, Michalak EM, Vandenberg CJ, Smyth GK, Strasser A, Cory S, Scott CL (2010) Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood. https://doi.org/10.1182/blood-2010-04-280818
Martin AG, Trama J, Crighton D, Ryan KM, Fearnhead HO (2009) Activation of p73 and induction of Noxa by DNA damage requires NF-kappa B. Aging (Albany NY). https://doi.org/10.18632/aging.100026
Pettitt SJ, Frankum JR, Punta M, Lise S, Alexander J, Chen Y, Yap TA, Haider S, Tutt ANJ, Lord CJ (2020) Clinical BRCA1/2 reversion analysis identifies hotspot mutations and predicted neoantigens associated with therapy resistance. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-19-1485
Zhu Y, Liu Y, Zhang C, Chu J, Wu Y, Li Y, Liu J, Li Q, Li S, Shi Q, Jin L, Zhao J, Yin D, Efroni S, Su F, Yao H, Song E, Liu Q (2018) Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat Commun. https://doi.org/10.1038/s41467-018-03951-0
Acknowledgements
We thank the Animal Experiment Centre of Zhengzhou University for providing the feeding and experimental platforms. This work was supported by the Collaborative Innovation Major Project of Zhengzhou (Grant No. 20XTZX08017)
Funding
This work was supported by the Collaborative Innovation Major Project of Zhengzhou (Grant No. 20XTZX08017). Funding for Scientific Research and Innovation Team of The First Affiliated Hospital of Zhengzhou University (ZYCXTD2023005). Wu Jieping Medical Foundation Special Fund for Targeted Cancer Research (No 320.6750.2023-02-1).
Author information
Authors and Affiliations
Contributions
Y. S. and L.W. contributions to conception and design and acquisition of data. Y. S., L. W., Y. Z., L. J., C. L., K. C. and L. L. are involved in data analysis and interpretation. Y. S., L. W., S. S., Y. W., Y. G., Y. Y., L. Z., Y. Z. involved in drafting the manuscript Y. S. and L. W. involved in revising it critically for important intellectual content. J. Z. Given final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
Under the pathogen-free environment, all animals were cared for and anesthetized before experimentation in accordance with the Guide for the Care and Use of Laboratory Animals. Animal experiments in this study have been approved by the Animal Welfare Review Committee of the First Afliated Hospital of Zhengzhou University. All methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org) for the reporting of animal experiments.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, Y., Wang, L., Zheng, Y. et al. Deubiquitinating enzyme USP28 inhibitor AZ1 alone and in combination with cisplatin for the treatment of non-small cell lung cancer. Apoptosis 29, 1793–1809 (2024). https://doi.org/10.1007/s10495-024-02008-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-024-02008-6