
Abstract
Metal ions play an important role in living organisms and are involved in essential physiological activities. However, the overload state of ions can cause excess free radicals, cell damage, and even cell death. Ferroptosis and cuproptosis are specific forms of cell death that are distinct from apoptosis, necroptosis, and other regulated cell death. These unique modalities of cell death, dependent on iron and copper, are regulated by multiple cellular metabolic pathways, including steady-state metal redox treatment mitochondrial activity of lipid, amino acid and glucose metabolism, and various signaling pathways associated with disease. Although the mechanisms of ferroptosis and cuproptosis are not yet fully understood, there is no doubt that ion overload plays a crucial act in these metal-dependent cell deaths. In this review, we discussed the core roles of ion overload in ferroptosis and cuproptosis, the association between metabolism imbalance and ferroptosis and cuproptosis, the extract the diseases caused by ion overload and current treatment modalities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Facts
-
Ferroptosis and cuproptosis are distinct forms of regulated programmed cell death that are characterized by their dependence on excessive accumulation of iron and copper ions, respectively.
-
The oxidation state of metal ions plays a crucial role in the mechanisms of ferroptosis and cuproptosis. Specifically, Fe2+ promotes ferroptosis, whereas Cu+ is a more potent form than Cu2+ in promoting cuproptosis.
-
Overload of iron ions can induce ferroptosis not only by triggering the Fenton reaction and causing direct peroxidation of PUFA-PIs, but also by serving as a necessary cofactor for enzymes involved in lipid peroxidation.
-
Intracellular copper accumulation results in the aggregation of mitochondria lipoylated proteins and destabilization of Fe-S cluster proteins, thereby triggering cuproptosis.
-
Both ferroptosis and cuproptosis affect mitochondria and exhibit crosstalk, owing to the close relationship between copper and iron metabolism.
Open questions
-
What are the precise morphological and biochemical changes associated with cuproptosis?
-
What is the exact quantitative relationship between ion overload and cell death? What is the threshold of ion overload that induces cell death?
-
How does iron metabolism regulate cuproptosis? How dose copper metabolism regulate ferroptosis? What are the specific interactions between ferroptosis and cuproptosis?
-
Dose the generation of reactive oxygen species (ROS) from copper-based Fenton reactions play a role in triggering ferroptosis?
-
Can other trace metal ions induce cell death through mechanisms distinct from the existent forms of cell death?
Introduction
Transition metal ions, including iron (Fe), copper (Cu), zinc (Zn), and manganese (Mn), are vital for life and are involved in many essential biochemical processes in living organisms, ranging from bacteria to human beings [1, 2]. These processes include carbon transformation, nucleic acid and protein synthesis, deoxyribonucleic acid (DNA) replication, the tricarboxylic acid (TCA) cycle, electron transport in mitochondria, glycolysis, and the metabolism of reactive oxidative species (ROS) [2, 3].
Maintaining metal ion homeostasis is crucial to achieve a proper balance of these ions in different cellular compartments. External or intrinsic alterations to metal ion metabolism can lead to ion deficiency or ion overload [1, 4]. Ion deficiency can limit ion availability for metalloenzyme synthesis and other biochemical pathways in multiple tissues, ultimately leading to reduced work capacity, developmental retardation, and various diseases such as hemochromatosis, cardiovascular disease [5, 6], iron deficiency anemia [7], Parkinson’s disease, and Friedreich’s ataxia and Pica [8,9,10]. Conversely, an excess of redox-active essential metals such as Fe and Cu can induce free radicals under certain conditions, causing inflammation, cell damage and cancerous changes, eventually leading to cardiovascular diseases, neurodegenerative diseases and metal ion overload disorders [11,12,13,14]. Each metal can react in a unique way and has different mechanisms of action. In parallel processes, excessive metal ions can induce oxidative stress, damage the antioxidant defense system, alter the redox balance, affect intracellular signaling pathways, and ultimately lead to cell damage or death.
Ferroptosis, driven by iron-dependent phospholipid peroxidation, has been recognized associated tightly with iron overload [15]. In addition to iron-dependent cell death, a recent study found that copper accumulation triggers the aggregation of mitochondrial lipoylated proteins and the destabilization of iron-sulfur (Fe-S) cluster proteins, leading to a particular type of cell death termed cuproptosis [16]. These metal-dependent cell death are distinctly different from apoptosis, necroptosis, and other regulated cell death (RCD) (Table 1). In this review, we discuss the central roles of iron and copper ions overload on metal-dependent cell death, summarize the regulation by multiple cellular metabolism pathways, present the interrelationship between these two forms of metal-dependent cell death and extract the diseases caused by ion overload.
Metal ions are deeply involved in cell metabolism
Metal ions play a vital role in cell metabolism, as they participate in various ionic states throughout processes such as absorption, distribution, storage, and elimination, at both the cellular and systemic levels in multicellular organisms.
Iron plays a crucial role in the formation and metabolism of ROS due to its ability to carry out one-electron reactions, determined by the electronic structure of the iron atom [2]. Iron is commonly found in ferric-reduced and ferrous-oxidized states, which it oscillates between under physiological conditions, and to a lesser extent in the highly oxidizing ferryl (Fe4+) form [2, 4]. Ferric iron (Fe3+) is bound to proteins, such as transferrin, to ensure good bioavailability and overcome solubility issues, as it is stable but poorly soluble in water [17]. This allows Fe3+ to be transported steadily in a redox-inactive state. In contrast, ferrous iron (Fe2+) is high reactivity and water-soluble and can generates ROS through the Fenton reaction [17]. Iron is essential for various type of biological processes in almost all forms of life, including being incorporated into multiple proteins as organic cofactors, such as iron-sulphur clusters (ISC) in matrix proteins like biotin synthase, aconitase, homoaconitase and ferredoxin [18]. Iron-containing proteins also play important roles in the catalytic centers of many enzymes, such as ribonucleotide reductase and DNA helicase during DNA replication, cytochrome oxidases in electron transport, and the TCA cycle for oxidative phosphorylation (OXPHOS) and energy production [19,20,21,22]. Iron levels are maintained within a range of 3–4 g by precise control of its absorption, mobilization, storage, and recycling in healthy individuals [23]. Given the importance of iron in fundamental physiological processes, sufficient iron is necessary for cell metabolism and development.
Copper is a crucial trace metal micronutrient that plays an essential role in various cell metabolism processes in mammals. Despite its lower abundance compared to other metals like iron and zinc, copper is widely used as a structural or catalytic cofactor for a diverse range of enzymes in mammals, aiding in cellular respiration, energy metabolism, iron homeostasis, ROS detoxification, and signaling in eukaryotic organisms [24]. Copper has the ability to switch between cuprous ion (Cu+) and cupric (Cu2+) oxidation states, conferring to cuproenzymes the ability to catalyze redox reactions [25]. With the trivalent form + 3 (Cu3+) exists, it is of relatively little biological significance [2]. Copper is a prosthetic group of complex IV (CIV) cytochrome c oxidase, one of the four components of the electron transport chain (ETC) in the process of TCA [25,26,27]. The ETC is a vital part of the inner mitochondrial membrane (IMM) that generates the proton motive force (PMF), empowering mitochondria to regulate cell metabolism and fate [28]. The proper assembly and function of ETC is dependent on copper. Additionally, copper directly aids in relieving ETC-generated ROS as a cofactor of the copper/zinc superoxide dismutase (SOD1), a protein located in both the cytosol and mitochondrial inner membrane space.
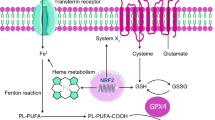
Although metal ions are necessary for cellular viability and growth, excessive accumulation of these ions can lead to poisoning and cell death. Various types of cell death can occur in response to different stresses, particularly oxidative stress. RCD involves tightly regulated signaling pathways and specific effector mechanisms, centered around cell death executioner proteins [29]. In contrast, metal-dependent cell death, including ferroptosis and cuproptosis, differs from other forms of RCD. When present in excess, ferrous ions and cuprous ions can be harmful due to their multiple roles in metabolism, resulting in cell damage and cell death. These effects are particularly pronounced in the liver, heart, pancreas, thyroid, and central nervous system. The major regulatory pathways of ferroptosis and cuproptosis are showed in the Fig. 1.

Major Regulatory Pathways in Ferroptosis and Cuproptosis. The regulatory pathways involved in ferroptosis can be roughly divided into three categories. The first one includes the GSH/GPX4 pathway, which encompasses the inhibition of system Xc¯ and glutamine pathway. The second one involves the regulation of iron metabolism, including the modulation of iron-regulatory proteins (IRPs) ACO1 and IREB2 related to ferritin metabolism and the regulatory pathways of FINO2 that impact iron levels. Excessive iron can induce ferroptosis by promoting reactive oxygen species (ROS) production through the Fenton reaction. The third category includes pathways related to lipid metabolism, such as acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), arachidonate lipoxygenases (ALOXs), etc., which effect lipid regulation and ferroptosis. Excess copper accumulation causes cuproptosis mainly through FDX1-mediated mitochondrial proteotoxic stress. On one hand, FDX1 reduces Cu2+ to Cu+, facilitating the lipoylation (LA) and aggregation of enzymes, especially dihydrolipoamide S-acetyltransferase (DLAT) involved in the regulation of mitochondrial TCA cycle. On the other hand, FDX1 causes the destabilization of Fe–S cluster proteins. Cu importers (SLC31A1) and exporters (ATP7A/B) regulate cuproptosis sensitivity by affecting intracellular Cu+ levels. GSH acts as a thiol-containing copper chelator that can block cuproptosis
Ferrous overload plays a key role in ferroptosis induction
Ferroptosis
Ferroptosis, a form of RCD triggered by iron-dependent lipid peroxidation on cellular membranes, was first identified and named as a distinct phenomenon by Dixon et al. [30] a decade ago. Morphologically and mechanistically, ferroptosis differs from apoptosis, necroptosis, and other types of cell death as outlined in Table 1 [30]. Unlike apoptosis, cells undergoing ferroptosis do not exhibit chromatin condensation and apoptotic body formation, but rather shrunken mitochondrial cristae [31, 32]. The execution of ferroptosis depends on three essential conditions, which are the main prerequisites for driving ferroptosis: (i) synthesis of polyunsaturated fatty acid (PUFA)-containing phospholipids (PLs) (PUFA-PLs) and iron-catalysed peroxidation which is the crux of ferroptosis execution; (ii) regulation of iron metabolism, which initiates the Fenton reaction and acts as an essential cofactor for enzymes that participate in lipid peroxidation; and (iii) modulation of mitochondrial metabolism, which drives ferroptosis through the multidimensional functions of mitochondria in bioenergetics, biosynthesis and ROS generation [15] (Fig. 1). This iron-dependent phospholipid peroxidation is regulated by various cellular metabolic ways, including reductive/oxidative (redox) homeostasis, iron metabolism, mitochondrial activity, metabolism of amino acid and lipid, and glycometabolism [33]. Among these pathways, iron metabolism plays a crucial role in the regulatory network of ferroptosis.
Iron and its metabolism
Iron is primarily obtained through dietary absorption and recycling in the body [30]. There are two forms of iron that can be absorbed from food: non-heme iron, which mainly exists as insoluble Fe3+ and needs to be reduced to Fe2+ before absorption and heme iron, which is mostly Fe2+ and can be directly absorbed by epithelial cells in the intestinal mucosal [34, 35]. The majority of iron is recycled from phagocytosis of senescent red blood cells, primarily by macrophages in the spleen, bone marrow, and liver [23]. The remaining iron is stored in hepatocytes, contributing to the regulation of systemic iron levels [17]. Iron absorption in duodenal enterocytes is another regulating mechanism for controlling iron levels. The water-soluble Fe2+ can be transported to several tissues by binding to transferrin (Tf), after it is transferred from enterocytes into the bloodstream by ferroprotein (FPN) [36]. Transferrin is a plasma protein that binds to iron and delivers it into cells via its receptor 1 (CD71), which is required for iron delivery in most cells and serves as a gatekeeper for regulating iron uptake [37]. Once inside the cell, iron can be utilized for various functions depending on the cellular and systemic conditions. After being released from Tf, iron is transferred to the cytosol by ferrous ion membrane transport protein DMT1 to join the labile iron pool (LIP) [38, 39]. Iron from the LIP can be transferred to mitochondria via DMT1, mitoferrin and siderofexin (SFXN1) [40]. Excess iron from the LIP is stored in ferritin, which can be transferred to and degraded in lysosomes, replenishes the LIP inturn [40]. Hepcidin, a 25 amino acid peptide, is the primary regulatory molecule of systemic iron homeostasis [41]. It binds to ferroportin, triggering the internalization, ubiquitination, and subsequent lysosomal degradation of the iron exporter, thereby reducing available circulating iron [42]. A relatively stable pool of labile iron is normally maintained through the organized regulation of iron absorption, storage, utilization, and export in cells [43]. It is evident that iron overload, which results from imbalanced regulation of iron metabolism, can trigger ferroptosis [15].
Iron ion overload triggers lipid peroxidation
Although the precise mechanisms by which iron contributes to ferroptosis are not yet fully understood, it is evident that iron overload is a crucial factor in this process. Recent studies have demonstrated that excessive iron ions can induce ferroptosis not only by triggering the Fenton reaction and causing direct peroxidation of PUFA-PLs [44], but also by serving as a necessary cofactor for enzymes involved in lipid peroxidation [45].
The non-enzymatic, iron-dependent Fenton reaction is a crucial step in the process of ferroptosis [13, 46]. Henry John Horstman proposed the reaction in which iron salts react with peroxides to produce hydroxyl radicals (Fe2+ + HOOH → Fe3+ + OH– + OH•), which now bears his name [47]. This reaction utilizes both iron and oxygen to catalyze a chain reaction, resulting in the propagation of phospholipid peroxidation, which is the hallmark of ferroptosis, leading to the formation of phospholipid hydroperoxides (PLOOHs) [33].
It should be noted that during the initiation phase of this iron-catalyzed chain reaction Fe2+ is likely to be more dominant, due to the poor solubility and limited bioavailability of Fe3+ in cells [33]. Fe2+ acts as a cofactor in the catalytic centers of many important enzymes that are causes for ferroptosis, several of these iron-containing enzymes can promote the lipid peroxidation that drives ferroptosis. For instance, 15-lipoxygenase can complex with phosphatidylethanolamine (PE) binding protein 1 (PEBP1), switching the substrate specificity of the enzyme from free PUFAs to PUFA tails of PLs [48]. Additionally, 12-lipoxygenase is required for p53-dependent ferroptosis [49]. While cytochrome P450 oxidoreductase (POR) and the NADPH oxidases (NOXs) also contribute to ROS production for lipid peroxidation during ferroptosis [50].
FINO2, the fourth class of ferroptosis-inducing compounds, was discovered in 2016 [51]. FINO2 represents an additional means of inducing ferroptosis, as it does not inhibit system Xc¯, deplete glutathione (GSH), directly inhibit glutathione peroxidase 4 (GPX4), or induce GPX4 degradation [15]. One possible mechanism by which FINO2 induces ferroptosis is through the oxidation of Fe2+ to Fe3+ via Fenton reaction, which generates alkoxyl radicals that initiate lipid peroxidation [15]. Another possible mechanism is that FINO2 binds to and activates lipoxygenases or other iron-dependent enzymes by oxidizing the non-heme iron cofactor; the active form of lipoxygenases is the ferric iron form [52].
Additional mechanisms that regulate cellular iron levels can affect the sensitivity of cells to ferroptosis: exporting iron through ferroprotein or ferritin-containing multivesicular bodies (MVBs) mediated by prominin-2 (prom2), as well as exosomes, can confer resistance to ferroptosis by reducing the intracellular pool of iron and its capacity to promote lipid peroxidation [53]. Ferrous iron is maintained in cells as the labile iron pool, which is bound to low molecular weight compounds, including GSH. The depletion of GSH can not only inactivate GPX4 but also mobilize Fe2+ for Fenton chemistry, promoting lipid peroxides propagations and ultimately leading to ferroptosis. In addition, iron storage in ferritin requires the formation of the GSH-iron complex, which is delivered to ferritin via the chaperone poly(rC) binding protein 1 (PCBP1) [54]. To sum up, many genes/proteins are involved in iron metabolic pathways which are involved in the execution of ferroptosis. A more extensive list of the main drugs and compounds implicated in ferroptosis is reported in Table 2.
Copper ion overload involved in cuproptosis
Cuproptosis
Cuproptosis is a distinct form of copper-induced regulated cell death, which was recently reported by Tsvetkov et al. in 2022 [16]. According to their findings, intracellular copper accumulation results in the aggregation of mitochondria lipoylated proteins and destabilization of Fe-S cluster proteins, thereby triggering cuproptosis. This novel mode of Cu-dependent, mitochondrially induced cell death differs from other oxidative stress-induced cell death, such as apoptosis, ferroptosis and necroptosis (Table 1) [16, 55,56,57]. Unlike these pathways, cuproptosis is triggered by mitochondrial stress, particularly the aggregation of lipoylated mitochondrial enzymes and the loss of Fe-S cluster proteins. Although the detailed mechanism of cuproptosis remains unclear, three critical processes have been identified to play a crucial role in its initiation. These include direct copper binding to lipoylated TCA cycle proteins, lipoylated DLAT oligomerization, and Fe-S cluster proteins destabilization with the ETC complex [55]. Furthermore, seven key genes that promote cuproptosis, with ferrodoxin-1 (FDX1) being the most significant have identified by using genome-wide CRISPSR-Cas 9 loss-of-function screens. FDX1 acts as the primary regulator of copper ionophore-induced cell death, which can reduce Cu2+ to its more toxic form Cu+. The remaining six genes encode either elements in the lipoic acid pathway (LIPT1, LIAS and DLD) or targets of lipoylation (DLAT, PDHA1 and PDHB).
Copper and its metabolism
Copper is an essential trace element as a cofactor for a diverse array of enzymes, critical for normal physiological function across various organisms, ranging from bacteria to human cells [11, 25, 58]. Given that copper cannot be synthesized or degraded through metabolic pathways, it must be acquired from external sources [59]. In the body, copper is predominantly stored in the liver, which serves as the main site for copper accumulation and distribution via the bloodstream or biliary excretion. The cellular of copper uptake in mammalian cells is predominantly mediated by CTR1 (SLC31A2), a copper transport protein that exhibits high affinity for the plasma membrane [60]. Although DMT1 (SLC11A2), a divalent metal transporter, has been implicated in copper uptake in certain cultured cell lines, its role in dietary copper absorption is not crucial, as demonstrated by the enterocyte-specific deletion of DMT1, which has no effect on intestinal copper absorption [61]. Intracellular copper is transported to the antioxidant copper chaperone 1 (ATOX1) and CuL (a low molecular weight copper ligand) before being transported into mitochondria, specifically to copper chaperone of cytochrome C oxidase (COX17) and copper chaperone of SOD1 (CCS1) [62]. Despite being an essential cofactor for all organisms, prolonged or chronic exposure to copper can lead to toxicity.
Copper overload and cuproptosis
Excessive copper ions can participate in the TCA cycle under the influence of FDX1, inducing DLAT aggregation and aberrant mitochondrial protein folding (Fig. 1). Subsequently, the loss of Fe-S protein leads to cell death due to energy metabolism defects [55]. Additionally, copper ion overload can result in redox imbalance, where the antioxidant defense of GSH and SOD is insufficient to counteract the damaging effects of ROS [63].
Cu2+ is reduced to Cu+ after enters into the mitochondria and then accompanied by the generation of ROS. Unlike ferroptosis, ROS generation is not essential for cuproptosis. Instead, Cu+ directly binds to lipoylated proteins and induces the oligomerization of lipoylated DLAT [16]. Tsvetkov et al. [16] discovered that Cu+ binding to lipoylated TCA cycle proteins does not simply lead to loss of function but can also result in toxic gain of function. They also found that elesclomol-mediated toxicity in cancer cells was linked to FDX1 levels, elevated mitochondrial respiration rate, and was dependent on Cu availability. Copper overload can also lead to the loss of Fe-S cluster proteins in an FDX1-dependent manner [16]. Fe-S cluster proteins play an important role in life activities, including electron chain transmission, genome stability maintenance, and gene expression regulation [64]. The hydrophilic antioxidant GSH can block the toxicity of Cu+ by chelating intracellular Cu, and depletion of glutathione can result in copper-dependent cell death [16].
Excess Cu+ can selectively disrupt a set of metabolic enzymes involved in protein lipoylation, which involves attaching the small sulfur-containing metabolite lipoic acid to a substrate protein, enabling catalysis by swinging between different subunits within an enzyme complex, and donating or accepting electrons [56]. Lipoylation is unique to TCA cycle enzymes, occurring in only 4 multimeric metabolic enzymes, all of which are found in the mitochondria, including dihydrolipoamide S-acetyltransferase (DLAT), a subunit of the pyruvate dehydrogenase complex [65]. Copper toxicity can be suppressed by genetically disrupting enzymes required for the synthesis of lipoic acid or of individual lipoylated enzymes themselves [56].
Metabolism is the core regulator in ferroptosis and cuproptosis
Metabolism pathways play a crucial role in regulating ferroptosis and cuproptosis, despite the distinct mechanisms underlying these two types of metal-dependent cell death. Proteins, lipids, and energy production as well as mitochondrial metabolism, all contribute to the regulation of these processes.
GSH suppresses ferroptosis and cuproptosis through different ways
Abnormal protein metabolism is crucial in the regulation of ferroptosis and cuproptosis, both of which are influenced by GSH. GSH is a thiol-containing tripeptide synthesized from glycine, glutamate, and cysteine, with cysteine availability being the main limiting factor [66, 67]. GSH exists in two states: reduced (GSH) and oxidized (GSSG) [35]. Reduced GSH is used by GPX4 to reduce reactive PUFA phospholipid hydroperoxides (PUFA-PL-OOH) to non-reactive and non-lethal PUFA phospholipid alcohols (PUFA-PL-OH), which ultimately suppresses ferroptosis. In addition, GSH can block cuproptosis by chelating intracellular Cu [55].
The GSH/GPX4 antioxidation system plays an important role in protecting cells from ferroptosis, which is regulated by the cystine/glutamate transporter (also known as system Xc¯) upstream. The system Xc¯ imports cystine into cells with a 1:1 counter-transport of glutamate. Once in cells, cystine can be oxidized to cysteine, which is used to synthesize GSH in a reaction catalyzed by glutamate–cysteine ligase (GCL) and glutathione synthetase (GSS). GSH, which acts as a reducing cofactor, using by GPX4, can reduce lipid hydroperoxides to lipid alcohols [68]. Depleting GSH can sensitize cells to ferroptosis. For example, the multidrug resistance gene MDR1 drives increased sensitivity to ferroptosis by causing efflux of GSH [69]. The cysteine catabolic enzyme cysteine dioxygenase 1 (CDO1) promotes ferroptosis by depleting cysteine and, in turn, GSH [70]. Synthesizing GSH can contribute to ferroptosis resistance, as shown in a recent report [71]. Glutamate-cysteine ligase, which synthesizes GSH, restrains ferroptosis not only by synthesizing GSH but also by limiting levels of glutamate through its conversion to gama-glutamyl peptides. Thus, the increased levels of glutamate can promote ferroptosis [71]. The role of GSH in suppressing cuproptosis differs from its role in suppressing ferroptosis. GSH chelates intracellular Cu to block cuproptosis. According to the study of Tsvetkov et al. [16], depletion of GSH by buthionine sulfoximine (BSO), a potent inhibitor of the enzyme gamma-glutamyl-cysteine synthetase, increased susceptibility to cuproptosis in A549 lung cancer cells by suppressing lipoylation and promoting DLAT oligomerization [72].
Destabilization of iron-sulfur cluster proteins promotes cell death
Fe-S clusters are widely distributed and constituted the largest class of metalloproteins across diverse biological systems. These clusters exist in various structures, which the most common being [2Fe-2 S], [3Fe–4 S] or [4Fe–4 S] [73]. Fe-S clusters have a broad range of biological role, including electron transfer within the mitochondrial respiratory chain, the generation of mitochondrial ROS, and DNA metabolism [73,74,75,76,77]. Additionally, Fe-S clusters also play a regulatory role in gene expression in response to oxidative stress, oxygen levels, and iron levels [78,79,80].
As a critical cofactor in redox maintenance and iron homeostasis, Fe-S clusters and their associated regulatory pathways contribute to ferroptosis evasion in cells by reducing the LIP. The activation of iron-responsive element (IRE)-binding proteins 1 and 2 (IRP1 and IRP2), which are the main regulators of cellular iron pool, occurs when cellular iron levels are low, leading to a rheostat-like iron-starvation response [81]. This response regulates iron uptake, storage, export and usage to increase the available iron pool. IRP1 is modulated by the occupancy of Fe-S clusters and can function as an iron-responsive protein when it loses its Fe-S clusters [82]. Disturbances in Fe-S clusters metabolism are thought to abnormally activate the iron-starvation response via IRP1, overloading cellular iron pools and triggering ferroptosis [46, 83, 84]. Several mitochondrial proteins including cysteine desulfurase (NSF1) [84, 85], iron-sulfur cluster assembly (ISCU) [86], CISD1 [87] and CISD2 [88] inhibit ferroptosis by increasing the biosynthesis of Fe-S. In contrast, frataxin, another protein involved in Fe-S clusters assembly, is upregulated in different cancer types and promotes cancer cell resistance to ferroptosis [89].
Excess copper ions inhibited the synthesis of Fe-S clusters under the regulation of FDX1, leading to a reduction in Fe-S cluster proteins [16]. Knockdown of FDX1 causes the loss of protein-lipid acylation, decreased mitochondrial respiration, accumulation of pyruvate and 𝛼-ketoglutarate, and loss of iron-sulfur cluster proteins [16]. However, the relationship between the reduction of Fe-S cluster proteins and cuproptosis remains to be explored. Additionally, protein aggregates could trigger cell death by disrupting the function of mitochondrial enzymes involved in the synthesis of iron-sulfur clusters, which are essential cofactors for other enzymes [64].
Lipid peroxidation, a hallmark of ferroptosis
Lipid peroxidation is a key feature of ferroptosis and affects PUFAs. Initially, it was believed that free PUFAs were the driving force behind ferroptosis. However, several studies have shown that free fatty acids are not the direct drivers of ferroptosis. Instead they must be esterified into membrane phospholipids and oxidized to transmit the ferroptosis signal. Specifically, PE containing arachidonic acid (AA) or its derivative adrenaline has been identified as the key phospholipid that induces ferroptosis. Acyl-coenzyme A (Acyl-CoA) synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are involved in the biosynthesis and remodeling of PE, activating PUFAs and influencing the transmembrane characteristics of PUFAs [90, 91]. Therefore, reducing the expression of ACSL4 and LPCAT3 can decrease the accumulation of lipid peroxide substrates in cells, leading to the inhibition of ferroptosis. Finally, PUFA-PE can undergo further oxidative reactions under the catalysis of lipoxygenases (ALOXs) and ultimately induce ferroptosis [45].
Glucose metabolism governs cell death through the energy regulation
The role of glucose metabolism in regulating ferroptosis and cuproptosis has not been extensively studied, unlike lipid and amino metabolism. Iron is essential in glucose metabolism, and both iron deficiency and overload can affect glucose metabolism. Iron overload is associated with decreased insulin sensitivity and promoted insulin resistance due to reduced glucose uptake, likely caused by increased ROS production and impaired autophagy [92].
Glucose is the primary fuel of the mitochondrial TCA cycle, and it has been shown to regulate ferroptosis [33]. Studies by Lee et al. [93] and Li et al. [94] have suggested that glucose starvation may suppresses ferroptosis due to the activation of AMP-activated kinase (AMPK) signaling, rather than modulation of TCA and mitochondrial respiration. AMPK activation leads to impaired biosynthesis of PUFAs, which are essential for ferroptosis, resulting in a protective effect against cell death [33, 45, 95]. Glucose starvation, however, can also increase ROS production, implying that it promotes ferroptosis [96]. Although the regulatory role of glucose metabolism in cuproptosis is not clear, cells that rely on mitochondrial respiration are more sensitive to copper ionophores, suggesting that cuproptosis is regulated by mitochondria respiration rather than glycolysis [16]. Further studies are needed to explore the details of how aerobic OXPHOS of glucose regulates cuproptosis.
Pathological contexts of kidney Diseases related to ferroptosis and cuproptosis
Metal ions are micronutrients that play critical roles in cellular functions and metabolic processes in the human body. However, excessive accumulation of metal ions can result in the generation of ROS, leading to cell death, tissue damage, and the development of various kidney diseases, including acute kidney injury, fibrosis, renal cell cancers and ischemia reperfusion injury of kidney transplantation.
Acute kidney injury
Acute kidney injury (AKI) is a common and serious clinical renal syndrome, which is caused by a variety of pathogenic factors and is an urgent disease to be solved. Recently, evidence has showed that ferroptosis plays an important role in AKI.
Studies have found many mechanisms by which iron death affects acute renal injury. ACSL4 is a promotor for ferroptosis and is negatively regulated by HIF-1a [97] but activated by HMGB1 [98]. When it comes to the ischemia reperfusion injury, HIF-1α downregulated and HMGB1 released from epithelial cells which leads to higher ACSL4 expression. Besides, ACSL4 is correlated with the infiltration of macrophages and that neutrophils are recruited by ferroptotic-cell induced macrophages. Lack of FSP1 or targeted manipulate the center of the selenoprotein glutathione peroxidase 4 (GPX4cys/-) makes it sensitive for tubular to ferroptosis and causes a special tubular necrosis [99]. Tripartite motif containing 21 (TRIM21) highly expresses in kidney with ischemia reperfusion injury (IRI) and affects ferroptosis by inducing ubiquitination degradation of GPX4 [100]. Polymyxin B (PMB) is widely used in multi-resistant gram-negative infections while its side-effects involve acute kidney injury. Recent study identified that PMB causes AKI by promoting ferroptosis with RNA-sequencing and further explored the mechanism that PMB decreases the expression of SLC7A11 and increase TFR1 through the activation of P53 [101]. On the other hand, some targets are capable of reversing ferroptosis. Activated Farnesoid X receptor (FXR) prevents against AKI by modulating transcription of ferroptosis-associated genes, like AIMD2, GGT6 and GSTA4 [102]. BNIP3 and PINK1-PINK2 related-mitophagy protect kidney from cisplatin-induced acute injury through ROS/HO1/GPX4 pathway [103].
In view of the prominent role of iron death in AKI, more and more studies have begun to try and develop drugs to inhibit ferroptosis to alleviate AKI. Quercetin decreases malondialdehyde and ROS and increases the level of GSH which turn to the blockage of morphologic changes of ferroptotic cells [104]. Vitamin K1 has been identified as an exciting inhibitor for ferroptosis and is prepared for AKI treatment [105]. Irisin alleviates AKI through activation of SIRT1/NRF2 pathway [106]. Leonurine protects kidney from cisplatin-induced injury by activating Nrf2/NRF2 pathway [107]. Paeoniflorin (PF) is a traditional Chinese medicine that protects kidney from AKI. Ma et al. identified in IRI model that PF prevents kidney from ferroptosis-induced injury by upregulating SLC7A11 [108].
Kidney fibrosis
Ferroptosis promotes interstitial fibrosis and inflammation in chronic kidney diseases or UUO or I/R mouse models. Ferroptosis inhibition ameliorated kidney fibrosis by reducing MCP-1 excretion and chemotaxis of macrophages [109]. Combination of Melatonin and Zileuton synergistically protect kidney from fibrosis in UUO model by upregulating AKT/mTOR/NRF2 signaling pathway which inhibits ferroptosis [110]. Formononetin inhibits Smad3/ATF3/SLC7A11 pathway, increases the expression of SLC7A11 and GPX4 and promotes Nrf2 nuclear accumulation, resulting in the amelioration of kidney fibrosis and ferroptosis [111].
IRI after kidney transplantation
IRI largely affect function recovery after kidney transplantation and studies have found that ferroptosis leads to delayed recovery of renal allograft. It is found that miR-20a-5p could reduce the kidney loss and structural damage by inhibiting ACSL4-dependent ferroptosis [112]. Tang et al. screened ferroptosis-related hub genes (IL-6, ATF3 and JUN) of IRI post-renal transplantation. They could be diagnostic biomarkers and target for preventing dysfunction of transplanted kidney [113].
Renal cancers
In recent years, it has been found that ferroptosis and other ion death are inhibited in the occurrence and progression of renal tumors. Exploring the mechanism of tumor inhibiting cell ion death and reactivating it may be a promising therapy for tumor treatment.
Clear cell renal cell carcinoma (ccRCC) is a main subtype among renal cancers. Protein disulfide isomerase A4 (PDIA4) confers resistance to ferroptosis via induction of ATF4/SLC7A11 in renal cell carcinoma. And Salinomycin exert anti-tumor ability by inhibiting PDIA4 [114]. The unique metabolic state of ccRCC is related to ferroptosis sensitivity and GPX4 plays a vital role in this progress [115]. Kruppel like factor 2 (KLF2) correlates to the expression of GPX4 in ccRCC and overexpressed KLF2 inhibits tumor growth and invasion by regulating ferroptosis [116]. Acyl-CoA synthetase long-chain family member 3 (ACSL3) regulates the accumulation of lipid droplets in ccRCC and is essential for tumor growth. In addition, ACSL3 also modulates ferroptosis sensitivity in a manner dependent on the composition of exogenous fatty acids. Both functions of ACSL3 could be exploited for ccRCC therapy [117]. AIM2 promotes progression and sunitinib resistance through FOXO3a-ACSL4 axis-regulated ferroptosis [118]. The Hippo Pathway Effector TAZ and ISCA2 are also contribute to ferroptosis in ccRCC [119]. What’s more, Lai et al. developed a predictive model with 8 ferroptosis-related LncRNA predicting prognosis of ccRCC [120]. Combined use of fatty acid amide hydrolase (FAAH) inhibitor URB597 and ferroptosis inducer (1 S,3R)-RLS3 shows strong synergetic inhibition of RCC through induction of G1 cell cycle arrest and promotion of ROS. The dual targets therapy modulates RCC sensitivity to ferroptosis [121].
FDX1, a key factor of cuprotosis, is found to de downregulated in ccRCC which shows correlation between cuproptosis and tumor mechanically [122]. Gang Luo et al. identified 6 cuproptosis-related ferroptosis genes (TRIB3, SLC2A3, PML, CD44, CDKN2A and MIOX) that correlate with worse survival [123]. Besides, they created a prognostic model incorporating these signatures and predict survival.
Renal medullary carcinoma (RMC) is a lethal malignant tumor derived from kidney medulla. It is characterized by the lack of SMARCB1 expression, which is a key subunit of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex. However, the origin of RMC still remains unclear. Recently, Bujamin H Vokshi et al. defined that the loss of transcription factors TFCP2L1, HOXB9 and MITF and the switch on of MYC and NFE2L2-related oncogenic and ferroptosis resistance programs involved in the convert from thick ascending limb (TAL) to RMC cells. And the transcription switch is reversed by SMARCB1 and leads to ferroptotic cell death. Ferroptosis resistance correlates TAL cells survival with high extracellular medullar iron concentrations with character of sickle cell, which possibly is an explanation for the reason why RMC is the only epithelial cell-derived SMARCB1-lost tumor [124].
Perspective and conclusion
To fully understand ferroptosis and cuproptosis, it is important to consider the following key points.
First, these are distinct forms of metal-dependent cell death that differ from other regulated programmed cell death. Ferroptosis is characterized by unique morphological and biochemical features, such as mitochondrial shrinkage and the accumulation of reactive oxygen species (ROS). In contrast, the exact changes in morphological and biochemical features of cuproptosis are still unclear.
Second, the accumulation of metal ions is not synonymous with ferroptosis or cuproptosis, as both iron and copper can have numerous effects beyond including cell death. The oxidation state of metal is also important, with Fe2+ promoting ferroptosis, whereas Fe3+ is generally inert and stored in ferritin. Similarly, Cu+ is a more toxic form than Cu2+ in promoting cuproptosis. Therefore, it is important to determine the redox state of these two metals and whether they contribute specifically to ferroptosis and cuproptosis in a given context.
Third, the production of ROS through iron-dependent Fenton reaction is a key mechanism that propagates lipid peroxidation, driving ferroptosis. ROS can also be generated from copper-based Fenton reaction, but it is unclear whether this is involved in ferroptosis.
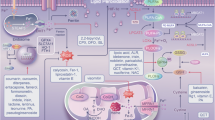
Finally, both ferroptosis and cuproptosis affect mitochondria but have some difference. Mitochondria can act as initiators and amplifiers of ferroptosis, while copper-dependent cell death is dependent on mitochondrial respiration. Nonetheless, our understanding of these forms of cell death remains incomplete, and the interactions between cuproptosis and ferroptosis need to be explored, given the close relationship between copper and iron metabolism (Fig. 2). (1) Copper is an essential cofactor for several enzymes and proteins, such as ceruloplasmin, which is responsible for iron export. It can oxidize Fe2+ to Fe3+, and is involved in the regulation of iron. When there is an excess of copper, ferroptosis may be inhibited. (2) Conversely, under conditions of copper overload, the ROS from copper-base Fenton reaction can increase, which may sensitize cell to ferroptosis. (3) Copper deficiency can inhibit iron absorption, transport and hemoglobin synthesis. (4) in cells of low Fe levels, the labile Cu+ content increases significantly. Even if iron intake is normal, a copper deficiency in the diet can cause anemia. (5) GSH is the principal substrate for GPX4, and the GSH synthase inhibitor BSO has similar cytotoxicity in cells supplemented with either copper or iron. These observations suggest a possibility of crosstalk between cuproptosis and ferroptosis in downstream events. Furthermore, it is still unclear whether other forms of cell death are involved in ferroptosis and cuproptosis. Future studies should aim to identify the details of metal-induced cell death and investigate whether other trace metal ion-induced forms of cell death exist.

Crosstalk Between Ferroptosis and Cuproptosis in the Context of Ion metalbolism. The interactions between cuproptosis and ferroptosis can be categorized into three main groups, considering the close relationship between copper and iron metabolism. First, copper is an essential cofactor for several enzymes and proteins, such as ceruloplasmin, which is responsible for iron export. It can oxidize Fe2+ to Fe3+, thus playing a role in iron regulation. Second, under conditions of copper overload, ROS production from copper-base Fenton reaction can increase. Third, GSH is the principal substrate for GPX4, and the GSH synthase inhibitor has similar cytotoxicity in cells supplemented with either copper or iron. Additionally, destabilization of Fe–S cluster proteins can also contribute to cuproptosis
In summary, there are many opportunities to elucidate the mechanisms of metal ions overload in both ferroptosis and cuproptosis. Such studies will illuminate the breadth of physiological and pathological roles related to ion overload of cell death. Moreover, targeting only one form of cell death, either ferroptosis or cuproptosis, may cause an imbalance in another metal ion. Therefore, it is important to consider the interactions between iron and copper overload when developing novel ferroptosis-or cuproptosis-based therapies for clinical use in the future.
Data availability
Not applicable.
References
Szentmihalyi K (2019) Metal element homeostasis and oxidative stress in pathological processes. Orv Hetil 160(36):1407–1416. https://doi.org/10.1556/650.2019.31499
Valko M et al (2016) Redox- and non-redox-metal-induced formation of free radicals and their role in human Disease. Arch Toxicol 90(1):1–37. https://doi.org/10.1007/s00204-015-1579-5
Wang X et al (2021) Mitochondrial Metal Ion Transport in Cell Metabolism and Disease. Int J Mol Sci 22(14). https://doi.org/10.3390/ijms22147525
Nelson N (1999) Metal ion transporters and homeostasis EMBO J, 18(16): p. 4361 – 71.10.1093/emboj/18.16.4361
Guertl B, Noehammer C, Hoefler G (2000) Metabolic cardiomyopathies Int J Exp Pathol, 81(6): p. 349 – 72.10.1046/j.1365-2613.2000.00186.x
Savarese G et al (2023) Iron Deficiency and Cardiovascular Disease. Eur Heart J 44(1):14–27. https://doi.org/10.1093/eurheartj/ehac569
Evstatiev R, Gasche C (2012) Iron sensing and signalling Gut, 61(6): p. 933 – 52.10.1136/gut.2010.214312
Babcock M et al (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin Science, 276(5319): p. 1709 – 12.10.1126/science.276.5319.1709
Andrews NC, Levy JE (1998) Iron is hot: an update on the pathophysiology of hemochromatosis. Blood 92(6):1845–1851
Askwith C, Kaplan J (1998) Iron and copper transport in yeast and its relevance to human disease Trends Biochem Sci, 23(4): p. 135 – 8.10.1016/s0968-0004(98)01192-x
Liu Y, Miao J (2022) An emerging role of defective copper metabolism in Heart Disease. Nutrients 14(3). https://doi.org/10.3390/nu14030700
Hsu CC et al (2022) Iron overload disorders Hepatol Commun, 6(8): p. 1842-1854.10.1002/hep4.2012
Dixon SJ, Stockwell BR (2014) The role of iron and reactive oxygen species in cell death Nat Chem Biol, 10(1): p. 9-17.10.1038/nchembio.1416
Papanikolaou G, Pantopoulos K (2005) Iron metabolism and toxicity Toxicol Appl Pharmacol, 202(2): p. 199-211.10.1016/j.taap.2004.06.021
Stockwell BR (2022) Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications Cell, 185(14): p. 2401-2421.10.1016/j.cell.2022.06.003
Tsvetkov P et al (2022) Copper induces cell death by targeting lipoylated TCA cycle proteins Science, 375(6586): p. 1254-1261.10.1126/science.abf0529
Roemhild K et al (2021) Iron metabolism: pathophysiology and pharmacology Trends Pharmacol Sci, 42(8): p. 640-656.10.1016/j.tips.2021.05.001
Sheftel A, Stehling O, Lill R (2010) Iron-sulfur proteins in health and disease Trends Endocrinol Metab, 21(5): p. 302 – 14.10.1016/j.tem.2009.12.006
Puig S et al (2017) The elemental role of iron in DNA synthesis and repair Metallomics, 9(11): p. 1483-1500.10.1039/c7mt00116a
Zhang C (2014) Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control Protein Cell, 5(10): p. 750 – 60.10.1007/s13238-014-0083-7
Morales M, Xue X (2021) Targeting iron metabolism in cancer therapy Theranostics, 11(17): p. 8412-8429.10.7150/thno.59092
Gozzelino R, Jeney V, Soares MP (2010) Mechanisms of cell protection by heme oxygenase-1 Annu Rev Pharmacol Toxicol, 50: p. 323 – 54.10.1146/annurev.pharmtox.010909.105600
Ganz T (2013) Systemic iron homeostasis Physiol Rev, 93(4): p. 1721 – 41.10.1152/physrev.00008.2013
Wang X, Wang WX (2021) Intracellular Biotransformation of Cu(II)/Cu(I) Explained High Cu Toxicity to Phytoplankton Chlamydomonas reinhardtii Environ Sci Technol, 55(21): p. 14772-14781.10.1021/acs.est.1c05408
Kim BE, Nevitt T, Thiele DJ (2008) Mechanisms for copper acquisition, distribution and regulation Nat Chem Biol, 4(3): p. 176 – 85.10.1038/nchembio.72
Mondola P et al (2016) The Cu, Zn Superoxide Dismutase: Not Only a Dismutase Enzyme Front Physiol, 7: p. 594.10.3389/fphys.2016.00594
Turski ML, Thiele DJ (2009) New roles for copper metabolism in cell proliferation, signaling, and disease J Biol Chem, 284(2): p. 717 – 21.10.1074/jbc.R800055200
Ryo Yonashiro AS, Miyachi M, Fukuda T, Matsushita N, Inatome R, Ogata Y (2009) Takehiro Suzuki, and a.S.Y. Naoshi Dohmae, Mitochondrial ubiquitin ligase MITOL ubiquitinates mutant SOD1 and attenuates mutant SOD1-induced reactive oxygen species generation 10.1091/mbc.E09
Tang D et al (2019) The molecular machinery of regulated cell death Cell Res, 29(5): p. 347-364.10.1038/s41422-019-0164-5
Dixon SJ et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death Cell, 149(5): p. 1060 – 72.10.1016/j.cell.2012.03.042
Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells Chem Biol, 15(3): p. 234 – 45.10.1016/j.chembiol.2008.02.010
Wolpaw AJ et al (2011) Modulatory profiling identifies mechanisms of small molecule-induced cell death Proc Natl Acad Sci U S A, 108(39): p. E771-80.10.1073/pnas.1106149108
Jiang X, Stockwell BR, Conrad M (2021) Ferroptosis: mechanisms, biology and role in disease Nat Rev Mol Cell Biol, 22(4): p. 266-282.10.1038/s41580-020-00324-8
Singh M et al (2006) Iron bioavailability: UK Food Standards Agency workshop report Br J Nutr, 96(5): p. 985 – 90.10.1017/bjn20061894
Ke K et al (2022) The crosstalk effect between ferrous and other ions metabolism in ferroptosis for therapy of cancer Front Oncol, 12: p. 916082.10.3389/fonc.2022.916082
Drakesmith H, Nemeth E, Ganz T (2015) Ironing out Ferroportin Cell Metab, 22(5): p. 777 – 87.10.1016/j.cmet.2015.09.006
Andrews NC, Schmidt PJ (2007) Iron homeostasis Annu Rev Physiol, 69: p. 69-85.10.1146/annurev.physiol.69.031905.164337
Dautry-Varsat A, Ciechanover A, Lodish HF (1983) pH and the recycling of transferrin during receptor-mediated endocytosis
Hentze NHaMW (2022) Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function
Zhang S et al (2022) Double-edge sword roles of iron in driving energy production versus instigating ferroptosis Cell Death Dis, 13(1): p. 40.10.1038/s41419-021-04490-1
Park CH et al (2001) Hepcidin, a urinary antimicrobial peptide synthesized in the liver J Biol Chem, 276(11): p. 7806 – 10.10.1074/jbc.M008922200
Nemeth E et al (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization Science, 306(5704): p. 2090 – 3.10.1126/science.1104742
CABANTCHIK OKaZI (2002) The labile iron pool: characterization, measurement, and participation in cellular processes(1)
Shah R, Shchepinov MS, Pratt DA (2018) Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis ACS Cent Sci, 4(3): p. 387-396.10.1021/acscentsci.7b00589
Yang WS et al (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis Proc Natl Acad Sci U S A, 113(34): p. E4966-75.10.1073/pnas.1603244113
Doll S, Conrad M (2017) Iron and ferroptosis: A still ill-defined liaison IUBMB Life, 69(6): p. 423-434.10.1002/iub.1616
Enric Brillas IS, Mehmet A, Oturan (2020) Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry
Wenzel SE et al (2017) PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals Cell, 171(3): p. 628–641.e26.10.1016/j.cell.2017.09.044
Chu B et al (2019) ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway Nat Cell Biol, 21(5): p. 579-591.10.1038/s41556-019-0305-6
Zou Y et al (2020) Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis Nat Chem Biol, 16(3): p. 302-309.10.1038/s41589-020-0472-6
Abrams RP, Carroll WL, Woerpel KA (2016) Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells ACS Chem Biol, 11(5): p. 1305 – 12.10.1021/acschembio.5b00900
Schilstra MJ, Veldink GA, Vliegenthart JF (1994) The dioxygenation rate in lipoxygenase catalysis is determined by the amount of iron (III) lipoxygenase in solution Biochemistry, 33(13): p. 3974 – 9.10.1021/bi00179a025
Brown CW et al (2019) Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export Dev Cell, 51(5): p. 575–586.e4.10.1016/j.devcel.2019.10.007
Patel SJ et al (2021) The iron chaperone and nucleic acid-binding activities of poly(rC)-binding protein 1 are separable and independently essential Proc Natl Acad Sci U S A, 118(25).10.1073/pnas.2104666118
Tang D, Chen X, Kroemer G (2022) Cuproptosis: a copper-triggered modality of mitochondrial cell death Cell Res, 32(5): p. 417-418.10.1038/s41422-022-00653-7
Kahlson MA, Dixon SJ (2022) Copper-induced cell death Science, 375(6586): p. 1231-1232.10.1126/science.abo3959
Cobine PA, Brady DC (2022) Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death Mol Cell, 82(10): p. 1786-1787.10.1016/j.molcel.2022.05.001
Pierson H, Yang H, Lutsenko S (2019) Copper Transport and Disease: What Can We Learn from Organoids? Annu Rev Nutr, 39: p. 75-94.10.1146/annurev-nutr-082018-124242
Shanbhag VC et al (2021) Copper metabolism as a unique vulnerability in cancer Biochim Biophys Acta Mol Cell Res, 1868(2): p. 118893.10.1016/j.bbamcr.2020.118893
Eisses JF, Chi Y, Kaplan JH (2005) Stable plasma membrane levels of hCTR1 mediate cellular copper uptake J Biol Chem, 280(10): p. 9635 – 9.10.1074/jbc.M500116200
Shawki A et al (2015) Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese Am J Physiol Gastrointest Liver Physiol, 309(8): p. G635-47.10.1152/ajpgi.00160.2015
Puig S, Thiele DJ (2002) Molecular mechanisms of copper uptake and distribution Curr Opin Chem Biol, 6(2): p. 171 – 80.10.1016/s1367-5931(02)00298-3
Tsang T, Davis CI, Brady DC (2021) Copper biology Curr Biol, 31(9): p. R421-r427.10.1016/j.cub.2021.03.054
Tsvetkov P et al (2019) Mitochondrial metabolism promotes adaptation to proteotoxic stress Nat Chem Biol, 15(7): p. 681-689.10.1038/s41589-019-0291-9
Rowland EA, Snowden CK, Cristea IM (2018) Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease Curr Opin Chem Biol, 42: p. 76-85.10.1016/j.cbpa.2017.11.003
Forman HJ, Zhang H, Rinna A (2009) Glutathione: overview of its protective roles, measurement, and biosynthesis Mol Aspects Med, 30(1–2): p. 1-12.10.1016/j.mam.2008.08.006
Aquilano K, Baldelli S, Ciriolo MR (2014) Glutathione: new roles in redox signaling for an old antioxidant Front Pharmacol, 5: p. 196.10.3389/fphar.2014.00196
Chen X et al (2021) Broadening horizons: the role of ferroptosis in cancer Nat Rev Clin Oncol, 18(5): p. 280-296.10.1038/s41571-020-00462-0
Cao JY et al (2019) A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity Cell Rep, 26(6): p. 1544–1556.e8.10.1016/j.celrep.2019.01.043
Hao S et al (2017) Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells Neoplasia, 19(12): p. 1022-1032.10.1016/j.neo.2017.10.005
Kang YP et al (2021) Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis Cell Metab, 33(1): p. 174–189.e7.10.1016/j.cmet.2020.12.007
Li S-R, Bu L-L, Cai L (2022) Cuproptosis: lipoylated TCA cycle proteins-mediated novel cell death pathway Signal Transduction and Targeted Therapy, 7(1).10.1038/s41392-022-01014-x
Lill R, Freibert SA (2020) Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis Annu Rev Biochem, 89: p. 471-499.10.1146/annurev-biochem-013118-111540
Ali ME et al (2014) The iron-sulfur core in Rieske proteins is not symmetric J Biol Inorg Chem, 19(8): p. 1287 – 93.10.1007/s00775-014-1185-7
Yankovskaya V et al (2003) Architecture of succinate dehydrogenase and reactive oxygen species generation Science, 299(5607): p. 700 – 4.10.1126/science.1079605
Stiban J et al (2014) The N-terminal domain of the Drosophila mitochondrial replicative DNA helicase contains an iron-sulfur cluster and binds DNA J Biol Chem, 289(35): p. 24032 – 42.10.1074/jbc.M114.587774
Mariotti L et al (2020) The iron-sulphur cluster in human DNA2 is required for all biochemical activities of DNA2 Commun Biol, 3(1): p. 322.10.1038/s42003-020-1048-4
Kobayashi K, Fujikawa M, Kozawa T (2014) Oxidative stress sensing by the iron-sulfur cluster in the transcription factor, SoxR J Inorg Biochem, 133: p. 87-91.10.1016/j.jinorgbio.2013.11.008
Kiley PJ, Beinert H (1998) Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster FEMS Microbiol Rev, 22(5): p. 341 – 52.10.1111/j.1574-6976.1998.tb00375.x
Hentze MW, Kühn LC (1996) Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress Proc Natl Acad Sci U S A, 93(16): p. 8175 – 82.10.1073/pnas.93.16.8175
Hentze MW et al (2010) Two to tango: regulation of Mammalian iron metabolism Cell, 142(1): p. 24-38.10.1016/j.cell.2010.06.028
Hirling H, Henderson BR, Kühn LC (1994) Mutational analysis of the [4Fe-4S]-cluster converting iron regulatory factor from its RNA-binding form to cytoplasmic aconitase Embo j, 13(2): p. 453 – 61.10.1002/j.1460-2075.1994.tb06280.x
Tong WH, Rouault TA (2006) Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis Cell Metab, 3(3): p. 199-210.10.1016/j.cmet.2006.02.003
Alvarez SW et al (2017) NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis Nature, 551(7682): p. 639-643.10.1038/nature24637
Lei G, Zhuang L, Gan B (2022) Targeting ferroptosis as a vulnerability in cancer Nat Rev Cancer, 22(7): p. 381-396.10.1038/s41568-022-00459-0
Du J et al (2019) DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin Free Radic Biol Med, 131: p. 356-369.10.1016/j.freeradbiomed.2018.12.011
Yuan H et al (2016) CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation Biochem Biophys Res Commun, 478(2): p. 838 – 44.10.1016/j.bbrc.2016.08.034
Kim EH et al (2018) CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer Cancer Lett, 432: p. 180-190.10.1016/j.canlet.2018.06.018
Du J et al (2020) Identification of Frataxin as a regulator of ferroptosis Redox Biol, 32: p. 101483.10.1016/j.redox.2020.101483
Dixon SJ et al (2015) Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death ACS Chem Biol, 10(7): p. 1604 – 9.10.1021/acschembio.5b00245
Doll S et al (2019) FSP1 is a glutathione-independent ferroptosis suppressor Nature, 575(7784): p. 693-698.10.1038/s41586-019-1707-0
Sha W et al (2021) Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus J Diabetes Res, 2021: p. 9999612.10.1155/2021/9999612
Lee H et al (2020) Energy-stress-mediated AMPK activation inhibits ferroptosis Nat Cell Biol, 22(2): p. 225-234.10.1038/s41556-020-0461-8
Li C et al (2020) LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis Signal Transduct Target Ther, 5(1): p. 187.10.1038/s41392-020-00297-2
Kagan VE et al (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis Nat Chem Biol, 13(1): p. 81-90.10.1038/nchembio.2238
Hay N (2016) Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer, 16(10): p. 635 – 49.10.1038/nrc.2016.77
Wang Y et al (2022) ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury Redox Biol, 51: p. 102262.10.1016/j.redox.2022.102262
Zhao Z et al (2023) Cytoplasmic HMGB1 induces renal tubular ferroptosis after ischemia/reperfusion Int Immunopharmacol, 116: p. 109757.10.1016/j.intimp.2023.109757
Tonnus W et al (2021) Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury Nat Commun, 12(1): p. 4402.10.1038/s41467-021-24712-6
Sun X et al (2023) TRIM21 ubiquitylates GPX4 and promotes ferroptosis to aggravate ischemia/reperfusion-induced acute kidney injury Life Sci, 321: p. 121608.10.1016/j.lfs.2023.121608
Li H et al (2023) Ferroptosis is involved in polymyxin B-induced acute kidney injury via activation of p53 Chem Biol Interact, 378: p. 110479.10.1016/j.cbi.2023.110479
Kim DH et al (2022) Farnesoid X receptor protects against cisplatin-induced acute kidney injury by regulating the transcription of ferroptosis-related genes Redox Biol, 54: p. 102382.10.1016/j.redox.2022.102382
Lin Q et al (2023) Mitophagy alleviates cisplatin-induced renal tubular epithelial cell ferroptosis through ROS/HO-1/GPX4 axis Int J Biol Sci, 19(4): p. 1192-1210.10.7150/ijbs.80775
Wang Y et al (2021) Quercetin alleviates acute kidney injury by inhibiting ferroptosis J Adv Res, 28: p. 231-243.10.1016/j.jare.2020.07.007
Kolbrink B et al (2022) Vitamin K1 inhibits ferroptosis and counteracts a detrimental effect of phenprocoumon in experimental acute kidney injury Cell Mol Life Sci, 79(7): p. 387.10.1007/s00018-022-04416-w
Qiongyue Z et al (2022) Post-treatment With Irisin Attenuates Acute Kidney Injury in Sepsis Mice Through Anti-Ferroptosis via the SIRT1/Nrf2 Pathway Front Pharmacol, 13: p. 857067.10.3389/fphar.2022.857067
Hu J et al (2022) Leonurine alleviates ferroptosis in cisplatin-induced acute kidney injury by activating the Nrf2 signalling pathway Br J Pharmacol, 179(15): p. 3991-4009.10.1111/bph.15834
Ma L et al (2023) Paeoniflorin alleviates ischemia/reperfusion induced acute kidney injury by inhibiting Slc7a11-mediated ferroptosis Int Immunopharmacol, 116: p. 109754.10.1016/j.intimp.2023.109754
Zhou L et al (2022) Targeting Ferroptosis Attenuates Interstitial Inflammation and Kidney Fibrosis Kidney Dis (Basel), 8(1): p. 57-71.10.1159/000517723
Jung KH et al (2023) Synergistic Renoprotective Effect of Melatonin and Zileuton by Inhibition of Ferroptosis via the AKT/mTOR/NRF2 Signaling in Kidney Injury and Fibrosis Biomol Ther (Seoul), 10.4062/biomolther.2023.062
Zhu B et al (2023) Formononetin ameliorates ferroptosis-associated fibrosis in renal tubular epithelial cells and in mice with chronic kidney disease by suppressing the Smad3/ATF3/SLC7A11 signaling Life Sci, 315: p. 121331.10.1016/j.lfs.2022.121331
Shi L et al (2023) MiR-20a-5p alleviates kidney ischemia/reperfusion injury by targeting ACSL4-dependent ferroptosis Am J Transplant, 23(1): p. 11-25.10.1016/j.ajt.2022.09.003
Tang Q et al (2023) Identification and verification of hub genes associated with ferroptosis in ischemia and reperfusion injury during renal transplantation Int Immunopharmacol, 120: p. 110393.10.1016/j.intimp.2023.110393
Kang L et al (2023) PDIA4 confers resistance to ferroptosis via induction of ATF4/SLC7A11 in renal cell carcinoma Cell Death Dis, 14(3): p. 193.10.1038/s41419-023-05719-x
Zou Y et al (2019) A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis Nat Commun, 10(1): p. 1617.10.1038/s41467-019-09277-9
Lu Y et al (2021) KLF2 inhibits cancer cell migration and invasion by regulating ferroptosis through GPX4 in clear cell renal cell carcinoma Cancer Lett, 522: p. 1-13.10.1016/j.canlet.2021.09.014
Klasson TD et al (2022) ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma Cancer Metab, 10(1): p. 14.10.1186/s40170-022-00290-z
Wang Q et al (2023) AIM2 promotes renal cell carcinoma progression and sunitinib resistance through FOXO3a-ACSL4 axis-regulated ferroptosis Int J Biol Sci, 19(4): p. 1266-1283.10.7150/ijbs.79853
Yang WH et al (2019) The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma Cell Rep, 28(10): p. 2501–2508.e4.10.1016/j.celrep.2019.07.107
Lai J, Miao S, Ran L (2023) Ferroptosis-associated lncRNA prognostic signature predicts prognosis and immune response in clear cell renal cell carcinoma Sci Rep, 13(1): p. 2114.10.1038/s41598-023-29305-5
Hao J et al (2023) Combination treatment with FAAH inhibitors/URB597 and ferroptosis inducers significantly decreases the growth and metastasis of renal cell carcinoma cells via the PI3K-AKT signaling pathway Cell Death Dis, 14(4): p. 247.10.1038/s41419-023-05779-z
Xu J et al (2022) Multi-omics pan-cancer study of cuproptosis core gene FDX1 and its role in kidney renal clear cell carcinoma Front Immunol, 13: p. 981764.10.3389/fimmu.2022.981764
Luo G et al (2023) Cuproptosis-Related Ferroptosis genes for Predicting Prognosis in kidney renal clear cell carcinoma Eur J Med Res, 28(1): p. 176.10.1186/s40001-023-01137-z
Vokshi BH et al (2023) SMARCB1 regulates a TFCP2L1-MYC transcriptional switch promoting renal medullary carcinoma transformation and ferroptosis resistance Nat Commun, 14(1): p. 3034.10.1038/s41467-023-38472-y
Gaschler MM et al (2018) FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation Nat Chem Biol, 14(5): p. 507-515.10.1038/s41589-018-0031-6
Ooko E et al (2015) Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells Phytomedicine, 22(11): p. 1045 – 54.10.1016/j.phymed.2015.08.002
Hassannia B et al (2018) Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma J Clin Invest, 128(8): p. 3341-3355.10.1172/jci99032
Lin PL et al (2020) Saponin Formosanin C-induced Ferritinophagy and Ferroptosis in Human Hepatocellular Carcinoma Cells Antioxidants (Basel), 9(8).10.3390/antiox9080682
Wang ZX et al (2021) Quercetin induces p53-independent cancer cell death through lysosome activation by the transcription factor EB and Reactive Oxygen Species-dependent ferroptosis Br J Pharmacol, 178(5): p. 1133-1148.10.1111/bph.15350
Liu JL et al (2018) Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications Front Neurosci, 12: p. 632.10.3389/fnins.2018.00632
Naito Y et al (2015) Association between renal iron accumulation and renal interstitial fibrosis in a rat model of chronic kidney disease Hypertens Res, 38(7): p. 463 – 70.10.1038/hr.2015.14
Li J et al (2020) Ferroptosis: past, present and future Cell Death Dis, 11(2): p. 88.10.1038/s41419-020-2298-2
Ikeda Y et al (2021) Role of ferroptosis in cisplatin-induced acute nephrotoxicity in mice J Trace Elem Med Biol, 67: p. 126798.10.1016/j.jtemb.2021.126798
Funding
This study was supported by National Natural Science Foundation of China (grants 82170765 to CY), the 2019 Shanghai Youth Talent Development Program (to CY) and Shanghai Municipal Key Clinical Specialty (shslczdzk05802).
Author information
Authors and Affiliations
Contributions
TC and CY conceived and designed the review. TC and LL collected, analyzed literatures, and wrote the draft. YW and XL helped to revise the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, T., Liang, L., Wang, Y. et al. Ferroptosis and cuproptposis in kidney Diseases: dysfunction of cell metabolism. Apoptosis 29, 289–302 (2024). https://doi.org/10.1007/s10495-023-01928-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-023-01928-z