
Abstract
Autophagy is a cellular self-catabolic process in which cytoplasmic constituents are sequestered in double membrane vesicles that fuse with lysosomes where they are degraded. As this catabolic activity generates energy, autophagy is often induced under nutrient limiting conditions providing a mechanism to maintain cell viability and may be exploited by cancer cells for survival under metabolic stress. However, progressive autophagy can be cytotoxic and autophagy can under certain settings substitute for apoptosis in induction of cell death. Moreover, loss of autophagy is correlated with tumorigenesis and several inducers of autophagy are tumor-suppressor genes. Thus, the relation of autophagy to cancer development is complex and depends on the genetic composition of the cell as well as on the extra-cellular stresses a cell is exposed to. In this review we describe the intricate nature of autophagy and its regulators, particularly those that have been linked to cancer. We discuss the multifaceted relation of autophagy to tumorigenesis and highlight studies supporting a role for autophagy in both tumor-suppression and tumor-progression. Finally, various autophagy-targeting therapeutic strategies for cancer treatment are presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy is a self-digestive process wherein bulk cytoplasmic components and intra-cellular organelles are sequestered in double membrane vesicles named autophagosomes. Upon maturation, autophagosomes fuse with lysosomes where their contents are degraded by the lysosomal proteases. The process of autophagy was initially described as a mechanism of cell survival under nutrient limiting conditions. Specifically, the autophagic catabolic activity enables cells to restore sufficient energy levels in the absence of nutrients and consequently promotes viability [1]. This feature of autophagy is of particular importance during developmental processes, as it is suggested to maintain normal metabolism by providing an alternative cellular source for energy. Incidentally, deficiency of Beclin 1 is embryonic lethal and Atg5 or Atg7 null mice fail to survive the neonatal starvation period and die perinatally [2, 3]. In addition, it has been established that basal autophagy is also essential for keeping cellular homeostasis by mediating the normal turnover of organelles and long-lived proteins. Disruption of this homeostasis can have severe effects. For example, deletion of Atg5 or Atg7 results in accumulation of polyubiquitinated proteins in the nervous system leading to neurodegeneration [4, 5]. A cyto-protective role of autophagy is also described during the immune response and includes defense against pathogens and T cell repertoire shaping [6, 7]. Paradoxically, many lines of evidence also describe a role for autophagy in antagonizing cell survival and promoting cell death, classified as autophagic, or type II, cell death. This discrepancy is partially resolved by the presumption that extensive activation of autophagy might lead to the disintegration of major cellular systems, such that “the point of no return” is crossed and recovery of the cell is unattainable. Other specific cellular settings, such as the integrity of other cell death systems, also influence the balance between the dual functions of autophagy.
The course of tumorigenesis involves many genetic alterations that collectively lead to cellular transformation and tumor growth. Intensive studies over the years delineated the contribution of loss of apoptotic (also named type I) cell death, responses to tumor formation [8]. Accordingly, numerous key regulators of apoptosis were described as tumor-suppressor genes and their cellular function was extensively studied. With the identification of autophagy as an alternative cell death program, its contribution to tumorigenesis was evaluated as well. Remarkably, the process of autophagy was revealed to be critical in certain scenarios of tumor formation. However, the link between autophagy and cancer appears to be multifaceted. Loss of autophagic responses is correlated with cancer development and furthermore, various inducers of autophagy including Beclin 1, p53, DAPk, p19ARF, TSC, LKB, and PTEN have been described as tumor-suppressors. Therefore, loss of autophagy might contribute to tumor-progression due to a failure to eliminate damaged cells through type II cell death, as in the case of loss of apoptotic responses. In addition, the cyto-protective features of autophagy can also contribute indirectly to its tumor-suppressive function such that loss of the autophagic-homeostasis maintenance might expose cells to various defects such as accumulation of damaged mitochondria. Damaged mitochondria produce high levels of free radicals that often cause damage to DNA resulting in genetic alterations that may promote tumor development. However, autophagy bears a different facet as well that correlates with promotion of cell survival under stress. As a corollary, autophagy might be advantageous to tumor development and its inhibition could therefore be required to block tumorigenesis.
In conclusion, autophagy is an intricate process with multiple biological aspects. Great effort is invested in deciphering its mechanistic features and their relevance to physiological processes, particularly to tumorigenesis. In contrast to the unambiguous role of the apoptotic network in tumor-suppression, the relation between autophagy and cancer is complex. This review will discuss the link between autophagy and cancer and will explore different autophagy-inducing drugs that are being used as anti-cancer therapy in the clinic.
The molecular mechanism of autophagy
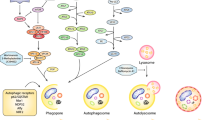
Deciphering the molecular mechanism of autophagy began in 1993 with the identification of the autophagy-related genes (Atg) in the yeast Saccharomyces cerevisiae [9]. Most of the Atg genes are conserved from yeast to humans and many orthologs of these genes have been identified in mammalian cells. The first steps in the formation of the autophagic vacuole are mediated by the class III phosphatidylinositol 3-kinase (PI3KC3), the ortholog of Vps34, the sole PI3K in yeast [10]. Among the key players in the autophagic machinery are those that form a complex with Vps34 and regulate its activity, such as Beclin 1 (the mammalian ortholog of Atg6) and the myristylated serine kinase Vps15/p150 [11]. UVRAG and Bif-1 are positive regulators of the interaction between Beclin 1 and Vps34, and thus promote Vps34 activation [12, 13]. Vps34 also binds Atg14 that directs the Vps34-complex to the organizing centers of autophagosome formation, known as the pre-autophagosomal structures (PAS) [14]. The mammalian ortholog of Atg14 has been recently identified [15]. Although the source of the autophagosomal membrane is still unidentified, the localization of Beclin 1 and Vps34 to the endoplasmic reticulum and trans-golgi network [16] suggests these sites as candidates for the autophagosome origin. The nucleation step of autophagosome biogenesis requires activation of Vps34 that produces phosphatidylinositol 3-phosphate (PI3P). PI3P presumably acts by recruiting proteins that contain PI3P-binding FYVE and PX domains, which are predicted to control membrane formation and elongation. An example is Atg18 that in complex with Atg2 is recruited to the PAS via phospholipids binding [17, 18].
The serine/threonine kinase Atg1 is essential for the induction of autophagy. In yeast, Atg1 activation requires its interaction with Atg13. TOR negatively regulates this step by phosphorylating Atg13 which reduces its affinity for Atg1 binding [19, 20]. Still, the specific function of Atg1 or of its mammalian ortholog, ULK1 (UNC-51-like kinase 1), is undefined, as well as the mechanism which couples its function to the activation of the Beclin 1/Vps34 complex.
Elongation of the autophagic vacuole membrane is mediated by two ubiquitin-like pathways. Atg12, a ubiquitin-like protein, is covalently conjugated to Atg5 by the action of the E1- and E2-like proteins Atg7 and Atg10, respectively [21–24]; Atg7 and Atg3 similarly act as E1- and E2-enzymes in the second ubiqutin-like pathway in which Atg8 (mammalian LC3) is lipidated by conjugation to phosphatidylethanolamine (PE) [25]. The Atg12–Atg5 dimer is recruited to the outer autophagosomal membrane, presumably via interaction with Atg16 [26, 27]. In accord, the site of LC3 lipidation is determined by the membrane localization of Atg16 [28]. Recently, the Atg12–Atg5 dimer was shown to have E3-like activity for Atg8 conjugation to PE [29]. This critical step in autophagosome expansion is mediated by the formation of an amide bond between the amino group of PE and the carboxyl-terminal glycine residue of Atg8, which is exposed following cleavage of the C-terminus of Atg8 by the cysteine protease Atg4 [25, 30]. Lipidation of Atg8, which occurs only under stimulation of autophagy, converts Atg8 from its soluble cytoplasmic form (LC3-I) to the membrane-bound, autophagosome-associated, form (LC3-II). The recruitment of Atg8 to autophagosomes is rate limiting in the process of membrane expansion. Atg8-PE mediates membrane tethering and hemifusion [31] and the amount of Atg8 at the PAS determines the size of the autophagosome [32]. Once the autophagosome is fully expanded, Atg8 is deconjugated from PE via the action of Atg4 and is released back to the cytosol [30]. Likewise, the Atg12–Atg5–Atg16 complex dissociates from the autophagosome membrane upon maturation.
Upon maturation, the autophagosome fuses with organelles of the endocytic compartment (early or late endosomes and lysosomes) to form the autolysosome. In yeast, this process was shown to require Ypt7p (the yeast homologue of Rab7) [33], Vam3p (a syntaxin homologue) [34], Sec18p (yeast homologue of N-ethylmaleimide sensitive factor, NSF), and Vti1p (a SNARE protein) [35]. A similar role for Rab7 and Vti1p was demonstrated in mammalian cells [36–38]. Interestingly, UVRAG, which facilitates Vps34 activation in the early steps of autophagosome formation, has a second role in later stages of autophagy wherein it regulates autophagosome maturation in a Beclin 1-independent manner. UVRAG facilitates the recruitment of the class C vacuolar protein sorting (C-Vps) complex to autophagosomes. The C-Vps complex has been extensively studied in fusion events between the endosome and vacuole in yeast. The interaction between UVRAG and the C-Vps complex stimulates Rab7–GTPase activity and results in autophagosome to lysosome fusion [39]. Fusion takes place in the perinuclear region which is enriched in lysosomes. For this to occur, autophagosomes must move from the cytosol towards the nucleus. The movement of autophagosomes occurs on microtubules [40, 41] and is mediated by the motor protein dynein [42]. Finally, the engulfed cytoplasmic proteins and organelles within the autolysosome are degraded by lysosomal proteins.
Tumor-suppressor genes and oncogenes that regulate/execute autophagy
One of the strong indications which link autophagy to cancer is the finding that some of the autophagic genes, or the genes which regulate the autophagic process, function as tumor-suppressors or oncogenes. Below we describe a few major examples.
Beclin 1 and its regulators
Beclin 1 was originally identified as a novel Bcl-2-interacting, coiled-coil protein, which has structural similarity to yeast Atg6. The autophagic function of Beclin 1 is evolutionary conserved and has been demonstrated in various species including yeast [43], dictyostelium [44], plants [45], C. elegans [46], mice [2, 47], and humans [48, 49]. The identification of Beclin 1 as a tumor-suppressor gene in 1999 established the first connection between autophagy and cancer [48]. The Beclin 1 locus (17q21) is frequently subjected to monoallelic deletions in human breast, ovarian, and prostate cancer as well as in brain tumors, signifying Beclin 1 as a haplo-insufficient tumor-suppressor gene in humans [48, 50, 51]. Studies in mice confirmed the findings in human tumors. Heterozygous gene disruption in mice resulted in the development of spontaneous breast and lung tumors, lymphoma, and hepatocellular carcinoma [2, 47]. Incidentally, Beclin 1 gene transfer induced autophagy and inhibited the tumorigenesis of human breast carcinoma cells in clonigenicity assays and in mouse xenograft models [48]. These findings suggested that the tumor-suppressive functions of Beclin 1 are associated with its positive regulation of autophagy. As detailed above, Beclin 1 acts as a platform for the recruitment and activation of the Vps34 complex, which is essential for the initiation of the early stages of autophagy (Fig. 1). An evolutionary conserved region of human Beclin 1, spanning from amino acids 244–337, is responsible for Vps34 binding and activation, and is thus required for the induction of autophagy. Remarkably, this region is also necessary for the inhibition of tumorigenicity in mice [49], supportive of the notion that the tumor-suppressive and autophagic functions of Beclin 1 are interrelated.

Signaling pathways that regulate autophagy. Positive regulators of autophagy are illustrated in green while negative regulators are illustrated in red. The Beclin 1-Vps34 complex is required for induction of autophagy and its activity is negatively regulated by binding of Beclin 1 to Bcl-2 and by JNK, which releases Bcl-2 inhibition through phosphorylation. Other negative regulators of autophagy include growth factor receptors (GFR), which lead to activation of mTOR through AKT and the Ras pathway. However, erk has also been demonstrated to act under certain settings as an inducer of autophagy. p53 has a dual function and can either induce or inhibit the induction of autophagy
Several regulators of Beclin 1 have been implicated in tumorigenesis. UVRAG, a Beclin 1-binding protein, is a tumor-suppressor candidate which maps to chromosome 11q13. Disruption of this locus is frequently associated with development of different human malignancies, including breast and colon cancers [52]. Monoallelic deletions or mutations in UVRAG have been reported in numerous human malignancies [53–55]. UVRAG and Beclin 1 directly interact via their coiled-coil domains and this interaction is suggested to promote Vps34 binding to and activation by Beclin 1 [12, 56]. The expression of UVRAG was shown to be required for Beclin 1-induced autophagy in human colon and breast cancer cells. Furthermore, UVRAG suppressed cell proliferation and tumor formation in vivo [12]. Bif-1 (also known as endophilin B1) is another Beclin 1-binding protein that suppresses tumorigenesis. Bif-1 interacts with Beclin 1 via UVRAG and promotes Vps34 activation and autophagosome formation. Bif-1 knock-out mice develop normally, unlike Beclin 1 null mice which are embryonic lethal, but demonstrate a high rate of tumor incidence. About 89.7% of Bif-1−/− mice developed spontaneous tumors at 12 months of age, compared to 14.3% of wild-type mice [13]. Moreover, reduced Bif-1 expression was observed in gastric carcinomas [57], invasive urinary bladder and gallbladder cancers [58], and a homozygous deletion of the Bif-1 gene was identified in mantle cell lymphomas [59]. Taken together, these findings suggest that two of the positive regulators of Beclin 1, UVRAG and Bif-1, function as potential tumor-suppressor genes. Bcl-2, an oncogenic negative regulator of Beclin 1, is discussed in the section below.
Bcl-2
The Bcl-2 family includes proteins containing at least one Bcl-2 homology (BH) region. In mammals, the Bcl-2 family is subdivided into anti-apoptotic members (such as Bcl-2 and Bcl-XL), which contain four BH domains, pro-apoptotic members (such as Bax and Bak), which contain three BH domains, and the typically pro-apoptotic BH3-only members. The Bcl-2 gene was initially found to be disrupted in most cases of follicular lymphomas carrying the t(14;18) chromosomal translocation [60]. Transgenic mice carrying a Bcl-2 immunoglobulin minigene fusion, that recapitulates the t(14;18) translocation, were found to develop follicular hyperplasia and lymphoma [61, 62]. Interestingly, unlike other previously characterized oncogenes, what accounted for Bcl-2’s oncogenic properties was inhibition of cell death rather than acceleration of proliferation.
The anti-cell death function of Bcl-2 was for years attributed solely to the inhibition of apoptosis. However, the identification of Beclin 1 as a Bcl-2 binding protein [48] was the first in the line of studies connecting Bcl-2 to a different program of cell death, namely autophagy. The first suggestion that Bcl-2 is involved in non-apoptotic cell death came with the finding that downregulation of Bcl-2 results in increased caspase-independent cell death in human leukemic HL60 cell line [63]. An anti-autophagic role of Bcl-2 was demonstrated in vivo where transgenic cardiac-expression of Bcl-2 in mice inhibited starvation-induced autophagy in the cardiac muscle [64]. Additionally, targeted silencing of Bcl-2 expression in human breast cancer cells with RNA-interference has been shown to promote autophagic cell death and thus presents a therapeutic potential [65].
The mechanism by which Bcl-2 inhibits autophagy is reliant on its interaction with Beclin 1. Beclin 1 contains a functional BH3 domain through which it binds to the BH3-binding groove in Bcl-2/Bcl-XL [66–68]. In contrast to wild-type Bcl-2, mutants that are defective in Beclin 1 binding failed to inhibit autophagy following nutrient starvation in Beclin 1 expressing human breast carcinoma cells [64]. The binding of Bcl-2 to Beclin 1 inhibits autophagy by negatively regulating the autophagy-promoting Beclin 1–Vps34 complex. For example, binding of Bcl-2 to Beclin 1 in autophagy-competent colon carcinoma cells decreased the association of Beclin 1 with Vps34 and consequently inhibited autophagy [64]. Recent findings further resolved the manner by which Bcl-2 inhibits the formation of the Beclin 1-Vps34 complex. Beclin 1 forms a dimer that is stabilized by Bcl-2. Interaction with UVRAG disrupts the dimerization interface of Beclin 1 and induces its monomerization. Bcl-2 and Bcl-XL reduce the binding affinity of UVRAG to Beclin 1 and thereby prevent its monomerization [56]. As UVRAG and Bcl-2 are known to have opposing functions on Beclin 1-induced Vps34 activation, it is plausible that monomerization of Beclin 1 is required for Vps34 binding and activation. The association of Bcl-2 with Beclin 1 is regulated in a stress-dependent manner. Endogenous Bcl-2 is found constitutively bound to Beclin 1 in HeLa cells. Elevated levels of interaction are observed under nutrient-rich (autophagy inhibitory) conditions, whereas following nutrient withdrawal (autophagy stimulatory conditions) the interaction is significantly reduced to minimal levels, correlating with activation of autophagy [64].
Different mechanisms have been proposed to mediate the dissociation of Beclin 1 from Bcl-2. One model depicts that other BH3-containing proteins will competitively displace the Beclin 1 BH3 domain from Bcl-2/Bcl-XL. This model is supported by evidence showing that BH3-mimetics and BH3-only proteins disrupt the association of Beclin 1 with Bcl-2/Bcl-XL and induce autophagy [66, 69]. Interestingly, a viral form of Bcl-2, such as that encoded by the tumorigenic murine γ-herpesvirus 68, binds Beclin 1 with a much higher affinity than cellular Bcl-2. This high affinity binding renders the viral Bcl-2 resistant to competition by BH3-only proteins and to displacement from Beclin 1 [70]. Therefore, inhibition of Beclin 1 by constitutive binding to viral Bcl-2 prevents the induction of autophagy and is a plausible contributor to the tumor-promoting properties of such viruses.
A second mechanism for the dissociation of Beclin 1 from Bcl-2 was suggested to be dependent on Bcl-2 phosphorylation. Bcl-2 can undergo phosphorylation by c-Jun N-terminal Kinase (JNK) on residues in the non-structured loop (T69, S70, and S87). Recently, this phosphorylation event was shown to regulate Beclin 1 binding to Bcl-2 and was linked to autophagy [71]. Following nutrient starvation, JNK was readily activated and led to the phosphorylation of the non-structured loop of Bcl-2 (T69, S70, and S87). A non-phosphorylatable Bcl-2 mutant, in which these residues were substituted with alanines, did not dissociate from Beclin 1 under starvation and inhibited the induction of autophagy. Loss of active JNK, either by pharmacological inhibition or by knock-out, had a similar inhibitory effect on Beclin 1 binding and autophagy. Conversely, expression of a constitutively active JNK was sufficient for the induction of Bcl-2 phosphorylation, release from Beclin 1, and stimulation of autophagy [71]. Thus, JNK positively regulates autophagy through direct phosphorylation of Bcl-2 and this leads to the release of the inhibitory lock on Beclin 1. Interestingly, only the ER-localized pool of Bcl-2 is subjected to regulation by these two mechanisms [66, 71], suggesting that spatial regulation is critical for the initiation of autophagy.
In conclusion, the inhibitory effect of Bcl-2 on autophagy suggests novel approaches for anti-cancer drug design. Targeting Bcl-2 binding to Beclin 1 will stabilize the monomeric, UVRAG-bound form which actively promotes autophagy. This can be achieved by strategies including competitive displacement of the BH3-domain of Beclin 1 from Bcl-2 by BH3-mimetics, or by promoting JNK activation. Alternatively, agents that disrupt the dimerization interface of Beclin 1 or stabilize its monomeric form might favor Vps34 binding and activation of autophagy.
mTOR and the signaling pathways which regulate its activity
One of the major regulators of autophagy is the target of rapamycin (TOR) which sends the main inhibitory signals to autophagy when nutrients and energy are prevalent. TOR, a conserved serine/threonine kinase, acts as a sensor of growth factors, nutrients, and energy-availability and its activation is associated with protein synthesis, cell growth, and with the inhibition of autophagy [72, 73]. TOR exists in two distinct complexes, TORC1 and TORC2 that are conserved from yeasts to mammals. Mammalian TORC1 (mTORC1) contains GβL and raptor while mTORC2 contains GβL, rictor, SIN1, and protor [74]. However, only TORC1 is sensitive to inhibition by rapamycin [74]. The two central substrates of mTORC1 are 4E-BP1 and p70S6K which link mTOR to the control of protein synthesis. Phosphorylation of 4E-BP1 leads to its dissociation from the RNA cap-binding protein eIF4E. This allows the assembly of the eIF4F complex that enhances cap-dependent mRNA translation [75]. Phopshorylation and activation of p70S6K leads to increased translation of TOP mRNAs that encode components of the translational apparatus such as ribosomal proteins and elongation factors [76, 77]. An example for a p70S6K target involved in translation is eEF-2 kinase. eEF-2 kinase phosphorylates eEF-2 and this blocks the elongation step of translation. Phosphorylation of eEF-2 kinase by p70S6K inhibits its activity, therefore preventing eEF-2 phosphorylation. This removes the block on eEF-2 and promotes protein translation [78].
mTOR activity is directly stimulated by Rheb, a small GTP-binding protein which binds to and activates mTOR in its GTP-bound form [79]. The tuberous sclerosis complex (TSC)1/TSC2 heterodimer acts as the GTPase-activating protein for Rheb, thus negatively regulating Rheb and mTOR activity [80]. The TSC1/TSC2 complex which integrates input from numerous upstream signals plays a central role in the control of mTOR signaling (Fig. 1). Phosphorylation by AKT, ERK, or RSK, all of which are stimulated by growth factors, inactivates the TSC1/TSC2 complex and allows Rheb activation of mTOR [79, 81, 82]. In contrast, phosphorylation by AMP-activated kinase (AMPK), which is activated under nutrient and energy depletion by high AMP/ATP ratios, activates the TSC1/TSC2 complex and leads to inactivation of mTOR [83]. Notably, TSC1 and TSC2 act as tumor-suppressors and mutations in these genes lead to development of the tuberous sclerosis syndrome, a pathogenesis characterized by the formation of benign tumors in various organs [84].
The inhibitory role of TOR in regulation of autophagy is conserved from yeast to humans. In yeast, under conditions that block TOR activation such as nutrient deprivation or rapamycin treatment, dephosphorylated Atg13 binds Atg1 and autophagy is induced. The precise manner by which mTOR inhibits autophagy in mammalian cells needs to be further elucidated. S6K has also been suggested to be involved in regulation of autophagy by TOR through feedback inhibition of class I PI3K-dependent insulin signaling pathway [85, 86]. As was described above, class I PI3K signaling activates mTOR and thus inhibits autophagy. Notably, class I PI3K, as well as its downstream targets AKT, ERK, and RSK1, all of which activate mTOR, are all oncogenes associated with aberrant uncontrolled cell growth [87]. On the other hand, PTEN, a protein and phospholipid phosphatase that negatively regulates PI3K signaling, is a known tumor-suppressor that is subject to deletions and mutations in numerous malignancies [88]. In summary, numerous signaling molecules that control mTOR activation have been implicated in tumor development. Whilst each of these molecules has multiple cellular targets, it is plausible that modulation of autophagy through mTOR regulation contributes to their oncogenic or tumor-suppressive properties. Implications of mTOR-targeted cancer therapy are further discussed below.
p53
Nearly 50% of all human malignancies harbor deletions or mutations of p53. As a pivotal regulator of the cell cycle checkpoints and an inducer of cell cycle arrest, senescence and apoptosis, p53 is the best-characterized tumor-suppressor to date. Recently, autophagy was added to the list of p53-regulated events. Interestingly, different reports describe p53 as either a positive or a negative regulator of autophagy. This section will summarize the main findings thus far and allude to the paradox of p53’s opposing roles in autophagy.
A role for p53 as a positive regulator of autophagy was described upon exposure of cells to genotoxic stress. Activation of p53 following treatment with the DNA-damaging agent etoposide led to inhibition of mTOR and resulted in the induction of autophagy. mTOR inhibition was dependent on AMPK activation by p53 and was mediated by TSC1 and TSC2 since their deletion abrogated p53’s ability to inhibit mTOR [89]. In addition to DNA-damage, oncogenic stress as recapitulated by overexpression of p19ARF also provided a trigger for p53-induced autophagy [90]. Interestingly, p53-induced autophagy is mediated not only by the inhibition of mTOR but also through p53’s transcriptional activity. Under genotoxic stress, p53 was shown to upregulate the transcription of DRAM (damage-regulated modulator of autophagy) [91]. DRAM, a 238 a.a. protein, highly conserved in higher eukaryotes, is localized to the lysosomal membrane. Knock-down of DRAM expression promoted survival after exposure to DNA-damage and DRAM was shown to be required for p53-induced autophagy and cell death. Knock-down of Atg5 expression inhibited this effect indicating that the DRAM-mediated p53-induced cell death involves the autophagic machinery. Interestingly, DRAM was found to be down-regulated in squamous cancers suggesting a role for DRAM as a tumor-suppressor gene [91].
In contrast to the abovementioned, loss of p53 function (by pharmacological inhibition or deletion) can trigger the onset of autophagy as well [92]. The lack of p53 expression alone was sufficient to induce high levels of basal autophagy. This enhancement of basal autophagy could not, however, be further augmented by different stimulators of autophagy such as nutrient starvation, rapamycin or ER-stress [93]. Interestingly, cytoplasmic localization of p53 mediates its inhibitory function towards autophagy. Restoration of p53 expression in p53−/− cells with mutants that are restricted to the cytoplasm (lacking the nuclear import sequence) effectively inhibited the autophagic response initiated by the loss of p53. In contrast, nuclear mutants of p53 (lacking the nuclear export sequence) failed to block autophagy. Therefore, regulation of autophagy by p53 is tightly regulated at the level of p53 localization with a nuclear localization favoring induction of autophagy while a cytoplasmic localization serves to hinder basal autophagy. Remarkably, several inducers of autophagy (starvation, rapamycin and ER-stress) were demonstrated to induce MDM2-dependent degradation of p53 [92]. Thus, p53 not only regulates autophagy but is also regulated by it at the level of protein stability.
It is a well-established notion that inactivation of p53 is advantageous for cancer cell survival. How then can the enigma of the dual role of p53 in autophagy regulation be explained? A suggestion might be that it is the particular cellular environment and specific stress that a cell is exposed to, or the precise stage in tumor formation, that dictates the final outcome. In early stages of tumorigenesis, genotoxic insults that activate autophagy through p53 could possibly be part of the “gate-keeping” function of p53. In such scenarios, autophagy may act as a cell death mechanism aimed at eliminating defective cells. However, once a tumor is formed, inactivation of p53 by deletion or mutation, or degradation of p53 following nutrient deprivation inside the tumor mass, might activate autophagy as an energy providing mechanism. In this case, autophagy might provide a survival advantage by continuously supplying energy to the tumor cells. Indeed, p53 null cells were shown to maintain ATP levels even when nutrients were withdrawn. Furthermore, cell viability under those conditions was shown to require the autophagic system [92].
Death associated protein kinase (DAPk)
Death associated protein kinase (DAPk), a calcium-calmodulin activated serine/threonine kinase, is a tumor-suppressor whose expression is lost in numerous human malignancies including B and T cell lymphomas, breast cancer, lung carcinoma, head and neck cancer, gastric cancer, cervical cancer, and prostate cancer [94, 95]. Loss of DAPk expression is principally due to epigenetic silencing by promoter hyper-methylation. In addition, a germline mutation in DAPk was found in cases of familial chronic lymphocytic leukemia (CLL), where a single nucleotide exchange increases the binding affinity of the HOXB7 transcription factor to the DAPk promoter, leading to silencing of the gene and predisposition to CLL [96]. Notably, loss of expression of DAPk is now being used as a diagnostic tool for cancer detection in tumor samples. DAPk functions at several stages of tumor development. At the early stages of tumorigenesis, DAPk blocks oncogenic transformation by activating a p53/p19ARF-dependent apoptotic checkpoint [97]. DAPk activation was shown in this context to increase p53 expression and transcriptional activity, leading to caspase-dependent cell death. Another major step in tumorigenesis that is inhibited by DAPk is tumor metastasis. Injection to mice of highly metastatic lung carcinoma cells, in which the DAPk gene was not expressed, resulted in excessive formation of metastases. However, reintroduction of DAPk in physiological expression levels in these cells significantly reduced metastases formation in the injected mice. This was by and large due to the sensitization of the cells to various death stimuli and to the induction of anoikis (cell death induced by detachment from the matrix) [98]. In fact, adhesion signaling and migratory cues are tightly regulated by DAPk. Activation of integrins and integrin-mediated survival signaling are suppressed by DAPk, which promotes p53-dependent apoptosis upon loss of cell adhesion [99]. Moreover, DAPk blocks tumor cell migration and invasion, both being critical steps in the process of metastasis. By interfering with the association of talin with the beta-integrin tail, DAPk prevents CDC42 activation and thus inhibits cell polarization and migration. Interestingly, this inhibitory effect on cell migration and invasion is independent of the apoptotic function of DAPk and is observed even in p53-deficient cells. Thus, the multifaceted nature of DAPk activity contributes to its competent tumor-suppressive function.
DAPk was originally identified as a gene involved in IFN-γ-induced cell death in HeLa cervical cancer cells [100]. Interestingly, IFN-γ induced caspase-independent cell death in these cells that was reminiscent of autophagy. Moreover, overexpression of DAPk was by itself sufficient to induce autophagosome accumulation [101]. Interestingly, DAPk was shown to also act as an inducer of caspase-dependent apoptotic cell death in different cellular scenarios [102] as well as of programmed necrosis through the phosphorylation of PKD and activation of JNK [103]. The involvement of DAPk in the regulation of different programs of cell death raised the intriguing possibility that DAPk functions at the cross road of necrosis, apoptosis and autophagy, switching between or integrating these pathways under specific settings. In this regard, it was recently shown that DAPk is activated under ER-stress leading to both apoptotic and autophagic death in the same cell [104]. The fact that DAPk is an integrator of various stimuli that induces distinct cell fates suggests that more than a single mechanism would account for its mode of action. A clue to one possible mechanism by which DAPk induces autophagy arose from a screen of peptide libraries aimed at isolating novel proteins which bind to the kinase domain of DAPk [105]. This screen identified the interaction between DAPk and MAP1B, a protein mainly implicated in neuronal differentiation and neurite growth. Interestingly, MAP1B interacts with high affinity with LC3 and phosphorylated MAP1B associates with autophagosomes [106]. Although, as to date, MAP1B was not recognized as a substrate of DAPk, their interaction was suggested to regulate membrane blebbing during a caspase-independent death process [105]. Further studies are required to delineate the downstream signals through which DAPk induces autophagy and to decipher the regulation of the switch between apoptosis and autophagy.
Further substantiation of DAPk as a significant regulator of autophagy came from studies in C. elegans which investigated autophagy at the multicellular organism level. In C. elegans, starvation induces autophagy that, depending on its level, has opposing roles promoting either survival or death of the organism. During starvation, the muscarinic acetylcholine pathway plays a central role in the induction of autophagy in the pharyngeal muscle whose activity is critical for the recovery from starvation. Remarkably, DAPk was identified as a mediator of starvation-induced autophagy in C. elegans [107]. Over-activation of the muscarinic signaling pathway causes excessive autophagy in the pharyngeal muscle and leads to death of the organism, presumably by malfunction of the muscle. Mutation in DAPk or knock-down of its expression partially rescued the phenotype of muscarinic signaling over-activation by reducing autophagy and promoting survival of the animal. Whether DAPk acts downstream or in parallel to muscarinic signaling remains to be elucidated in future studies.
The paradox of autophagy and its implications in suppressing/activating tumor development
Studying autophagy presents a paradox; it is a homeostatic cyto-protective mechanism that under metabolic stress promotes survival but can nevertheless, under certain settings, act as a program of caspase-independent cell death (Fig. 2). This section will focus on the ambiguity of the autophagic function and its relation to other cell death programs, and will consider its implications in suppressing/activating tumor development.

The paradox of autophagy. A scheme representing the different links between autophagy and tumorigenesis. Autophagy can act either as a tumor-promoting or tumor-suppressing mechanism. Induction of autophagy by oncogenes and under metabolic stress serves to fulfill the cell’s energy requirement and hence maintain viability and induce tumor formation. On the other hand, autophagy suppresses tumorigenesis by blocking angiogenesis and necrotic inflammation, which promote tumor growth. Autophagy is also important for tumor-suppression through immuno-surveillance by mediating tumor antigen presentation. Finally, sustained autophagy leads to cell death
The cross talk between autophagy and apoptosis
Many lines of evidence now indicate that common death stimuli, previously attributed to the induction of apoptosis solely, trigger autophagy as well. Examples of these include etoposide, an inducer of p53-dependent apoptosis, that was shown to induce autophagy in mouse embryonic fibroblasts in a p53-dependent manner [89]; Ligation of the TRAIL receptor-2 by human scFv antibody triggered autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells [108]; smARF, a short, mitochondrial isoform of the ARF tumor-suppressor, induces autophagy and cell death that is blocked by Beclin 1 or Atg5 RNAi [109]; the cellular redox state also effects autophagy as reactive oxygen species regulate the activity of Atg4 through oxidation and thus induce autophagy [110]. Transcriptional positive regulation of autophagy by an accepted apoptotic regulator was also described. The E2F1 transcription factor was shown to induce autophagy by upregulating the expression of four different autophagy genes LC3, Atg1, Atg5, and DRAM [111]. In conclusion, it is now established that various acknowledged stimulators of cell death entail the autophagic machinery.
Another layer of complexity stems from the finding that there exists a cross talk between autophagy and apoptosis. Moreover, this cross talk varies from positive to negative feedbacks in different scenarios. Under some circumstances, autophagy contributes to apoptosis and consequently augments caspase-dependent cell death. For example, Atg5 was shown to play a crucial role in IFN-γ-induced cell death by interacting with FADD [112]. Additionally, activation of autophagy by TNFα in Erwing sarcoma cells, in which NF-κB (which activates mTOR) was blocked, was required for induction of apoptosis and cell death [113]. On the other hand, under certain settings autophagy is activated only when apoptosis is blocked, indicative of a negative feedback between the two processes. A switch to autophagy when apoptosis is inactivated could presumably provide a cellular back-up mechanism for inducing cell death. For example, such a negative feedback was observed under caspase-8 inhibition which induced autophagic cell death that was dependent on activation of the receptor-interacting protein (a serine-threonine kinase) and JNK [114]. In a different case, treatment with etoposide, thapsigargin (inducer of ER-stress), or staurosporine induced autophagy and cell death in Bax/Bak double knock-out fibroblasts which are apoptosis-deficient. Notably, cell death was inhibited when autophagy was blocked either by Atg5 knock-down, Beclin 1 knock-out, or pharmacological inhibition of PI3 K activity with 3-MA, indicating that autophagy was required for cell death in these cells [115]. Likewise, lipopolysaccharides (LPS) triggered macrophage cell death in the presence of the caspase inhibitor zVAD, which showed features of autophagy and was inhibited by Beclin 1 RNAi and chemical inhibitors of autophagy [116]. Caspase inhibition was also shown to trigger autophagy-mediated selective degradation of the ROS scavenger catalase, leading to accumulation of reactive oxygen species and cell death, which was inhibited by Atg7 or Atg8 knock-down [117].
In vivo documentation of autophagic cell death
Notably, although autophagic cell death was demonstrated mostly in studies performed in cell lines, a physiological role for the importance of this program of cell death has been demonstrated as well. In drosophila, autophagic cell death is essential during the development of the salivary gland [57]. Massive cell death induced by autophagy is also associated with progression of neurodegenerative diseases such as Alzheimer, Parkinson, and others [118–121].
Autophagy in tumor development
Of importance is the understanding of the circumstances under which autophagy promotes either cell death or cell survival in the context of tumorigenesis, as this may have implications in cancer therapy. For the most part, autophagy has been shown to suppress tumor growth. Accordingly, and as previously discussed, various tumor-suppressors (e.g., PTEN, TSC1-TSC2, p53, and DAPk) are inducers of autophagy, while some inhibitors of autophagy (e.g., Akt, Ras) possess oncogenic activity. In contrast, there is also evidence that autophagy can positively contribute to tumor-progression. This discrepancy might be explained by the dual-functionality of autophagy, which depends on both the cellular and extra-cellular settings, as will be discussed below. Thus, deciphering this controversy demands the investigation of autophagy in each cellular scenario individually.
During tumor development, transformed cells undergo a sequence of events that include intra-cellular alterations as well as changes in their microenvironment. These changes seem to be the decisive factors in the autophagy-induced cell fate. One of the first stages in tumorigenesis is oncogenic transformation, which often corresponds to aberrant growth factor signaling and constitutive PI3K/AKT/mTOR activation. Such conditions, in which protein synthesis and proliferation are enhanced, create an increased energy demand in the cell. However, as autophagy, which is the main alternative source for energy, is blocked by the constitutive activation of mTOR, transformed cells often reach the state of metabolic catastrophe. Additionally, as the transformed cells grow and form a solid tumor, nutrients become unavailable to cells inside the tumor mass that then become metabolically stressed. Only in later stages of tumor-progression angiogenesis is completed and sufficient blood supply reaches the tumor cells. Interestingly, it was shown that the metabolically stressed regions of the tumor mass, but not the vascularized ones, activate the autophagic machinery [122, 123]. Similarly, 3D morphogenesis assays of mammary epithelial cells showed that autophagy is activated only in the central acinar cells, which are under increased metabolic stress [123]. Therefore, autophagy is induced in transformed cells undergoing metabolic stress, a feature commonly associated with human solid tumors [124]. Activation of autophagy might confer a growth advantage to these cells through the restoration of energy levels, in agreement with a role for autophagy in promoting tumor survival.
In contrast to its suggested role in promoting the survival of metabolically stressed tumor cells, autophagy has also been widely implicated in suppression of tumorigenesis. In apoptosis-deficient cells, inactivation of the autophagic machinery, coincidently with metabolic stress, results in the induction of necrotic cell death [122]. During necrosis, cells release pro-inflammatory factors such as the high mobility group B1 (HMGB1) protein to the extra-cellular milieu. These factors recruit macrophages, which produce cytokines and chemokines that impact proliferation and angiogenesis at the site of the inflammatory infiltrate. For simplification, this could be analogous to the process of wound healing, where inflammatory factors induce cell proliferation. Thus, the initiation of an inflammatory response to stress-mediated tumor necrosis may act to stimulate angiogenesis and promote tumor growth. Such a phenomenon was demonstrated in apoptosis-deficient cells where inactivation of autophagy-induced necrosis that stimulated an inflammatory response and was associated with tumor growth [122]. Thus, in the case of apoptosis-deficient cells, induction of autophagy during metabolic stress suppresses necrosis and acts as a tumor-suppressive mechanism. It should be noted, however, that stimulation of an immunogenic response can also help remove stressed cells, and cancer cells often evade this step leading to their uncontrolled proliferation. Antigen presentation by tumor cells enables the immune system to recognize and eliminate primary tumors in a process known as immunosurveillance [125]. The first connection between autophagy and the immune system came from reports showing that autophagy can be regulated by different immune factors such as IFN-γ, which positively regulates autophagy [101, 126] and IL-13, which inhibits autophagy through class I PI3K [127]. Autophagy was also shown to promote MHC class II presentation of peptides [128]. Additionally, recent findings suggest a role of autophagy in tumor antigen presentation and autophagosomes were suggested to act as antigen carriers for cross-presentation in vitro. Moreover, it was demonstrated in melanoma tumor cells that autophagy is essential for cross-presentation in vivo [129]. The involvement of autophagy in the acquisition of adaptive immunity against tumors could present new approaches in development of anti-cancer vaccines.
An additional characteristic of autophagy in tumor-suppression is the maintenance of genome integrity. Under metabolic stress, impairment of the autophagic machinery resulted in genomic instability that included DNA double-strand breaks, centrosome abnormalities, and increased DNA content [130]. Gene amplification is the major mechanism of oncogene activation in cellular transformation. Enhanced chromosomal instability and gene amplification was observed in Beclin 1 heterozygote cells overexpressing Bcl-2 [130]. The increased gene amplification and mutation rates enables these cells to overcome the loss of autophagy-mediated survival under metabolic stress. Similar findings were also demonstrated in vivo where autophagy was induced in metabolically-stressed regions of mice tumors. Allelic loss of Beclin 1 resulted in enhanced genomic damage in these mice [123]. Genome integrity might be maintained via autophagy-mediated elimination of damaged organelles and proteins. Depolarized mitochondria are a source for genotoxic free radicals. In the absence of autophagy as a scavenger mechanism, DNA mutations accumulate readily and may lead to cellular transformation. Of note, the susceptibility of autophagy-compromised cancer cells to DNA-damage in response to metabolic stress might be of therapeutic significance. Pharmacologic depletion of pyrimidines with the chemotherapeutic agent PALA, which depletes pyrimidine nucleotide (dNTP) pools, triggers the onset of autophagy that is required to limit gene amplification, suggesting that autophagy-deficient cells may be particularly prone to this specific metabolic stress [130].
Autophagy also regulates angiogenesis through inhibition of vascular endothelial growth factor (VEGF) signaling. In response to metabolic stress, the pro-angiogenic protein neuropilin 1, which binds VEGF in both endothelial and carcinoma cells, is rapidly degraded by autophagy [131]. mTOR-inhibitors were also demonstrated to effectively block angiogenesis in vivo in correlation with induction of autophagy [132, 133]. Therefore, stimulating autophagy can inhibit tumor-progression by blocking angiogenesis. However, under certain circumstances autophagy may have an opposite effect. Autophagy was induced by the anti-angiogenic drug kringle 5 in human endothelial cells. However, blocking autophagy enhanced the anti-angiogenic activity of kringle 5 by inducing apoptotic cell death of the endothel [134].
In summary, autophagy possesses both tumor-promoting and tumor-suppressive properties (Fig. 2). It is the specific intra- and extra-cellular context that determines in which cell fate induction of autophagy will culminate. Exploitation of autophagy for anti-cancerous purposes should take into account such variables including the stage in tumorigenesis, the integrity of the apoptotic machinery in the tumor, and the consequences of a necrotic inflammatory response. Some examples of how autophagy is manipulated in cancer therapy are discussed in the following section.
Autophagy-inducing drugs in cancer therapy
An emerging difficulty in the treatment of cancer patients is the development of tumor resistance to radiation and drug therapy. In particular, various malignant cells acquire mutations and/or deletions in genes that are crucial for the induction of apoptosis, thus rendering them incapable of executing apoptotic cell death. Such tumors display resistance to apoptosis-inducing drugs and are challenging to treat. Unfortunately, these tumors also often correlate with poor prognosis. The increasing amount of data that have accumulated in the last years signifying autophagy as a mechanism of programmed cell death presents new opportunities for developing alternative anti-cancer therapies. Accordingly, activation of autophagy in apoptosis-resistant tumors, possibly in combination with inhibitors of necrosis, could provide a way to induce cell death and impede tumor growth. Indeed, several autophagy-inducing drugs are already being used in the clinic for the treatment of different malignancies and numerous other compounds are now in clinical trials. The following section describes some of the interesting examples of autophagy-inducing drugs in the treatment of malignancy.
Arsenic trioxide (As2O3)
Prevalently used in the clinic for the treatment of hematological malignancies, arsenic trioxide is a powerful therapeutic tool that induces high rates of complete remission with no reported severe side effects [135]. The cytotoxic effects of arsenic trioxide have been attributed to the induction of apoptosis following cytochrome c release and caspase activation. However, recently, treatment of human T-lymphocytic leukemia cells with arsenic trioxide was shown to cause cytotoxicity through induction of autophagy as well [136]. A remarkable feature of arsenic trioxide is its success in the treatment of malignant glioma cells, which in contrast to hematological malignancies display resistance to many types of therapy including radiation, chemotherapy and a range of drugs. Interestingly, arsenic trioxide was shown to be effective in the growth inhibition and induction of cell death in malignant glioma cells through activation of autophagy. Treatment with clinically used low doses of arsenic trioxide (2 μM) resulted in the formation of autophagic vacuoles and autophagy was shown to be the cause of cell death in these cells [137]. The Bcl-2 family member, Bcl-2-adenovirus E1B 19-kDa-interacting protein 3 (BNIP3), plays a major role in arsenic trioxide-induce autophagic cell death in malignant glioma cells. BNIP3 expression is upregulated following arsenic trioxide treatment and it is required for the induction of autophagy and cell death in these settings [138]. Interestingly, when autophagy was inhibited by Bafilomycin A1, apoptosis was alternatively induced by arsenic trioxide [137], suggestive of a complex cross-regulation of the apoptotic and autophagic networks. In conclusion, induction of autophagic cell death by arsenic trioxide provides promising new opportunities for the treatment of resistant malignant gliomas and is now in clinical trials.
Imatinib (Gleevac)
The competitive Bcr-Abl tyrosine kinase inhibitor imatinib is widely used for the treatment of chronic myelogenous leukemia (CML) as well as other malignancies. Recent data suggest that imatinib induces autophagy in human cell lines as part of its mode of action [139]. Administration of imatinib to multi-drug-resistant Kaposi’s sarcoma cells also resulted in the induction of autophagy and reduced survival of these drug resistant cells [140]. In rat C6 chemo-resistant glioma cells, combinatorial treatment of imatinib with Anafranil, an anti-depressant shown to have anti-neoplastic activity, significantly induced apoptosis and autophagy in a synergistic effect and resulted in growth inhibition and cell death [141]. Additionally, imatinib was found to be effective in the treatment of glioblastomas in several clinical trials.
Histone deacetylase inhibitor (HDAC) suberoylanilide hydroxamic acid (SAHA)
HDAC inhibitors, such as SAHA, demonstrate an anti-proliferative function and lead to growth arrest and cell death in various transformed cells both in vitro and in vivo. Through the regulation of histone acetylation, HDAC’s influence multiple targets and have an effect on cell fate. SAHA treatment, currently in clinical trials, proves to be effective in the treatment of both hematologic and solid tumors [142]. Evidence shows that SAHA-induced cell death in Hela cells is caused by the induction of autophagy and is independent of caspase activation and apoptosis [143]. In addition, SAHA treatment successfully inhibited growth of chondrosarcoma cells that show high resistance to conventional therapy and is usually efficiently treated only by surgical procedures [144]. Activation of autophagy by SAHA in these cells was shown to induce cell death and remarkably resulted in inhibition of tumor growth in mice xenograft models [144]. Thus, initiation of autophagic cell death by SAHA has clear therapeutic implications for apoptosis-defective tumors.
SAHA treatment has also been shown to be effective in the treatment of imatinib-resistant primary cells from CML patients bearing mutations in Bcr-Abl that correlate with a poor clinical prognosis [145]. However, contrary to the aforementioned, in this case the anticancer activity of SAHA was enhanced when autophagy was blocked with 3-MA and chloroquine. Importantly, p53 is not required for the induction of cell death and therefore this combinatorial treatment could be a promising for the treatment of refractory CML with p53 alterations [145].
EB1089 (vitamin D3 analog)
The vitamin D3 analog EB1089 is effective in the growth arrest and induction of cell death in various malignant cells [146]. Cell death following EB1089 administration in breast cancer cells is associated with autophagy. The monoallelic deletion of Beclin 1 in these cells confers partial resistance to EB1089 and restoration of Beclin 1 expression to normal levels sensitizes to EB1089 and reduces tumor growth [147]. EB1089 also potentiated the response of breast cancer cells to irradiation and led to the induction of autophagic cell death [148]. Vitamin D3 analogs are now in clinical trials for the treatment of various malignancies.
mTOR-inhibitors
Rapamycin, a macrolide antibiotic produced by Streptomyces hygroscopicus, was initially approved by the Food and Drug Administration as an immunosuppressant in 1999. Rapamycin binds the 12 kDa immunophilin FK506-binding protein (FKBP12) and inhibits the mTORC1 complex. As mTOR is a major regulator of cell growth that has been implicated in tumorigenesis, rapamycin treatment has anti-cancerous properties and is now in advanced clinical trials for the treatment of various malignancies. Rapamycin derivatives, CCI-779 and RAD001, also demonstrate promising results in advanced clinical trials and hold the advantage of showing no immunosuppressive effects at intermittent dosing. The anti-cancer properties of rapamycin were initially attributed to the downregulation of AKT signaling. Nonetheless, a growing collection of evidence suggests that tumor-suppression following rapamycin administration is associated with the induction of autophagic cell death.
Here are some examples linking efficient anti-cancer intervention by mTOR-inhibitors to autophagy. mTOR-inhibitors sensitize various tumor cells to radiation therapy. Combined treatment of RAD001 with the caspase-3 inhibitor DEVD radiosensitized non-small cell lung cancer cells in mice models and resulted in enhanced cytotoxicity and delayed tumor growth. The increased cytotoxicity was linked to the induction of autophagy [133]. Autophagy was required for susceptibility to cell death in irradiated breast and prostate cancer cells after rapamycin and RAD001 treatment, respectively [149, 150]. Treatment of mantle cell lymphoma with CCI-779 and RAD001 induced tumor remission with a 30–40% response rate [151–153]. Likewise, the efficacy of rapamycin and its analogs in inhibiting cell growth and inducing cell death was demonstrated in mantle cell lymphoma cell lines and primary tumor cells [154]. These studies demonstrated an apoptosis-independent cell death mechanism in response to mTOR-inhibition and induction of autophagy was demonstrated in CCI-779-treated cells [155]. Efficient cytotoxicity caused by mTOR-inhibitors was also demonstrated in malignant gliomas and correlated with induction of autophagy [156, 157]. Interestingly, inhibiting mTOR activity with these agents has also been shown to effect tumor vasculature. Block of angiogenesis and enhanced radiosensitization was observed in malignant glioma cells treated with rapamycin and RAD001 [132], and in apoptosis-inhibited lung cancer cells as well as lung cancer xenograft model with the combination of Z-DEVD and RAD001 [133]. Thus, mTOR-inhibitors could be used as anti-angiogenic drugs when combined with radiation.
In conclusion, inhibition of mTOR by rapamycin or its derivatives provides a powerful therapeutic tool for the treatment of various malignancies. The recognition that autophagy has a prominent role in the cytotoxic effects of these compounds is therapeutically beneficial and could lead to the development of new combinatorial treatments whereby autophagy will be specifically induced to fight tumorigenesis.
Conclusions and perspectives
Exploitation of autophagy for cancer treatment presents novel therapeutic opportunities. However, the multifaceted nature of autophagy and its diverse cross talk with other programs of cell death must be concerned when targeting the autophagic system. As the outcome of inducing autophagy varies from conferring a growth advantage up to promoting cell death, and as the function of autophagy is determined by the complete genetic makeup of every cell as well as by the environmental cues a cell is exposed to, no simple rule can be drawn as to the outcome of targeting autophagy. Thus, identification of a “fingerprint”, which includes these parameters, for every cancer type is essential for selecting an effective treatment. Moreover, with various autophagy-inducing drugs, such as those discussed above, already in clinical use, the main challenge today is to determine when to apply each drug, depending on the biological understanding of the status of the cancer cells. Importantly, precaution must be taken in application of these drugs as they may have multiple effects other than autophagy. In the last years the field of autophagy has expanded greatly and with it our understanding of this intricate process. Still, further studies, which will investigate the manipulation of autophagy in vivo, specifically in mammals, are necessary for projecting the available knowledge onto the clinic. As key findings continuously arise, it is anticipated that autophagy will be increasingly employed in fighting tumorigenesis.
Abbreviations
- Atg:
-
Autophagy related genes
- PI3K:
-
Phosphatidylinositol 3-kinase
- TOR:
-
Target of rapamycin
- PAS:
-
Pre-autophagosomal structure
- DAPk:
-
Death associated protein kinase
References
Huang WP, Klionsky DJ (2002) Autophagy in yeast: a review of the molecular machinery. Cell Struct Funct 27:409–420. doi:10.1247/csf.27.409
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor-suppressor. Proc Natl Acad Sci USA 100:15077–15082. doi:10.1073/pnas.2436255100
Kuma A, Hatano M, Matsui M et al (2004) The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036. doi:10.1038/nature03029
Komatsu M, Waguri S, Chiba T et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. doi:10.1038/nature04723
Hara T, Nakamura K, Matsui M et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. doi:10.1038/nature04724
Orvedahl A, Levine B (2008) Eating the enemy within: autophagy in infectious diseases. Cell Death Differ 16(1):57–69
Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L (2008) Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455(7211):396–400
Lowe SW, Lin AW (2000) Apoptosis in cancer. Carcinogenesis 21:485–495. doi:10.1093/carcin/21.3.485
Tsukada M, Ohsumi Y (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 333:169–174. doi:10.1016/0014-5793(93)80398-E
Kihara A, Noda T, Ishihara N, Ohsumi Y (2001) Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 152:519–530. doi:10.1083/jcb.152.3.519
Stack JH, DeWald DB, Takegawa K, Emr SD (1995) Vesicle-mediated protein transport: regulatory interactions between the Vps15 protein kinase and the Vps34 PtdIns 3-kinase essential for protein sorting to the vacuole in yeast. J Cell Biol 129:321–334. doi:10.1083/jcb.129.2.321
Liang C, Feng P, Ku B et al (2006) Autophagic and tumour suppressor activity of a novel Beclin 1-binding protein UVRAG. Nat Cell Biol 8:688–699. doi:10.1038/ncb1426
Takahashi Y, Coppola D, Matsushita N et al (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9:1142–1151. doi:10.1038/ncb1634
Obara K, Sekito T, Ohsumi Y (2006) Assortment of phosphatidylinositol 3-kinase complexes—Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell 17:1527–1539. doi:10.1091/mbc.E05-09-0841
Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19(12):5360–5372
Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T (2001) Beclin-phosphatidylinositol 3-kinase complex functions at the trans-golgi network. EMBO Rep 2:330–335. doi:10.1093/embo-reports/kve061
Suzuki K, Kubota Y, Sekito T, Ohsumi Y (2007) Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12:209–218. doi:10.1111/j.1365-2443.2007.01050.x
Obara K, Sekito T, Niimi K, Ohsumi Y (2008) The Atg18–Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem 283:23972–23980. doi:10.1074/jbc.M803180200
Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y (2000) Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150:1507–1513. doi:10.1083/jcb.150.6.1507
Scott SV, Nice DC 3rd, Nau JJ et al (2000) Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J Biol Chem 275:25840–25849. doi:10.1074/jbc.M002813200
Mizushima N, Noda T, Yoshimori T et al (1998) A protein conjugation system essential for autophagy. Nature 395:395–398. doi:10.1038/26506
Mizushima N, Sugita H, Yoshimori T, Ohsumi Y (1998) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 273:33889–33892. doi:10.1074/jbc.273.51.33889
Tanida I, Mizushima N, Kiyooka M et al (1999) Apg7p/Cvt2p: a novel protein-activating enzyme essential for autophagy. Mol Biol Cell 10:1367–1379
Shintani T, Mizushima N, Ogawa Y, Matsuura A, Noda T, Ohsumi Y (1999) Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J 18:5234–5241. doi:10.1093/emboj/18.19.5234
Ichimura Y, Kirisako T, Takao T et al (2000) A ubiquitin-like system mediates protein lipidation. Nature 408:488–492. doi:10.1038/35044114
Mizushima N, Yamamoto A, Hatano M et al (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152:657–668. doi:10.1083/jcb.152.4.657
Mizushima N, Kuma A, Kobayashi Y et al (2003) Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12–Apg5 conjugate. J Cell Sci 116:1679–1688. doi:10.1242/jcs.00381
Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19:2092–2100. doi:10.1091/mbc.E07-12-1257
Hanada T, Noda NN, Satomi Y et al (2007) The Atg12–Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 282:37298–37302. doi:10.1074/jbc.C700195200
Kirisako T, Ichimura Y, Okada H et al (2000) The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 151:263–276. doi:10.1083/jcb.151.2.263
Nakatogawa H, Ichimura Y, Ohsumi Y (2007) Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130:165–178. doi:10.1016/j.cell.2007.05.021
Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9:1102–1109. doi:10.1038/ncb1007-1102
Kirisako T, Baba M, Ishihara N et al (1999) Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol 147:435–446. doi:10.1083/jcb.147.2.435
Darsow T, Rieder SE, Emr SD (1997) A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J Cell Biol 138:517–529. doi:10.1083/jcb.138.3.517
Ishihara N, Hamasaki M, Yokota S et al (2001) Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell 12:3690–3702
Jager S, Bucci C, Tanida I et al (2004) Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117:4837–4848. doi:10.1242/jcs.01370
Gutierrez MG, Munafo DB, Beron W, Colombo MI (2004) Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci 117:2687–2697. doi:10.1242/jcs.01114
Atlashkin V, Kreykenbohm V, Eskelinen EL, Wenzel D, Fayyazi A, Fischer von Mollard G (2003) Deletion of the SNARE vti1b in mice results in the loss of a single SNARE partner, syntaxin 8. Mol Cell Biol 23:5198–5207. doi:10.1128/MCB.23.15.5198-5207.2003
Liang C, Lee JS, Inn KS et al (2008) Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10:776–787. doi:10.1038/ncb1740
Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z (2006) Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem 281:36303–36316. doi:10.1074/jbc.M607031200
Kochl R, Hu XW, Chan EY, Tooze SA (2006) Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 7:129–145. doi:10.1111/j.1600-0854.2005.00368.x
Jahreiss L, Menzies FM, Rubinsztein DC (2008) The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 9:574–587. doi:10.1111/j.1600-0854.2008.00701.x
Kametaka S, Okano T, Ohsumi M, Ohsumi Y (1998) Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J Biol Chem 273:22284–22291. doi:10.1074/jbc.273.35.22284
Otto GP, Wu MY, Kazgan N, Anderson OR, Kessin RH (2004) Dictyostelium macroautophagy mutants vary in the severity of their developmental defects. J Biol Chem 279:15621–15629. doi:10.1074/jbc.M311139200
Yoshimoto K, Hanaoka H, Sato S et al (2004) Processing of ATG8s, ubiquitin-like proteins, and their deconjugation by ATG4s are essential for plant autophagy. Plant Cell 16:2967–2983. doi:10.1105/tpc.104.025395
Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301:1387–1391. doi:10.1126/science.1087782
Qu X, Yu J, Bhagat G et al (2003) Promotion of tumorigenesis by heterozygous disruption of the Beclin 1 autophagy gene. J Clin Invest 112:1809–1820
Liang XH, Jackson S, Seaman M et al (1999) Induction of autophagy and inhibition of tumorigenesis by Beclin 1. Nature 402:672–676. doi:10.1038/45257
Furuya N, Yu J, Byfield M, Pattingre S, Levine B (2005) The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor-suppressor function. Autophagy 1:46–52
Aita VM, Liang XH, Murty VV et al (1999) Cloning and genomic organization of Beclin 1, a candidate tumor-suppressor gene on chromosome 17q21. Genomics 59:59–65. doi:10.1006/geno.1999.5851
Miracco C, Cosci E, Oliveri G et al (2007) Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol 30:429–436
Bekri S, Adelaide J, Merscher S et al (1997) Detailed map of a region commonly amplified at 11q13→q14 in human breast carcinoma. Cytogenet Cell Genet 79:125–131. doi:10.1159/000134699
Goi T, Kawasaki M, Yamazaki T et al (2003) Ascending colon cancer with hepatic metastasis and cholecystolithiasis in a patient with situs inversus totalis without any expression of UVRAG mRNA: report of a case. Surg Today 33:702–706. doi:10.1007/s00595-002-2567-y
Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK (2004) Manipulation of nonsense mediated decay identifies gene mutations in colon cancer cells with microsatellite instability. Oncogene 23:639–645. doi:10.1038/sj.onc.1207178
Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ (2008) Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol 39:1059–1063. doi:10.1016/j.humpath.2007.11.013
Noble CG, Dong JM, Manser E, Song H (2008) BCL-XL and UVRAG cause a monomer-dimer switch in Beclin 1. J Biol Chem 283(38):26274–26282
Lee JW, Jeong EG, Soung YH et al (2006) Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology 38:312–315. doi:10.1080/00313020600820880
Kim SY, Oh YL, Kim KM et al (2008) Decreased expression of Bax-interacting factor-1 (Bif-1) in invasive urinary bladder and gallbladder cancers. Pathology 40:553–557. doi:10.1080/00313020802320440
Balakrishnan A, von Neuhoff N, Rudolph C et al (2006) Quantitative microsatellite analysis to delineate the commonly deleted region 1p22.3 in mantle cell lymphomas. Genes Chromosomes Cancer 45:883–892. doi:10.1002/gcc.20352
Tsujimoto Y, Cossman J, Jaffe E, Croce CM (1985) Involvement of the bcl-2 gene in human follicular lymphoma. Science 228:1440–1443. doi:10.1126/science.3874430
McDonnell TJ, Deane N, Platt FM et al (1989) Bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57:79–88. doi:10.1016/0092-8674(89)90174-8
McDonnell TJ, Korsmeyer SJ (1991) Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 349:254–256. doi:10.1038/349254a0
Saeki K, Yuo A, Okuma E et al (2000) Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ 7:1263–1269. doi:10.1038/sj.cdd.4400759
Pattingre S, Tassa A, Qu X et al (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939. doi:10.1016/j.cell.2005.07.002
Akar U, Chaves-Reyez A, Barria M et al (2008) Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 4:669–679
Maiuri MC, Le Toumelin G, Criollo A et al (2007) Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 26:2527–2539. doi:10.1038/sj.emboj.7601689
Oberstein A, Jeffrey PD, Shi Y (2007) Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem 282:13123–13132. doi:10.1074/jbc.M700492200
Feng W, Huang S, Wu H, Zhang M (2007) Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol 372:223–235. doi:10.1016/j.jmb.2007.06.069
Erlich S, Mizrachy L, Segev O et al (2007) Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3:561–568
Ku B, Woo JS, Liang C, et al (2008) Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog 4:e25. doi:10.1371/journal.ppat.0040025
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30:678–688. doi:10.1016/j.molcel.2008.06.001
Arsham AM, Neufeld TP (2006) Thinking globally and acting locally with TOR. Curr Opin Cell Biol 18:589–597. doi:10.1016/j.ceb.2006.09.005
Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17:596–603. doi:10.1016/j.ceb.2005.09.009
Loewith R, Jacinto E, Wullschleger S et al (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10:457–468. doi:10.1016/S1097-2765(02)00636-6
Gingras AC, Raught B, Sonenberg N (2001) Regulation of translation initiation by FRAP/mTOR. Genes Dev 15:807–826. doi:10.1101/gad.887201
Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G (1997) Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J 16:3693–3704. doi:10.1093/emboj/16.12.3693
Meyuhas O (2000) Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem 267:6321–6330. doi:10.1046/j.1432-1327.2000.01719.x
Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG (2001) Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20:4370–4379. doi:10.1093/emboj/20.16.4370
Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657. doi:10.1038/ncb839
Inoki K, Li Y, Xu T, Guan KL (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834. doi:10.1101/gad.1110003
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121:179–193. doi:10.1016/j.cell.2005.02.031
Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor-suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA 101:13489–13494. doi:10.1073/pnas.0405659101
Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL (2004) Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz–Jeghers syndrome. Genes Dev 18:1533–1538. doi:10.1101/gad.1199104
Kwiatkowski DJ, Manning BD (2005) Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 14(2):R251–R258
Codogno P, Meijer AJ (2005) Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 12(suppl 2):1509–1518. doi:10.1038/sj.cdd.4401751
Klionsky DJ, Meijer AJ, Codogno P (2005) Autophagy and p70S6 kinase. Autophagy 1:59–60 discussion 60–51
Samuels Y, Ericson K (2006) Oncogenic PI3K and its role in cancer. Curr Opin Oncol 18:77–82. doi:10.1097/01.cco.0000198021.99347.b9
Kim RH, Mak TW (2006) Tumours and tremors: how PTEN regulation underlies both. Br J Cancer 94:620–624
Feng Z, Zhang H, Levine AJ, Jin S (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 102:8204–8209. doi:10.1073/pnas.0502857102
Abida WM, Gu W (2008) p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res 68:352–357. doi:10.1158/0008-5472.CAN-07-2069
Crighton D, Wilkinson S, O’Prey J et al (2006) DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126:121–134. doi:10.1016/j.cell.2006.05.034
Tasdemir E, Maiuri MC, Galluzzi L et al (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10:676–687. doi:10.1038/ncb1730
Tasdemir E, Chiara Maiuri M, Morselli E et al (2008) A dual role of p53 in the control of autophagy. Autophagy 4:810–814
Bialik S, Kimchi A (2006) The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem 75:189–210. doi:10.1146/annurev.biochem.75.103004.142615
Bialik S, Kimchi A (2004) DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin Cancer Biol 14:283–294. doi:10.1016/j.semcancer.2004.04.008
Raval A, Tanner SM, Byrd JC et al (2007) Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 129:879–890. doi:10.1016/j.cell.2007.03.043
Raveh T, Droguett G, Horwitz MS, DePinho RA, Kimchi A (2001) DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol 3:1–7. doi:10.1038/35050500
Inbal B, Cohen O, Polak-Charcon S et al (1997) DAP kinase links the control of apoptosis to metastasis. Nature 390:180–184. doi:10.1038/36599
Wang WJ, Kuo JC, Yao CC, Chen RH (2002) DAP-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. J Cell Biol 159:169–179. doi:10.1083/jcb.200204050
Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A (1995) Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev 9:15–30. doi:10.1101/gad.9.1.15
Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A (2002) DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol 157:455–468. doi:10.1083/jcb.200109094
Shani G, Marash L, Gozuacik D et al (2004) Death-associated protein kinase phosphorylates ZIP kinase, forming a unique kinase hierarchy to activate its cell death functions. Mol Cell Biol 24:8611–8626. doi:10.1128/MCB.24.19.8611-8626.2004
Eisenberg-Lerner A, Kimchi A (2007) DAP kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death Differ 14:1908–1915. doi:10.1038/sj.cdd.4402212
Gozuacik D, Bialik S, Raveh T et al (2008) DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 15(12):1875–1886
Harrison B, Kraus M, Burch L et al (2008) DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing. J Biol Chem 283:9999–10014. doi:10.1074/jbc.M706040200
Halpain S, Dehmelt L (2006) The MAP1 family of microtubule-associated proteins. Genome Biol 7:224
Kang C, You YJ, Avery L (2007) Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev 21:2161–2171. doi:10.1101/gad.1573107
Park KJ, Lee SH, Kim TI et al (2007) A human scFv antibody against TRAIL receptor 2 induces autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells. Cancer Res 67:7327–7334. doi:10.1158/0008-5472.CAN-06-4766
Reef S, Zalckvar E, Shifman O et al (2006) A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell 22:463–475. doi:10.1016/j.molcel.2006.04.014
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26:1749–1760. doi:10.1038/sj.emboj.7601623
Polager S, Ofir M, Ginsberg D (2008) E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 27:4860–4864. doi:10.1038/onc.2008.117
Pyo JO, Jang MH, Kwon YK et al (2005) Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 280:20722–20729. doi:10.1074/jbc.M413934200
Djavaheri-Mergny M, Amelotti M, Mathieu J et al (2006) NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem 281:30373–30382. doi:10.1074/jbc.M602097200
Yu L, Alva A, Su H et al (2004) Regulation of an ATG7-Beclin 1 program of autophagic cell death by caspase-8. Science 304:1500–1502. doi:10.1126/science.1096645
Shimizu S, Kanaseki T, Mizushima N et al (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 6:1221–1228. doi:10.1038/ncb1192
Xu Y, Kim SO, Li Y, Han J (2006) Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem 281:19179–19187. doi:10.1074/jbc.M513377200
Yu L, Wan F, Dutta S et al (2006) Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 103:4952–4957. doi:10.1073/pnas.0511288103
Cataldo AM, Hamilton DJ, Nixon RA (1994) Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res 640:68–80. doi:10.1016/0006-8993(94)91858-9
Anglade P, Vyas S, Javoy-Agid F et al (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol 12:25–31
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721. doi:10.1126/science.290.5497.1717
Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S (2003) Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res 73:341–350. doi:10.1002/jnr.10663
Degenhardt K, Mathew R, Beaudoin B et al (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10:51–64. doi:10.1016/j.ccr.2006.06.001
Karantza-Wadsworth V, Patel S, Kravchuk O et al (2007) Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 21:1621–1635. doi:10.1101/gad.1565707
Folkman J (2003) Angiogenesis and apoptosis. Semin Cancer Biol 13:159–167. doi:10.1016/S1044-579X(02)00133-5
Smyth MJ, Godfrey DI, Trapani JA (2001) A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol 2:293–299. doi:10.1038/86297
Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766. doi:10.1016/j.cell.2004.11.038
Arico S, Petiot A, Bauvy C et al (2001) The tumor-suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem 276:35243–35246. doi:10.1074/jbc.C100319200
Dengjel J, Schoor O, Fischer R et al (2005) Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 102:7922–7927. doi:10.1073/pnas.0501190102
Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM (2008) Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res 68:6889–6895. doi:10.1158/0008-5472.CAN-08-0161
Mathew R, Kongara S, Beaudoin B et al (2007) Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 21:1367–1381. doi:10.1101/gad.1545107
Bae D, Lu S, Taglienti CA, Mercurio AM (2008) Metabolic stress induces the lysosomal degradation of neuropilin-1 but not neuropilin-2. J Biol Chem 283(42):28074–28080
Shinohara ET, Cao C, Niermann K et al (2005) Enhanced radiation damage of tumor vasculature by mTOR-inhibitors. Oncogene 24:5414–5422. doi:10.1038/sj.onc.1208715
Kim KW, Hwang M, Moretti L, Jaboin JJ, Cha YI, Lu B (2008) Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy 4:659–668
Nguyen TM, Subramanian IV, Kelekar A, Ramakrishnan S (2007) Kringle 5 of human plasminogen, an angiogenesis inhibitor, induces both autophagy and apoptotic death in endothelial cells. Blood 109:4793–4802. doi:10.1182/blood-2006-11-059352
Miller WH Jr, Schipper HM, Lee JS, Singer J, Waxman S (2002) Mechanisms of action of arsenic trioxide. Cancer Res 62:3893–3903
Qian W, Liu J, Jin J, Ni W, Xu W (2007) Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res 31:329–339. doi:10.1016/j.leukres.2006.06.021
Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I (2003) Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res 63:2103–2108
Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S (2005) Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 24:980–991. doi:10.1038/sj.onc.1208095
Ertmer A, Huber V, Gilch S et al (2007) The anticancer drug imatinib induces cellular autophagy. Leukemia 21:936–942
Basciani S, Vona R, Matarrese P et al (2007) Imatinib interferes with survival of multi drug resistant Kaposi’s sarcoma cells. FEBS Lett 581:5897–5903. doi:10.1016/j.febslet.2007.11.069
Bilir A, Erguven M, Oktem G et al (2008) Potentiation of cytotoxicity by combination of imatinib and chlorimipramine in glioma. Int J Oncol 32:829–839
Marks PA (2007) Discovery and development of SAHA as an anticancer agent. Oncogene 26:1351–1356. doi:10.1038/sj.onc.1210204
Shao Y, Gao Z, Marks PA, Jiang X (2004) Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA 101:18030–18035. doi:10.1073/pnas.0408345102
Yamamoto S, Tanaka K, Sakimura R et al (2008) Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res 28:1585–1591
Carew JS, Nawrocki ST, Kahue CN et al (2007) Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 110:313–322. doi:10.1182/blood-2006-10-050260
Hansen CM, Binderup L, Hamberg KJ, Carlberg C (2001) Vitamin D and cancer: effects of 1, 25(OH)2D3 and its analogs on growth control and tumorigenesis. Front Biosci 6:D820–D848. doi:10.2741/Hansen
Hoyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jaattela M (2005) Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ 12:1297–1309. doi:10.1038/sj.cdd.4401651
Demasters G, Di X, Newsham I, Shiu R, Gewirtz DA (2006) Potentiation of radiation sensitivity in breast tumor cells by the vitamin D3 analogue, EB 1089, through promotion of autophagy and interference with proliferative recovery. Mol Cancer Ther 5:2786–2797. doi:10.1158/1535-7163.MCT-06-0316
Cao C, Subhawong T, Albert JM et al (2006) Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res 66:10040–10047. doi:10.1158/0008-5472.CAN-06-0802
Paglin S, Lee NY, Nakar C et al (2005) Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res 65:11061–11070. doi:10.1158/0008-5472.CAN-05-1083
Witzig TE, Geyer SM, Ghobrial I et al (2005) Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 23:5347–5356. doi:10.1200/JCO.2005.13.466
Haritunians T, Mori A, O’Kelly J, Luong QT, Giles FJ, Koeffler HP (2007) Antiproliferative activity of RAD001 (everolimus) as a single agent and combined with other agents in mantle cell lymphoma. Leukemia 21:333–339. doi:10.1038/sj.leu.2404471
Yee KW, Zeng Z, Konopleva M et al (2006) Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 12:5165–5173. doi:10.1158/1078-0432.CCR-06-0764
Younes A (2008) Therapeutic activity of mTOR-inhibitors in mantle cell lymphoma: clues but no clear answers. Autophagy 4:707–709
Yazbeck VY, Buglio D, Georgakis GV et al (2008) Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol 36:443–450. doi:10.1016/j.exphem.2007.12.008
Iwamaru A, Kondo Y, Iwado E et al (2007) Silencing mammalian target of rapamycin signaling by small interfering RNA enhances rapamycin-induced autophagy in malignant glioma cells. Oncogene 26:1840–1851. doi:10.1038/sj.onc.1209992
Yokoyama T, Iwado E, Kondo Y et al (2008) Autophagy-inducing agents augment the antitumor effect of telerase-selve oncolytic adenovirus OBP-405 on glioblastoma cells. Gene Ther 15:1233–1239. doi:10.1038/gt.2008.98
Acknowledgments
We apologize to those authors whose work could not be cited due to space limitations. This work was supported by the Center of Excellence grant from the Flight Attendant Medical Research Institute (FAMRI) and by the grant from Israel Science Foundation administered by the Israel Academy of Sciences and Humanities. AK is the incumbent of Helena Rubinstein Chair of Cancer Research.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This review is dedicated to the memory of Dr. Avner Eisenberg 1953–2004.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Eisenberg-Lerner, A., Kimchi, A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis 14, 376–391 (2009). https://doi.org/10.1007/s10495-008-0307-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0307-5