
Abstract
To analyze the underlying cellular mechanisms of adaptation to ischemia-induced apoptosis through short acidic pretreatment, i.e. acidic preconditioning (APC), Wistar rat coronary endothelial cells (EC) were exposed for 40 min to acidosis (pH 6.4) followed by a 14 h recovery period (pH 7.4) and finally treated for 2 h with simulated in vitro ischemia (glucose-free anoxia at pH 6.4). APC led to a transient activation of p38 and Akt kinases, but not of JNK and ERK1/2 kinases, which was accompanied by significant reduction of the apoptotic cell number, caspase-12/-3 cleavage and Bcl-xL overexpression. These effects of APC were completely abolished by prevention of Akt- or p38-phosphorylation during APC. Furthermore, knock-down of Bcl-xL by siRNA-transfection also abolished the anti-apoptotic effect of APC. Therefore, APC leads to protection of EC against ischemic apoptosis by activation of Akt and p38 followed by overexpression of Bcl-xL, which is a key anti-apoptotic mechanism of APC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Apoptosis of endothelial cells (EC) may be responsible for acute and chronic coronary artery diseases, e.g. through endothelial dysfunction [1], atherogenesis [2] or thrombosis [3]. Myocardial ischemia has been shown to be an important trigger of apoptosis [4, 5]. Among several protection strategies directed against ischemic injury, ischemic preconditioning, i.e. adaptation to ischemia through one or more short preceding ischemic episodes, demonstrated a powerful protective potential against ischemic cell death [6]. Since pretreatment with ischemia is clinically not applicable, many attempts have been undertaken to achieve the protective effect of ischemic preconditioning either pharmacologically [7] or by pretreatment with moderate environmental stress, e.g. hypoxia [8], glucose deprivation [9] or acidosis [10, 11], which are components of ischemia.
Previously acidosis was found to be an important stress factor triggering apoptosis in coronary EC under ischemic conditions through activation of caspase-12 [12]. Accordingly, short acidic pretreatment, i.e. acidic preconditioning (APC), can protect EC against ischemic apoptosis [11]. The underlying initial cellular mechanism leading to this protection, however, remained unknown. Several previous reports suggested that acidosis may lead to activation of MAP-kinases and PI3-Akt-kinases pathways [13, 14], which play an essential role in regulation of apoptosis [15]. To examine whether these kinases may be responsible for the anti-apoptotic effect of APC was the aim of the present study. We found that APC-induced activation of p38- and Akt-kinases leads to overexpression of Bcl-xL and protection against apoptosis.
Methods
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Cell culture
Coronary EC were isolated from 250 to 300 g male Wistar rats and maintained in Earle’s minimal essential medium 199 supplemented with 10% fetal calf serum and 10% newborn calf serum till two passages as previously described [16]. The purity of the cell culture (>95% EC) was confirmed by immunochemical staining with antibodies against vWF and by uptake of DiI-ac-LDL as previously described [17]. Experiments were performed with monolayers reaching 80–90% confluence and 18 h prior to experiments serum content in culture medium was reduced from 20 to 5%.
In vitro simulated ischemia
To simulate ischemic conditions cells were treated with anoxia in combination with glucose-deprivation and acidosis as described previously [12]. Dishes were incubated for 2 h at 37°C in a gas-tight chamber under continuous flush with a humidified gas mixture (95% N2 + 5% CO2). Analysis of the buffer pH after 2 h of simulated ischemia did not reveal any significant alteration.
Acidic preconditioning
Before simulated ischemia, cells were exposed to acidosis in cell culture medium (pH 6.4) for 40 min followed by a recovery period for 14 h in cell culture medium at pH 7.4. This protocol of preconditioning with acidosis was chosen from several preconditioning protocols, i.e. variation in time of APC (20–40 min) and in time of recovery period (6–24 h) as well as comparison of single versus repetitive (2 and 3 periods) acidic pretreatment, since it provides the maximal protection against ischemic apoptosis. In control group, similar treatment, i.e. change of medium, was performed at pH 7.4. In some experiments treatment with inhibitors of p38 kinase (SB203580, Calbiochem) and PI3 kinase (LY294002, Calbiochem) during APC was performed.
Hoechst-33342 and propidium iodide staining
To distinguish between apoptotic and necrotic cells a staining of nuclei with Hoechst-33342 and propidium iodide was applied as described previously [12]. Briefly, cells were trypsinized, washed with PBS and incubated for 10 min with 1 μg/ml Hoechst-33342 and 5 μg/ml propidium iodide. Stained nuclei were visualized with a converted fluorescence microscope at a magnification of 700× using excitation light at 350 nm for Hoechst-33342 and 540 nm for propidium iodide.
For quantitative assay, a blind analysis of 200–300 nuclei from randomized 4–5 fields was applied. Cells were scored as apoptotic, when nuclei stained with Hoechst-33342 produced unequivocal bright blue fluorescence due to chromatin condensation [12]. Propidium iodide stained nuclei with normal nuclear morphology, i.e. without signs of chromatin condensation, were scored as necrotic. Cells exhibiting both chromatin alteration and propidium iodide stained nuclei (i.e. “late-stage apoptotic cells”) were included in the apoptotic population. The number of these cells did not exceed 5% of all cells.
TUNEL staining
TUNEL staining using the In Situ Cell Death Detection Kit, TMR red (Roche Diagnostic GmbH, Mannheim) was performed according to the manufacturer’s instruction. Samples were analyzed with a Leica TCS SP2 confocal microscope. Four culture dishes (each 200–300 cells) per group were used for the quantification of TUNEL positive cells.
Western blot
Primary antibodies were: phospho-ERK1/2, ERK1/2, phospho-SAPK/JNK, SAPK/JNK phospho-Akt, Akt, phospho-p38, p38, Bcl-xL and caspase-3 (Cell Signaling), phospho-p38alpha (Millipore), p38beta (Santa Cruz), actin (Chemicon International), and caspase-12 (Calbiochem). Specific bands were visualized after incubation with peroxidase-linked/HRP-labeled secondary antibodies by chemiluminescence using Super-Signal kit (Thermo Scientific). For multiple analyses of different proteins, western blot was performed from the same cell extract. Equivalent sample loading (60 μg proteins/well) was confirmed by stripping membranes with the Blot Restore Membrane Stripping buffer (Thermo Scientific) followed by treatment with antibodies against actin. Since the loading control was similar in all membranes, only one blot was shown in representative figures.
siRNA transfection
Knock-down of Bcl-xL was achieved by treatment of EC with small interfering RNA (siRNA) duplexes, corresponding to separate regions within the rat Bcl-xL RNA sequence (Accession number NM_001033671, Cat. Nr: L-080091-01, Dharmacon Research, Lafayette, CO, USA). As a control, non-targeting siRNA duplexes (Cat. Nr: D-001810-01) were applied. Cells were transfected according to manufacturer’s instructions. Briefly, cells were seeded 1 day before transfection in MEM containing 10% fetal bovine serum without antibiotics. Bcl-xL siRNA or non-targeting siRNA were mixed with oligofectamine (Invitrogen) in OptiMEM (Gibco BRL) for 15 min at room temperature and then added to the culture medium at a final concentration of 60 nM. Cells were incubated at 37°C for 72 h. Protein expression was determined by Western blot using a specific antibody to Bcl-xL, which revealed ≥90% reduction of Bcl-xL after 72 h.
Statistical analysis
Data are given as mean ± SEM. The comparison of means between the groups was performed by one-way analysis of variance followed by Bonferroni post-hoc test. Statistical significance was accepted when P < 0.05.
Results
APC-induced protection is accompanied by phosphorylation of p38 and Akt kinases
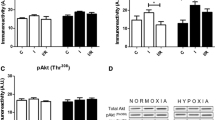
Exposure of EC to simulated ischemia for 2 h led to cleavage of caspase-3 and a rise of apoptotic cell number (28.8 ± 1.4, n = 5) (Fig. 1a). Short pretreatment with acidosis (pH 6.4) in the cell culture medium followed by a recovery period at pH 7.4, i.e. APC, significantly reduced the apoptosis rate (13.0 ± 1.1, n = 6, P < 0.05 vs. ischemia alone) and caspase-3 cleavage. To examine whether this effect is due to activation of Akt or MAP kinases, phosphorylation of Akt, p38, ERK1/2 and JNK kinases was analyzed. Acidic treatment in cell culture medium did not increase the phosphorylation of ERK1/2 and JNK kinases, whereas the phosphorylation of p38 kinase and Akt kinase was markedly increased after 10 min of acidic treatment (Fig. 1b). This effect was transient and nearly disappeared at the end of acidic pretreatment, i.e. after 40 min (Fig. 1c). Change of medium pH from 6.4 to 7.4 after 40 min of APC had no effect on phosphorylation of investigated kinases (data not shown).

APC suppressed apoptosis and induced activation of p38 and Akt kinases. a Number of apoptotic EC (Hoechst-33342 staining) and western blot analysis of cleaved caspase-3 under control conditions (no treatment) or after 2 h of simulated ischemia (SI) without or with APC treatment. Values are mean ± SEM, n = 5–6. *P < 0.05 versus SI. b Western blot analysis of phospho-ERK1/2, phospho-SAPK/JNK, phospho-p38 and phospho-Akt kinases performed with whole cell lysates prepared from control EC (Con) or from EC after 10 min of APC. APC led to a marked increase in phosphorylation of Akt and p38, but not ERK1/2 or SAPK/JNK kinases. c Western blot analysis of phospho-p38 and phospho-Akt at different time (10–40 min) during APC. d Western blot analysis of p38-alpha and phospho-p38-alpha in control EC (Con) or in EC after 10 min of APC. e Western blot analysis of p38-beta in control rat cardiomyocytes (CM) or in control rat coronary EC. Note that no p38-beta protein could be detected in EC. Western blot data are representative of three to five independent experiments with similar results
It has been shown that two isoforms of p38 kinase, i.e. p38-alpha and p38-beta, are expressed in the myocardium [18]. Therefore, to find out which isoform was activated by APC western blot analysis was performed. A marked expression of p38-alpha was found in EC (Fig. 1d). APC significantly increased the phosphorylation of p38-alpha. Surprisingly, no p38-beta expression could be found by western blot analysis in rat coronary EC, whereas in rat ventricular cardiomyocytes, used as a positive control, a marked expression p38-beta was found (Fig. 1e).
Inhibition of p38 and Akt kinases activation abolishes the anti-apoptotic effect of APC
To find out whether activation of p38 and Akt kinases during APC plays a causal role in the anti-apoptotic effect of preconditioning, treatment with 3 μmol/l SB203580, a specific inhibitor of p38 kinase, or with 10 μmol/l LY294002, a specific inhibitor of PI3 kinase, was applied during APC. These concentrations were used as they abolished kinases phosphorylation without cross-reaction on Akt and p38 phosphorylation, respectively (Fig. 2a). The finding that treatment with SB203580 prevented acidosis-induced p38 phosphorylation indicates the involvement of the acidosis-induced autophosphorylation of p38 kinase [19].

Inhibition of p38 or Akt kinases prevented the anti-apoptotic effect of APC. a Inhibition of p38 kinase with SB203580 (SB) or PI3 kinase with LY294002 (LY) during APC abolished the APC-induced phosphorylation of p38 and Akt kinases without significant cross-effect on Akt and p38 kinases, respectively. Data are representative of three independent experiments with similar results. b Number of apoptotic (Hoechst-33342 staining) and necrotic EC (propidium iodide staining) under control conditions (no treatment) or after 2 h of simulated ischemia (SI) without or with APC treatment. Inhibition of p38 kinase with 3 μmol/l SB203580 (SB) or PI3 kinase with 10 μmol/l LY294002 (LY) during APC abolished the anti-apoptotic effect of APC. Values are mean ± SEM, n = 6–8. *P < 0.05 versus SI. c Representative images of the TUNEL-staining in control EC or in EC after 2 h of simulated ischemia (SI) without or with APC treatment. F-actin was stained with specific antibodies (green) and nuclei were stained DAPI (blue). Arrows show nuclei of apoptotic cells. d Statistical analysis of the number of TUNEL-positive apoptotic EC. Similar conditions as in b. Values are mean ± SEM, n = 4. *P < 0.05 versus SI
Analysis of apoptotic cell number revealed, that inhibition of p38 or Akt kinases activation during acidic pretreatment abolished the anti-apoptotic effect of APC, i.e. reduction of the apoptotic cell number (Hoechst-33342 staining), whereas treatment with inhibitors alone, i.e. without APC, had no effects on ischemia-induced apoptosis. No effect on necrotic cell death could be detected (Fig. 2b). Similarly, APC alone had no effects on apoptotic or necrotic cell death during 14 h recovery period in our model [11].
To substantiate the apoptosis analysis, TUNEL staining of the nuclei was performed (Fig. 2c). This apoptosis detection method confirmed our data with Hoechst-33342 staining and demonstrated prevention of the anti-apoptotic effect of APC by inhibition of p38 and Akt kinases (Fig. 2d).
It has been shown that exposure of coronary EC to simulated ischemia leads to apoptosis mainly through activation of ER dependent pathway, i.e. due to cleavage of ER-bound caspase-12 [12]. Similarly, in the present study a cleavage of caspase-12 was found (Fig. 3), which was attenuated by APC. This effect of APC was p38- and Akt-dependent, since inhibition of these kinases during acidic pretreatment abolished the APC-effect on caspase-12 cleavage (Fig. 3).

Western blot analysis of cleaved caspase-12 prepared from extracts of control EC (Con) or EC exposed to simulated ischemia (SI) without or with APC treatment. Note that treatment with 3 μmol/l SB203580 (SB) or with 10 μmol/l LY294002 (LY) during APC abolished the inhibitory effect of APC on caspase-12 cleavage during SI. Data are representative of three independent experiments with similar results
Acidic preconditioning-induced overexpression of Bcl-xL is p38- and Akt-dependent
To understand more about the underlying downstream anti-apoptotic mechanisms induced by p38 and Akt activation, the expression of Bcl-2 family proteins, i.e. Bax, Bak, Bcl-xL and Bcl-2, was investigated. Although APC did not change the expression of Bax, Bak and Bcl-2 (data not shown), the pronounced over-expression of Bcl-xL was detected by western blot analysis (Fig. 4a). The APC-induced Bcl-xL overexpression was p38- and Akt-dependent, since inhibition of p38 or Akt kinases during APC prevented Bcl-xL overexpression (Fig. 4a).

Importance of Bcl-xL overexpression in protective mechanism of APC. a Western blot analysis of Bcl-xL prepared from extracts of control EC (Con) or EC after 40 min APC followed by 14 h recovery period (APC). Note that treatment with SB203580 (SB) or LY294002 (LY) abolished the APC-induced overexpression of Bcl-xL. Data are representative of three independent experiments with similar results. b Western blot analysis of Bcl-xL prepared from extracts of control EC (Con) or EC after treatment with Bcl-xL targeting siRNA (siRNA, 60 nmol/l, 72 h). Data are representative of three independent experiments with similar results. c Western blot analysis of Bcl-xL in EC pre-treated with Bcl-xL targeting siRNA followed by APC (siRNA + APC) or no treatment (siRNA). Data are representative of three independent experiments with similar results. d Effect of pre-treatment for 72 h with targeting siRNA (siRNA) and acidic preconditioning (APC) on the number of apoptotic EC after 2 h of simulated ischemia (SI).Values are mean ± SEM, n = 4–5. Note that no reduction of the apoptotic cell number by APC could be detected after Bcl-xL knock-down
To prove whether the Akt- and p38-dependent overexpression of Bcl-xL might be a key mechanism in the APC-induced protection against ischemic apoptosis, knock-down of Bcl-xL was performed before simulated ischemia by treatment of EC with Bcl-xL targeting siRNA. After 72 h treatment the reduction of Bcl-xL by about 90% was found (Fig. 4b). Under siRNA pre-treatment, no rise of Bcl-xL expression could be detected after APC (Fig. 4c). Subsequent analysis of apoptosis revealed that the Bcl-xL knock-down abolished the anti-apoptotic effect of APC (Fig. 4d).
Discussion
The aim of the present study was to find out which signaling mechanisms participate in the anti-apoptotic effect of APC. The main findings are the following: (1) The anti-apoptotic effect of APC was associated with transient phosphorylations of p38 and Akt kinases during acidic pretreatment followed by overexpression of Bcl-xL. (2) Prevention of the phosphorylation of these kinases during APC abolished the APC-induced overexpression of Bcl-xL and the anti-apoptotic effect of APC.
In the present study we used the model of simulated in vitro ischemia, i.e. combination of glucose-free anoxia with extracellular acidosis (pH 6.4), which was characterized previously in details for endothelial cells and cardiomyocytes [11, 20, 21]. Such an extent of extracellular acidification is comparable to an in vivo myocardial ischemia, where extracellular pH can drop to as low as 6.0 [22]. It has been shown that acidification is an initial trigger for the cleavage and activation of the endoplasmic reticulum-bound caspase-12 and subsequent apoptosis of coronary EC under simulated ischemia [12]. Several reports from other groups confirmed a pro-apoptotic potential of severe acidosis in vitro and in vivo [23–25]. Thus, acidosis seems to be an important stress factor leading to apoptosis under ischemic environment. According to the preconditioning paradigm, “almost any stress factor that is potentially harmful for cells can elicit a preconditioned state, i.e. increased resistance to the damaging stress, when applied in small quantities” [6]. Indeed, protective effect of APC was demonstrated in cell culture [11] and in the whole heart [10]. However, the underlying cellular mechanisms of this protection, specifically against ischemic apoptosis, were poorly understood. In the present study the phosphorylation of several MAP kinases and Akt kinase was investigated, since these kinases (1) can be activated by extra- or intracellular acidification [13, 14, 26] and (2) may modulate the processing of apoptosis [15]. The kinase-pattern activated by acidosis is dependent on the cell type. Indeed, a similar degree of acidosis (pH 6.5–6.6) leads to activation of p38 kinase in rat cardiomyocytes [27], whereas it induces activation of PI3 kinase in human neutrophils [14] or ERK1/2 kinase in human glioblastoma cells [13]. In the present study, applying coronary endothelial cells, a significant increase in phosphorylation of Akt and p38 kinases, but not ERK1/2 or SAPK/JNK kinases, during acidic treatment was found. The activation of these kinases seems to be an initial mechanism in the anti-apoptotic effect of APC, since inhibition of Akt and p38 activation during APC completely abolished the APC-induced protection against ischemic apoptosis.
In agreement with these findings, several studies demonstrated the involvement of both p38 and Akt kinases in the protection against apoptosis induced by various protocols of preconditioning [27–29]. The cellular mechanisms of the anti-apoptotic action of Akt and p38 are complex. In the present study we found that the overexpression of Bcl-xL is an important link between Akt-/p38-activation and the protection against apoptosis. In agreement with this finding, the regulation of Bcl-xL expression by Akt and p38 kinases has been previously shown [30, 31]. The transcriptional regulation of the bcl-xl gene involves participation of several transcriptional factors, e.g. STATs, Rel/NF-kB, Ets and AP-1 [32]. Since Akt and p38 kinases are potential up-stream kinases regulating these transcriptional factors, it can be suggested that the acidosis-induced activation of Akt and p38 kinases leads to a transcriptional up-regulation of bcl-xl gene. Bcl-xL overexpression seems to be the key mechanism down-stream of p38 and Akt kinases responsible for the anti-apoptotic effect of APC, since Bcl-xL knock-down completely abolished the APC effect on apoptosis (Fig. 4).
Previously we found that cleavage of the endoplasmic reticulum-bound caspase-12 represents a main apoptotic pathway in coronary EC under simulated ischemia [12]. These data and the finding of the present study that inhibition of Akt and p38 kinases abolished both (1) overexpression of Bcl-xL and (2) APC-induced suppression of caspase-12 cleavage argue for Bcl-xL as an essential mechanism in the suppression of ischemia-induced caspase-12 cleavage. Indeed, recent reports demonstrated that overexpression of Bcl-xL prevents endoplasmic reticulum stress and caspase-12 cleavage [33, 34].
In conclusion, preconditioning of coronary EC with acidosis leads to activation of Akt and p38 kinases followed by overexpression of Bcl-xL and suppression of caspase-12 cleavage, which is a key mechanism in the APC-induced protection against ischemic apoptosis. Further understanding of the pH-sensing mechanism responsible for this adaptation phenomenon may lead to the discovery of new therapeutic approaches for treatment of diseases accompanied by apoptosis, e.g. tumor, neurodegenerative and ischemic diseases.
Abbreviations
- EC:
-
Endothelial cells
- APC:
-
Acidic preconditioning
- siRNA:
-
Small interfering RNA
References
Werner N, Wassmann S, Ahlers P et al (2006) Circulating CD31+/annexin V+ apoptotic microparticles correlate with coronary endothelial function in patients with coronary artery disease. Arterioscler Thromb Vasc Biol 26:112–116. doi:10.1161/01.ATV.0000191634.13057.15
Chen J, Mehta JL, Haider N et al (2004) Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ Res 94:370–376. doi:10.1161/01.RES.0000113782.07824.BE
Bombeli T, Karsan A, Tait JF et al (1997) Apoptotic vascular endothelial cells become procoagulant. Blood 89:2429–2442
Chakrabarti S, Hoque AN, Karmazyn M (1997) A rapid ischemia-induced apoptosis in isolated rat hearts and its attenuation by the sodium–hydrogen exchange inhibitor HOE 642 (cariporide). J Mol Cell Cardiol 29:3169–3174. doi:10.1006/jmcc.1997.0561
Scarabelli T, Stephanou A, Rayment N et al (2001) Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation 104:253–256
Bolli R (2007) Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol 292:H19–H27. doi:10.1152/ajpheart.00712.2006
Zhao TC, Kukreja RC (2002) Late preconditioning elicited by activation of adenosine A(3) receptor in heart: role of NF-kappa B, iNOS and mitochondrial K(ATP) channel. J Mol Cell Cardiol 34:263–277. doi:10.1006/jmcc.2001.1510
Engelman DT, Chen CZ, Watanabe M et al (1995) Hypoxic preconditioning enhances functional recovery after prolonged cardioplegic arrest. Ann Thorac Surg 59:428–432. doi:10.1016/0003-4975(94)00846-Y
Ebel D, Redler S, Preckel B et al (2005) Moderate glucose deprivation preconditions myocardium against infarction. Horm Metab Res 37:516–520. doi:10.1055/s-2005-870321
Lundmark JA, Trueblood N, Wang LF et al (1999) Repetitive acidosis protects the ischemic heart: implications for mechanisms in preconditioned hearts. J Mol Cell Cardiol 31:907–917. doi:10.1006/jmcc.1998.0931
Kumar S, Reusch HP, Ladilov Y (2008) Acidic preconditioning suppresses apoptosis and increases expression of Bcl-xL in coronary endothelial cells under simulated ischemia. J Cell Mol Med 12:1584–1592. doi:10.1111/j.1582-4934.2007.00172.x
Kumar S, Kasseckert S, Kostin S et al (2007) Ischemic acidosis causes apoptosis in coronary endothelial cells through activation of caspase-12. Cardiovasc Res 73:172–180. doi:10.1016/j.cardiores.2006.09.018
Xu L, Fukumura D, Jain RK (2002) Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. J Biol Chem 277:11368–11374. doi:10.1074/jbc.M108347200
Martinez D, Vermeulen M, Trevani A et al (2006) Extracellular acidosis induces neutrophil activation by a mechanism dependent on activation of phosphatidylinositol 3-kinase/Akt and ERK pathways. J Immunol 176:1163–1171
Cross TG, Scheel-Toellner D, Henriquez NV et al (2000) Serine/threonine protein kinases and apoptosis. Exp Cell Res 256:34–41. doi:10.1006/excr.2000.4836
Piper HM, Spahr R, Mertens S et al (1990) Microvascular endothelial cells from heart. In: Piper HM (ed) Cell culture techniques in hearth and vessel research. Springer, Heidelberg, pp 158–177
Ando H, Kubin T, Schaper W et al (1999) Cardiac microvascular endothelial cells express alpha-smooth muscle actin and show low NOS III activity. Am J Physiol 276:H1755–H1768
Sugden PH, Clerk A (1998) “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ Res 83:345–352
Ge B, Gram H, Di Padova F et al (2002) MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science 295:1291–1294. doi:10.1126/science.1067289
Ladilov Y, Schäfer C, Held A et al (2000) Mechanism of Ca(2+) overload in endothelial cells exposed to simulated ischemia. Cardiovasc Res 47:394–403. doi:10.1016/S0008-6363(00)00108-5
Ladilov YV, Balser C, Piper HM (1998) Protection of rat cardiomyocytes against simulated ischemia and reoxygenation by treatment with protein kinase C activator. Circ Res 82:451–457
Ferrari R, Cargnoni A, Bernocchi P et al (1996) Metabolic adaptation during a sequence of no-flow and low-flow ischemia. A possible trigger for hibernation. Circulation 94:2587–2596
Aoyama K, Burns DM, Suh SW et al (2005) Acidosis causes endoplasmic reticulum stress and caspase-12-mediated astrocyte death. J Cereb Blood Flow Metab 25:358–370. doi:10.1038/sj.jcbfm.9600043
Webster KA, Discher DJ, Kaiser S et al (1999) Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J Clin Investig 104:239–252. doi:10.1172/JCI5871
Thatte HS, Rhee JH, Zagarins SE et al (2004) Acidosis-induced apoptosis in human and porcine heart. Ann Thorac Surg 77:1376–1383. doi:10.1016/j.athoracsur.2003.07.047
Zheng M, Reynolds C, Jo SH et al (2005) Intracellular acidosis-activated p38 MAPK signaling and its essential role in cardiomyocyte hypoxic injury. FASEB J 19:109–111
Zhao TC, Taher MM, Valerie KC et al (2001) p38 Triggers late preconditioning elicited by anisomycin in heart: involvement of NF-kappaB and iNOS. Circ Res 89:915–922. doi:10.1161/hh2201.099452
Han H, Wang H, Long H et al (2001) Oxidative preconditioning and apoptosis in L-cells. Roles of protein kinase B and mitogen-activated protein kinases. J Biol Chem 276:26357–26364. doi:10.1074/jbc.M011136200
Izuishi K, Tsung A, Hossain MA et al (2006) Ischemic preconditioning of the murine liver protects through the Akt kinase pathway. Hepatology 44:573–580. doi:10.1002/hep.21298
Bachelor MA, Bowden GT (2004) Ultraviolet A-induced modulation of Bcl-XL by p38 MAPK in human keratinocytes: post-transcriptional regulation through the 3′-untranslated region. J Biol Chem 279:42658–42668. doi:10.1074/jbc.M406626200
Hatano E, Brenner DA (2001) Akt protects mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis through NK-kappa B activation. Am J Physiol Gastrointest Liver Physiol 281:G1357–G1368
Sevilla L, Zaldumbide A, Pognonec P et al (2001) Transcriptional regulation of the bcl-x gene encoding the anti-apoptotic Bcl-xL protein by Ets, Rel/NFkappaB, STAT and AP1 transcription factor families. Histol Histopathol 16:595–601
Murakami Y, Aizu-Yokota E, Sonoda Y et al (2007) Suppression of endoplasmic reticulum stress-induced caspase activation and cell death by the overexpression of Bcl-xL or Bcl-2. J Biochem 141:401–410. doi:10.1093/jb/mvm044
Morishima N, Nakanishi K, Tsuchiya K et al (2004) Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J Biol Chem 279:50375–50381. doi:10.1074/jbc.M408493200
Acknowledgments
The technical help of G. Scheibel and K. Rezny is gratefully acknowledged. This study is a part of the thesis of J.-P. Flacke submitted in fulfillment of the requirements for the degree of Doctor of Medicine at the Ruhr-University Bochum (Germany).
Funding
This study was supported by Forum Grant F591-2007 from the Ruhr-University Bochum and by Grant LA 1159/6-1 from the Deutsche Forschungsgemeinschaft.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Flacke, JP., Kumar, S., Kostin, S. et al. Acidic preconditioning protects endothelial cells against apoptosis through p38- and Akt-dependent Bcl-xL overexpression. Apoptosis 14, 90–96 (2009). https://doi.org/10.1007/s10495-008-0287-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-008-0287-5