
Abstract
Primary cell cultures (n = 16) were initiated from tissues of embryonic and neonatal larval Ornithodoros moubata following methods developed for hard ticks. After maintenance for 20–25 months in vitro, cell multiplication commenced in surviving cultures, leading to the establishment of six cell lines designated OME/CTVM21, 22, 24, 25, 26 and 27. All lines are maintained at 28°C, with subculture at 2–8 week intervals. The cultures comprise heterogeneous populations of large cells of 15–100 μm in diameter, often with finger-like protrusions and/or intracellular crystals, rarely attached, predominantly floating and forming clumps or hollow multicellular vesicles up to 1 mm in diameter. Attempts to cryopreserve the cells are described. Tick-borne encephalitis virus has been serially passaged ten times in OME/CTVM21 cells without significant decrease in virus production and with no change in its biological properties as shown by the size and morphology of plaques produced in porcine kidney cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Argasid (soft) ticks of the Ornithodoros moubata species complex, known as eyeless tampans, live in close association with humans and their domestic animals in East, Central and Southern Africa (Walker et al. 2003). As well as feeding on pigs and poultry, O. moubata will readily attach to and feed on humans (Estrada-Pena and Jongejan 1999). Ticks of the O. moubata complex transmit the spirochaete Borrelia duttoni, causative agent of relapsing fever in humans (Felsenfeld 1965). They have also been incriminated as reservoirs and vectors of African swine fever virus (ASFV) which they transmit readily to domestic pigs under experimental conditions (Plowright et al. 1969).
While tick tissue culture systems and continuous cell lines have made important contributions to research into the biology, genomics and control of ixodid ticks and the pathogens they transmit (Bell-Sakyi et al. 2007), progress in development of in vitro culture systems for argasid ticks has been much slower. The earliest report of soft tick primary cell cultures was that of Sidorov (1960), cited by Rehacek (1976), who obtained survival for several days of haemocytes in autologous haemolymph from Ornithodoros lahorensis, Ornithodoros papillipes and Argas persicus. Fujisaki et al. (1975) cultured haemocytes from engorged female O. moubata for up to 8 months, but did not report significant cell multiplication, and subcultures were unsuccessful. Moulton (1978) achieved survival of viable haemocytes of late-stage nymphal and adult Ornithodoros coriaceus for up to 9 months; the cells retained their phagocytic capability and multiplication was occasionally observed. Yunker (1987) reported cultivation of Argas arboreus haemocytes which formed confluent monolayers surviving for over 2 months.
The first continuous cell lines from a soft tick species were established using embryonic tissues from the seabird tick Carios capensis (Mattila et al. 2007). Four of the five lines were infected with rickettsial endosymbionts (Mattila et al. 2007), and one line has been used to propagate an ixodid tick symbiont, Rickettsia peacockii (Kurtti et al. 2005). There are no other published reports of in vitro cultivation of symbiotic or pathogenic bacteria in argasid cells, but recently two O. moubata cell lines were used to propagate tick-borne encephalitis virus (TBEV; Růžek et al. 2008). Here we describe the establishment of these and additional O. moubata cell lines in more detail, and report the adaptation of TBEV, naturally transmitted by hard ticks of the genus Ixodes, to growth in the soft tick cells by serial passage.
Materials and methods
Origin and maintenance of Ornithodoros moubata ticks
Ornithodoros moubata ticks, originally from Kenya, were maintained for >5 years in a laboratory colony at CEH Oxford following standard methods for argasid tick maintenance (Jones et al. 1988).
Ornithodoros moubata primary cell cultures
Primary cell cultures were initiated following a method used for ixodid ticks (Bell-Sakyi 1991). Engorged female O. moubata were transferred to Edinburgh University, where they were surface-sterilised in 0.1% benzalkonium chloride (5 min) and 70% ethanol (1 min), rinsed twice in sterile deionised water and dried on sterile filter paper. The female ticks were placed in groups of 3–4 in sterile plastic petri dishes and allowed to oviposit at 25°C, 85% r.h. After 6–19 days the resultant egg batches, comprising eggs at all stages of development between oviposition and eclosion, plus newly-hatched inactive larvae in all egg batches >7 days-old, were used to initiate primary cell cultures. The eggs and larvae were transferred to a fresh sterile plastic Petri dish, surface-sterilised individually or in groups of 3 or 4 batches by immersion in 70% ethanol (1 min) and rinsed twice with sterile deionised water. The eggs and larvae were crushed gently in 2–3 ml Hanks BSS using the flattened end of a glass rod, and the resultant cell and tissue suspensions were washed in HBSS with or without prior filtration through plastic gauze, pore size 300 μm. The HBSS was removed by centrifugation at 200g for 5 min; the cells and tissues were resuspended in complete culture medium and incubated in 2 ml volumes in flat-sided tubes (Nunc) at 28°C. Medium was changed weekly by removal and replacement of 1.5 ml; this was facilitated by first standing the tubes upright for 5 min to allow the floating cell and tissue clumps to sink to the bottom of the tube.
Cultures were initiated in four different media, all supplemented with 20% foetal calf serum (FCS), 2 mM l-glutamine, 100 units/ml penicillin and 1 μg/ml streptomycin: (1) L-15 (Leibovitz) medium with 10% tryptose phosphate broth (TPB; L-15), (2) Hanks balanced salt solution with 0.5% lactalbumin hydrolysate (H-Lac), (3) equal volumes of L-15 and H-Lac (L-15/H-Lac), and iv) equal volumes of L-15 (Leibovitz) medium and Minimal Essential Medium with Hanks salts, with 10% TPB (L-15/MEM). All medium components were obtained from Invitrogen (Paisley, UK) except the lactalbumin hydrolysate (Sigma, Poole, UK) and the FCS (PAA Laboratories, Yeovil, UK).
Subculture and low-temperature storage
Subculture was carried out as follows: during the medium change, the spent medium from a selected parent culture tube was transferred to a new daughter tube and incubated overnight at 28°C. Next day the spent medium was discarded from the daughter tube, 2 ml of fresh medium was added gently to the parent tube and half of the resultant cell suspension was transferred to the daughter tube with the minimum amount of mixing and disruption. A similar method was used to passage cultures in 6 ml volumes in 25 cm2 flasks (Nunc). Repeated pipetting and centrifugation, even at speeds as low as 50g, visibly damaged many of the cells, resulting in reduced viability.
Cultures used in attempts at cryopreservation were pooled in a plastic universal container and held upright for 20 min to allow the cells to settle to the bottom. Most of the supernatant medium was removed, leaving 1–2 ml, and the universal container was placed on ice. An equal volume of medium with 60% FCS and double-strength cryoprotectant (producing a final concentration of 10% dimethyl sulphoxide [DMSO] or 15% polyvinylpyrrolidine [PVP] or 7.5% DMSO/7.5% PVP) was added dropwise with gentle swirling and the cell suspension was held on ice for 5–25 min to equilibrate. Aliquots of 1 ml were dispensed into cryotubes and held on dry ice for 5 min–24 h before transferring to vapour-phase liquid nitrogen storage, or immediately diluted in 9 ml complete medium with 40% FCS in a flat-sided tube to check viability. Frozen aliquots were thawed rapidly by immersing the cryotube in a 37°C waterbath. As soon as the cell suspension was thawed, it was diluted in 9 ml ice-cold complete medium with 40% FCS in a flat-sided tube and held on ice for 0–15 min to equilibrate. Following dilution, all culture tubes were incubated at 28°C overnight before removal of 8 ml medium.
Cultures were also held at 15°C for 1–19 weeks or 4°C for 1–3 weeks without medium change before returning to 28°C.
Infection with TBEV
Tick-borne encephalitis virus European subtype prototype strain Neudoerfl, kindly provided by Professor F.X. Heinz, Institute of Virology, Medical University of Vienna, Austria, was used in the virus infection experiments. The growth rate of TBEV in the O. moubata cell lines OME/CTVM21 and 22, and in three cell lines from the natural vector Ixodes ricinus (IRE/CTVM18, 19 and 20) over a 10-day period was determined by plaque assay, as described previously (Růžek et al. 2008). To investigate if TBEV retains its infectivity even after serial subcultures of the virus in O. moubata cells, 10 serial passages of the virus were performed in OME/CTVM21 cells. Diluted cell-free supernatant culture medium was used for each subsequent round of infection (~105 cells; multiplicity of infection ~1). Supernatant media were collected at 10 days post infection (p.i; passages1–5) or 4–5 days p.i. (passages 6–10), and used to assess virus production by plaque assay on porcine kidney (PS) cells in 24-well plates using a 1.5% carboxymethylcellulose overlay as described previously (De Madrid and Porterfield 1969). PS cells were incubated at 37°C in an atmosphere of 0.5% CO2 in air for 5 days and the plates were then stained with naphthalene black. Infectivity was expressed as plaque-forming units (pfu) per ml.
Results
Primary cell cultures
Four primary cultures were initiated from four pooled egg batches in January 2003 (Table 1). Initially all the cultures comprised floating tissue lumps and some adherent cells (Fig. 1), but by day 50 all cells had detached and subsequently formed floating clumps of very large, vacuolated cells and multicellular vesicles. One culture was lost to fungal contamination and the remaining three survived for up to 21 months, but never showed significant cell multiplication; subculture attempts from two of the cultures were unsuccessful.

Primary cultures of Ornithodoros moubata tissues at day 30 after initiation. Live, inverted microscope; scale bar = 100 μm
Twelve primary cultures were initiated from six egg batches in February and March 2005 (Table 1). Two of these primary cultures developed fungal contamination and a further four were discarded between days 14 and 242, when metabolism ceased, while the remaining six are still alive and healthy at the time of writing (46 months), and all have been subcultured to at least passage 4. Initially the cells and tissues in the primary cultures behaved as described above; some cell clumps reattached to the tube surface in all six cultures between days 391 and 483 and have remained attached. All the cultures also contain floating cell clumps and multicellular vesicles. Both primary cultures and subcultures periodically display rafts of loosely associated heterogeneous cells floating on the surface of the culture medium, first observed on day 337.
Establishment of Ornithodoros moubata cell lines
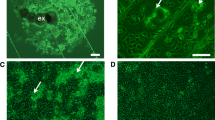
The primary cultures which gave rise to OME/CTVM21 and OME/CTVM22 were initiated from a single 10-day-old egg batch (Růžek et al. 2008). The appearance of the primary cultures during the first year was as described above. Some floating cell clumps reattached between days 390 and 450. Between days 600 and 650, clumps of smaller, actively dividing cells appeared in both cultures. OME/CTVM21 was first subcultured on day 459, and is currently at passage 18. OME/CTVM22 was first subcultured successfully on day 653, and is currently at passage 11. At the time of writing, both cell lines comprise heterogeneous populations of large cells 15–100 μm in diameter, often with finger-like protrusions and/or intracellular crystals, rarely attached, predominantly floating and forming clumps or hollow multicellular vesicles up to 1 mm in diameter (Fig. 2). Subcultures are carried out at 2–6 week intervals.

Ornithodoros moubata cell lines OME/CTVM21 (left) and OME/CTVM22 (right) at passage 6, 36 months after initiation. Live, inverted microscope; scale bar = 100 μm
All attempts to cryopreserve cells of lines OME/CTVM21 and OME/CTVM22, at passage levels between 3 and 6 and with DMSO, PVP or DMSO/PVP, were unsuccessful; there were no viable cells present in resuscitated cultures within 30 min of thawing. Cells mixed with cryprotectant but not frozen were unharmed and metabolised and grew normally after removal of the cryoprotectant solution. Cells held at 15°C resumed normal growth within 7 days when returned to 28°C after 14 weeks and within 16 days after 19 weeks at the lower temperature, whereas cells held at 4°C for 1 week showed reduced viability when returned to 28°C, and died after storage at 4°C for 2 or more weeks.
Four additional cell lines, OME/CTVM24, 25, 26 and 27, exhibited significant cell multiplication after 23–25 months (Table 1), and are currently at passage levels 11, 7, 7 and 4, respectively; these lines do not yet grow at a consistent rate, with subculture at 6–8 week intervals. The cell types and manner of growth of OME/CTVM24 and 27 resemble that of OME/CTVM22. OME/CTVM25 and 26 contain variable quantities of spiky crystals of unknown composition, which apparently form initially within the cells but are subsequently liberated, possibly by destruction of the host cell, into the supernatant medium where they continue to enlarge (Fig. 3).

Ornithodoros moubata cell line OME/CTVM26 at passage 3, 36 months after initiation. Live, inverted microscope; scale bar = 100 μm
Adaptation of TBEV to growth in Ornithodoros moubata cells
In comparison with cell lines from the vector tick Ixodes ricinus, TBEV initially replicated more slowly in the O. moubata cells, but by day 10 had achieved virus titres nearly as high as in cells of the natural vector (Fig. 4). The TBEV titre, and the size and morphology of plaques produced in PS cells by the passaged virus, were investigated over 10 serial passages in OME/CTVM21 cells. The average titre of the virus during consecutive passages was 4.2 × 106 pfu/ml. During the first five passages this titre was achieved in the supernatant medium at day 10 p.i. whereas in the subsequent five passages the titre was achieved by day 4–5 p.i. (Table 2), indicating that TBEV had become adapted to growth in the O. moubata cells. There was no difference in the size and morphology of plaques in PS cells produced by the TBEV before and after the serial passages (data not shown).

Growth kinetics of TBEV in Ornithodoros moubata and Ixodes ricinus cell lines
Discussion
Nearly 50 years after the first reported attempts to cultivate argasid cells in vitro, and over 30 years after the first ixodid tick cell lines were established, there are now several continuous cell lines from two soft tick species, representing different genera (Carios and Ornithodoros) and different geographical origins (New World, North America, and Old World, Africa). In both cases the materials and methods used to initiate the primary cultures that gave rise to the cell lines were based on techniques used successfully for establishment of ixodid tick cell lines. Moreover, in both cases the success rate for achievement of cell lines from primary cultures was relatively high (Bell-Sakyi et al. 2007), at 100% for C. capensis (Mattila et al. 2007; T. J. Kurtti, personal communication) and 37.5% for the O. moubata lines reported here. Therefore it is likely that these methods could be applied to other argasid tick species with the expectation of success, at least in the hands of experienced tick cell culturists.
Early workers apparently focussed on haemocytes as a potential source of proliferating cells (Fujisaki et al. 1975; Moulton 1978; Yunker 1987); isolated haemocytes survived for up to 9 months with occasional multiplication but successful subculture was not achieved. Varma (1989) regretted that, despite “numerous attempts” (no details given), there were at that time no continuous cell lines available from argasid ticks. In the present study, the primary cultures were initiated from eggs and neonate larvae, with no attempt to select particular tissues or cell types. Consequently they contained a wide range of cell types, presumably including haemocytes, and significant cell multiplication was not observed until the cultures were around 20 months old. It is possible that the heterogeneity of the O. moubata cultures provided a survival advantage over the haemocyte monocultures used by previous workers, supporting a long period of adaptation to the culture conditions. The C. capensis embryo-derived primary cultures and young cell lines also comprised a diversity of cell types (Mattila et al. 2007). As already seen with ixodid ticks, from which nearly 90% of cell lines are derived from eggs (Bell-Sakyi et al. 2007), it seems that use of whole embryos as starting material is still the most likely route to success.
The C. capensis primary cultures were incubated at 31°C, first passaged at 3 months, and had reached passage 40 after 36 months (Mattila et al. 2007). In contrast, the O. moubata cultures were maintained at 28°C, first passaged successfully at between 14.5 and 24 months, and at 46 months have only reached passage 4–18. O. moubata cells can be maintained at 31°C for several weeks but do not currently grow significantly faster at the higher temperature (data not shown). The difference in initial adaptation period and growth rate between cell lines of the two species could be due to intrinsic differences between the parent ticks, to incubation temperature and/or culture medium—the C. capensis lines are maintained at 32–34°C in antibiotic-free L-15B300 medium supplemented with 10% FCS, 5% TPB, HEPES and 0.1% bovine lipoprotein and (Mattila et al. 2007), or to a combination of these factors.
Apart from the longer adaptation period and slower growth rate exhibited by the O. moubata cells so far, there are many similarities between the O. moubata and C. capensis cell lines, especially line CCE3. These include the occurrence of clumped cells and floating hollow multicellular vesicles, the inability to disperse the cells for counting, and the difficulty experienced in cryopreserving the cells—Mattila et al (2007) reported that CCE3 could not be frozen and resuscitated using the procedure which was applied successfully to line CCE2. Monolayers of epithelial cells, as described for line CCE2, have not been seen so far in any of the O. moubata cell lines; indeed O. moubata cells have only attached to the surface of the culture vessel in around 2% of subcultures, and only several months after the passage was carried out. The six O. moubata cell lines are currently very heterogeneous, with a wide range of different cell sizes and morphologies identifiable in live cultures and Giemsa-stained cytocentrifuge smears. With live cell diameters between 15 and 100 μm, the O. moubata cells are on average much larger than cultured ixodid cells, and apparently more delicate since they can be disrupted by even gentle pipetting and very low speed centrifugation.
The previous lack of in vitro culture systems for soft ticks has undoubtedly held back research progress into these parasites and the pathogens they transmit. However, the C. capensis cell lines have already been applied usefully in studies on rickettsial symbionts both of the parent tick (Mattila et al. 2007) and of an ixodid tick species, Dermacentor andersoni (Kurtti et al. 2005). To our knowledge, there have been no published reports of propagation in tick cell or organ cultures of the two economically important pathogens transmitted by O. moubata, B. duttoni and the DNA arbovirus ASFV. However, the new O. moubata cell lines reported here open up possibilities for in vitro studies on the interaction between both of these pathogens and their tick vector. A different tick-borne arbovirus, the RNA flavivirus TBEV, has been propagated successfully in two of the O. moubata lines (Růžek et al. 2008) and, interestingly, the virus titre at day 10 was nearly as high as that achieved in cell lines derived from the vector tick I. ricinus. Immunofluorescence microscopy showed that TBEV infected less than 10% of the O. moubata cells but more than 75% of the I. ricinus cells (Růžek et al. 2008); the much larger size of the individual O. moubata cells could result in higher levels of virus production per cell resulting in overall virus titres per culture similar to those produced by the smaller I. ricinus cells. Moreover, we demonstrate here that the virus can be passaged up to ten times in O. moubata cells with no decrease in titre and no phenotypic change with respect to the size and morphology of plaques produced in PS cells.
Although TBEV is not transmitted by O. moubata in nature, as their distributions do not overlap, the predilection of O. moubata for feeding on humans raises the question whether or not this tick species could become infected with and transmit TBEV if it ingested an infected bloodmeal. Generally, argasid ticks are not considered to be competent vectors of TBEV on the basis of major differences in interactions between TBEV and ixodid and argasid species (Nuttall and Labuda 1994). However, Langat virus, a tick-borne flavivirus closely related to TBEV, has been successfully transmitted under laboratory conditions by Ornithodoros sonrai ticks both transstadially and transovarially (Turell and Durden 1994), and up to 3 years after initial oral exposure (Turell et al. 2004). Lawrie et al. (2004) reported experimental infection of O. moubata with another arthropod-borne flavivirus, West Nile virus, both systemically through feeding on a viraemic mouse, and non-systemically by co-feeding infected and uninfected ticks on an uninfected mouse. Moreover, infected O. moubata were able to transmit the virus to susceptible rodent hosts during feeding.
In conclusion, the O. moubata cell lines described here will be useful for studies on the biology and physiology of this tick species at the cellular and molecular levels as well as for in vitro propagation of the pathogens that it transmits both naturally and potentially. Worldwide, there is increasing pressure to reduce the use of animals for in vivo pathogenicity studies, and for the development of both vaccines and drug treatments including antivirals. The development of cell culture systems that might replace animals is an obvious route to choose. Today, most screening for antivirals is initiated in cell culture systems that are robust and can provide valuable efficacy and safety data prior to the development of in vivo models and ultimately clinical trials. As the replicative processes, evolution, epidemiology and pathogenesis of more exotic vector-borne pathogens are studied, appropriate vector cell lines need to be developed. Tick cell lines, both argasid and ixodid, will play an increasingly important role in this important area of veterinary and medical research.
References
Bell-Sakyi L (1991) Continuous cell lines from the tick Hyalomma anatolicum anatolicum. J Parasitol 77:1006–1008. doi:10.2307/3282757
Bell-Sakyi L (2004) Ehrlichia ruminantium grows in cell lines from four ixodid tick genera. J Comp Pathol 130:285–293. doi:10.1016/j.jcpa.2003.12.002
Bell-Sakyi L, Zweygarth E, Blouin EF, Gould EA, Jongejan F (2007) Tick cell lines: tools for tick and tick-borne disease research. Trends Parasitol 23:450–457. doi:10.1016/j.pt.2007.07.009
De Madrid AT, Porterfield JS (1969) A simple microculture method for the study of group B arboviruses. Bull World Health Organ 40:113–121
Estrada-Pena A, Jongejan F (1999) Ticks feeding on humans: a review of records on human-biting Ixodoidea with special reference to pathogen transmission. Exp Appl Entomol 23:685–715
Felsenfeld O (1965) Borreliae, human relapsing fever, and parasite-vector-host relationships. Bacteriol Rev 29:46–74
Fujisaki K, Kitaoka S, Morii T (1975) Hemocyte types and their primary cultures in the argasid tick, Ornithodoros moubata Murray (Ixodoidea). Appl Entomol Zool (Jpn) 10:30–39
Jones LD, Davies CR, Steele GM, Nuttall PA (1988) The rearing and maintenance of ixodid and argasid ticks in the laboratory. Anim Technol 39:99–106
Kurtti TJ, Simser JA, Baldridge GD, Palmer AT, Munderloh UG (2005) Factors influencing in vitro infectivity and growth of Rickettsia peacockii (Rickettsiales: Rickettsiaceae), an endosymbiont of the Rocky Mountain wood tick, Dermacentor andersoni (Acari, Ixodidae). J Invertebr Pathol 90:177–186. doi:10.1016/j.jip.2005.09.001
Lawrie CH, Uzcategui NY, Gould EA, Nuttall PA (2004) Ixodid and argasid tick species and West Nile virus. Emerg Infect Dis 10:653–657
Mattila JT, Burkhardt NY, Hutcheson HJ, Munderloh UG, Kurtti TJ (2007) Isolation of cell lines and a rickettsial endosymbiont from the soft tick Carios capensis (Acari: Argasidae: Ornithodorinae). J Med Entomol 44:1091–1101. doi:10.1603/0022-2585(2007)44[1091:IOCLAA]2.0.CO;2
Moulton JE (1978) Preparation of primary cultures of tick cells. Am J Vet Res 39:1558–1564
Nuttall PA, Labuda M (1994) Tick-borne encephalitis subgroup. In: Sonenshine DE, Mather TN (eds) Ecological dynamics of tick-borne zoonoses. Oxford University Press, Oxford, pp 351–391
Plowright W, Parker J, Pierce MA (1969) The epizootiology of African swine fever in Africa. Vet Rec 85:668–674
Rehacek J (1976) Tick tissue culture and arboviruses. In: Kurstak E, Maramorosch K (eds) Invertebrate tissue culture applications in medicine biology and agriculture. Academic Press, New York, pp 21–33
Růžek D, Bell-Sakyi L, Kopecký J, Grubhoffer L (2008) Growth of tick-borne encephalitis virus (European subtype) in cell lines from vector and non-vector ticks. Virus Res 137:142–146. doi:10.1016/j.virusres.2008.05.013
Turell MJ, Durden LA (1994) Experimental transmission of Langat (tick-borne encephalitis virus complex) virus by the soft tick Ornithodoros sonrai (Acari: Argasidae). J Med Entomol 31:148–151
Turell MJ, Mores CN, Lee JS, Paragas JJ, Shermuhenedova D, Endy TP, Khodjaev S (2004) Experimental transmission of Karshi and Langat (tick-borne encephalitis virus complex) viruses by Ornithodoros ticks (Acari: Argasidae). J Med Entomol 41:973–977
Varma MGR (1989) Progress in the study of human and animal pathogens in primary and established tick cell lines. In: Mitsuhashi J (ed) Invertebrate cell system applications, vol 2. CRC Press, Boca Raton, pp 119–128
Walker AR, Bouattour A, Camicas J-L, Estrada-Pena A, Horak IG, Latif AA, Pegram RG, Preston PM (2003) Ticks of domestic animals in Africa: a guide to identification of species. Bioscience Reports, Edinburgh
Yunker CE (1987) Preparation and maintenance of arthropod cell cultures: Acari, with emphasis on ticks. In: Yunker CE (ed) Arboviruses in arthropod cells in vitro, vol 1. CRC Press, Boca Raton, pp 35–51
Acknowledgments
This research was funded by the Wellcome Trust project 075799 (LBS), and the Grant Agency of the Czech Republic 524/08/1509 and Research Centre of the Ministry of Education, Youth and Sports of the Czech Republic LC06009 (DR); EAG is supported by the European Union 6th Framework grant VIZIER [Structural Genomics]. The authors would like to thank Paula Ruybal for the cell line photographs.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bell-Sakyi, L., Růžek, D. & Gould, E.A. Cell lines from the soft tick Ornithodoros moubata . Exp Appl Acarol 49, 209–219 (2009). https://doi.org/10.1007/s10493-009-9258-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10493-009-9258-y