
Abstract
The widespread use of high throughput genome sequencing technologies has resulted in a significant increase in the number of available sequences, creating new challenges for genome annotation and prediction of protein-coding genes in terms of error detection and quality control. Multiple Sequence Alignments (MSAs) of the predicted protein sequences provide important contextual information that can be used to distinguish errors (caused by artifacts in the raw genome data, badly predicted gene sequences, or the alignment methods themselves) from true biological events. This can be achieved either by human expertise or by statistical analysis of the sequence data. Here, we propose a new approach that uses visual representations of MSAs as inputs for Convolutional Neural Networks (CNN) to classify MSAs into erroneous and non-erroneous categories. The MSAs are extracted from a unique in-house dataset, in which errors are carefully identified. Our model, called De-MISTED (Deep learning for MultIple Sequence alignmenTs Error Detection) identifies MSAs containing erroneous sequences with high accuracy (87%) and sensitivity (92%). Visual explanation techniques show that our model correctly identifies the position of multiple errors of different types (insertions, deletions and mismatches). Close examination of the data showed that our model can also identify errors that were not previously annotated in the data. The De-MISTED method thus contributes to a more robust exploitation of the genome data.











Similar content being viewed by others


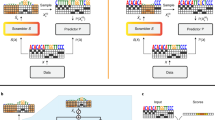
Data Availability
The dataset generated for the current study is available in the Zenodo repository: https://doi.org/10.5281/zenodo.6637475.
Notes
The filtering protocol is a simple in-house program that takes as input an erroneous MSA in XML format and filters out erroneous sequences which are defined by specific start and end tags
References
Aoki G, Sakakibara Y (2018) Convolutional neural networks for classification of alignments of non-coding RNA sequences. Bioinformatics 34:i237–i244
Carroll H, Beckstead W, O’Connor T et al (2007) Dna reference alignment benchmarks based on tertiary structure of encoded proteins. Bioinform (Oxford England) 23:2648–9. https://doi.org/10.1093/bioinformatics/btm389
Chatzou M, Magis C, Chang JM et al (2015) Multiple sequence alignment modeling: methods and applications. Brief Bioinform 2015. https://doi.org/10.1093/bib/bbv099
Chiner-Oms A, González-Candelas F (2016) Evalmsa: A program to evaluate multiple sequence alignments and detect outliers. Evol Bioinform 12:EBO.S40,583. https://doi.org/10.4137/EBO.S40583
Consortium TU (2018) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47 (D1):D506–D515. https://doi.org/10.1093/nar/gky1049
Corpet F, Servant F, Gouzy J et al (2000) ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res 28:267–9. https://doi.org/10.1093/nar/28.1.267
DeBlasio DF, Kececioglu J (2018) Adaptive local realignment of protein sequences. J Comput Biol J Comput Mol Cell Biol 25(7):780–793
Dragan MA, Moghul I, Priyam A et al (2016) Genevalidator: Identify problems with protein-coding gene predictions. Bioinform 32. https://doi.org/10.1093/bioinformatics/btw015
Edgar RC, Batzoglou S (2006) Multiple sequence alignment. Curr Opin Struct Biol 16 (3):368–73
Finn RD, Bateman A, Clements J et al (2014) Pfam: the protein families database. Nucleic Acids Res 42(D1):D222–D230. https://doi.org/10.1093/nar/gkt1223, https://arxiv.org/abs/https://academic.oup.com/nar/article-pdf/42/D1/D222/3643441/gkt1223.pdf
Gibbs R, Rogers J, Katze M et al (2007) Evolutionary and biomedical insights from the rhesus macaque genome. Science 316:222–34
He K, Zhang X, Ren S, Sun J (2016) Deep residual learning for image recognition. In: 2016 IEEE conference on computer vision and pattern recognition (CVPR). https://doi.org/10.1109/CVPR.2016.90, pp 770–778
Jafari R, Javidi M, Kuchaki Rafsanjani M (2019) Using deep reinforcement learning approach for solving the multiple sequence alignment problem. SN Appl Sci 1. https://doi.org/10.1007/s42452-019-0611-4
Jehl P, Sievers F, Higgins D (2015) OD-seq: Outlier detection in multiple sequence alignments. BMC Bioinforma 16:269. https://doi.org/10.1186/s12859-015-0702-1
Kanz C, Aldebert P, Althorpe N et al (2005) The embl nucleotide sequence database. Nucleic Acids Res 33:D29–33. https://doi.org/10.1093/nar/gki098
Katoh K, Standley D, Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30:772–780. Molecular biology and evolution 30. https://doi.org/10.1093/molbev/mst010
Katoh K, Misawa K, Ki Kuma et al (2002) MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res 30:3059–66
Khenoussi W, Vanhoutreve R, Poch O et al (2014) SIBIS: A Bayesian model for inconsistent protein sequence estimation. Bioinform (Oxford England) 30. https://doi.org/10.1093/bioinformatics/btu329
Krizhevsky A, Sutskever I, Hinton GE (2012) Imagenet classification with deep convolutional neural networks. Commun ACM 60:84–90
Larkin M, Blackshields G, Brown N et al (2007) Clustal W and clustal X version 2.0. Bioinformatics 23:2947–2948
Meyer C, Scalzitti N, Jeannin-Girardon A et al (2020) Understanding the causes of errors in eukaryotic protein-coding gene prediction: a case study of primate proteomes. BMC Bioinforma 21
Mircea IG, Bocicor I, Czibula G (2018a) A reinforcement learning based approach to based approach multiple sequence alignment. In: Balas VE, Jain LC, Balas MM (eds) Soft computing applications. Springer International Publishing, Cham, pp 54– 70
Mircea I-G, Bocicor M-I (2014) On reinforcement learning based multiple sequence alignment
Nagy A, Patthy L (2013) MisPred: A resource for identification of erroneous protein sequences in public databases. Database J Biol Databases Curation 2013:bat053. https://doi.org/10.1093/database/bat053
Nagy A, Patthy L (2014) Fixpred: a resource for correction of erroneous protein sequences. Database: The Journal of Biological Databases and Curation
Nagy A, Hegyi H, Farkas K et al (2008) Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinforma 9:353. https://doi.org/10.1186/1471-2105-9-353
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302(1):205–17
O’Leary NA, Wright MW, Brister JR et al (2015) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44(D1):D733–D745. https://doi.org/10.1093/nar/gkv1189. https://arxiv.org/abs/https://academic.oup.com/nar/article-pdf/44/D1/D733/9482930/gkv1189.pdf
Pearson W (2004) Finding protein and nucleotide similarities with fasta. Current protocols in bioinformatics / editoral board. Andreas D Baxevanis [others] Chapter 3. https://doi.org/10.1002/0471250953.bi0309s04
Prosdocimi F, Linard B, Pontarotti P et al (2011) Controversies in modern evolutionary biology: the imperative for error detection and quality control. BMC Genomics 13:5–5
Rajpurkar P, Irvin J, Zhu K et al (2017) Chexnet: Radiologist-level pneumonia detection on chest x-rays with deep learning. arXiv:http://arxiv.org/abs/1711.05225
Russakovsky O, Deng J, Su H et al (2015) Imagenet large scale visual recognition challenge. Int J Comput Vis 115:211–252
Scalzitti N, Jeannin-Girardon A, Collet P et al (2020) A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genomics 21:293. https://doi.org/10.1186/s12864-020-6707-9
Simonyan K, Zisserman A (2015) Very deep convolutional networks for large-scale image recognition. arXiv:http://arxiv.org/abs/1409.1556
Srivastava N, Hinton GE, Krizhevsky A et al (2014) Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res 15:1929–1958
Szegedy C, Liu W, Jia Y et al (2015) Going deeper with convolutions. In: 2015 IEEE conference on computer vision and pattern recognition (CVPR), pp 1–9. https://doi.org/10.1109/CVPR.2015.7298594
Szegedy C, Vanhoucke V, Ioffe S et al (2016) Rethinking the inception architecture for computer vision. In: 2016 IEEE conference on computer vision and pattern recognition (CVPR), pp 2818–2826
Tamura K, Stecher G, Peterson D et al (2013) MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30 https://doi.org/10.1093/molbev/mst197
Thompson J, Higgins D, Gibson T (1994) Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–80
Thompson J, Plewniak F, Poch O (1999) Balibase: A benchmark alignment database for the evaluation of multiple alignment programs. Bioinforma (Oxford England) 15:87–8. https://doi.org/10.1093/bioinformatics/15.1.87
Thompson J, Plewniak F, Ripp R et al (2001) Towards a reliable objective function for multiple sequence alignments. J Mol Biol 314:937–951. https://doi.org/10.1006/jmbi.2001.5187
Thompson J, Thierry JC, Poch O (2003) Rascal: Rapid scanning and correction of multiple sequence alignments. Bioinforma (Oxford England) 19:1155–61. https://doi.org/10.1093/bioinformatics/btg133
Thompson JD (2016) Statistics for bioinformatics : methods for multiple sequence alignment. iSTE Press
Thompson JD, Linard B, Lecompte O et al (2011) A comprehensive benchmark study of multiple sequence alignment methods: Current challenges and future perspectives. PLoS ONE 6
Tong J, Pei J, Otwinowski Z et al (2014) Refinement by shifting secondary structure elements improves sequence alignments. Proteins Struct Funct Bioinform 83. https://doi.org/10.1002/prot.24746
Vanhoutreve R, Kress A, Legrand B et al (2016) LEON-BIS: Multiple alignment evaluation of sequence neighbours using a bayesian inference system. BMC Bioinforma 17. https://doi.org/10.1186/s12859-016-1146-y
Wang H, Wang Z, Du M et al (2020) Score-CAM: Score-weighted visual explanations for convolutional neural networks. In: 2020 IEEE/CVF conference on computer vision and pattern recognition workshops (CVPRW), pp 111–119
Wang Y, Wu H, Cai Y (2018) A benchmark study of sequence alignment methods for protein clustering. BMC Bioinformatics 19. https://doi.org/10.1186/s12859-018-2524-4
Warnow T (2021) Revisiting evaluation of multiple sequence alignment methods. Humana Press Inc., pp 299–317. Methods in Molecular Biology, https://doi.org/10.1007/978-1-0716-1036-7_17
Xuyu X, Dafan Z, Qin J et al (2010) Ant colony with genetic algorithm based on planar graph for multiple sequence alignment. Inf Technol J 9. https://doi.org/10.3923/itj.2010.274.281
Yosinski J, Clune J, Bengio Y et al et al (2014) How transferable are features in deep neural networks?. In: Ghahramani Z, Welling M, Cortes C (eds) Advances in neural information processing systems. https://proceedings.neurips.cc/paper/2014/file/375c71349b295fbe2dcdca9206f20a06-Paper.pdf, vol 27. Curran Associates Inc
Zaal D, Nota B (2015) Adoma: A command line tool to modify clustalw multiple alignment output. Mol Inform 35. https://doi.org/10.1002/minf.201500083
Zhang C, Zheng W, Mortuza S et al (2019) DeepMSA: Constructing deep multiple sequence alignment to improve contact prediction and fold-recognition for distant-homology proteins. Bioinform (Oxford England) 36. https://doi.org/10.1093/bioinformatics/btz863
Acknowledgements
The authors would like to thank the BiGEst bioinformatics platform for technical support. This work was supported by the French Infrastructure Institut Français de Bioinformatique (IFB) ANR-11-INBS-0013, ANR ArtIC ANR-20-THIA-0006 and Institute funds from the French Centre National de la Recherche Scientifique and the University of Strasbourg.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
We include supplementary material providing additional Score-CAM [47] results obtained by our proposed models (A) and (B).
Pierre Collet, Julie D. Thompson and Anne Jeannin-Girardon contributed equally to this work.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khodji, H., Collet, P., Thompson, J.D. et al. De-MISTED: Image-based classification of erroneous multiple sequence alignments using convolutional neural networks. Appl Intell 53, 18806–18820 (2023). https://doi.org/10.1007/s10489-022-04390-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10489-022-04390-7