
Abstract
Streptomyces leeuwenhoekii strains C34T, C38, C58 and C79 were isolated from a soil sample collected from the Chaxa Lagoon, located in the Salar de Atacama in northern Chile. These streptomycetes produce a variety of new specialised metabolites with antibiotic, anti-cancer and anti-inflammatory activities. Moreover, genome mining performed on two of these strains has revealed the presence of biosynthetic gene clusters with the potential to produce new specialised metabolites. This review focusses on this new clade of Streptomyces strains, summarises the literature and presents new information on strain C34T.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Streptomyces is the most studied of the filamentous actinobacteria and a prolific source of specialised metabolites with biological activities of importance in human health and in agriculture (Kieser et al. 2000). The genomes of streptomycetes are gifted with sets of gene clusters encoding the biosynthetic pathways for a variety of specialised metabolites (Gomez-Escribano et al. 2016). Recently, the search for new pharmacologically active and agriculturally useful compounds has turned to streptomycetes from little-scrutinised and extreme biomes under the premise that extreme abiotic factors will select rare diversity with unique chemistry (Bull et al. 2016).
Since 2004, the Atacama Desert, considered the driest and oldest desert on Earth, has been the subject of extensive culture-dependent microbial surveys in soils of hyper-aridity (Salar de Atacama and Cerro Chajnantor), extreme hyper-aridity (Yungay) and the most extreme hyper-aridity (Lomas Bayas) with the conclusion that it is, surprisingly, an abundant source of soil-dwelling microorganisms (Okoro et al. 2009; Busarakam 2014; Idris 2016; Bull et al. 2018). A group of streptomycetes isolated from a soil sample collected from Chaxa Lagoon in the Salar de Atacama stands out for its ability to synthesise a variety of new specialised metabolites with potentially useful biological activities. These strains formed a distinct clade in the Streptomyces 16S rRNA gene tree and were subsequently identified as a novel species, named Streptomyces leeuwenhoekii, of which strain C34 is the type strain (DSM 42122, NRRL B-24963) (Busarakam 2014; Busarakam et al. 2014; Elsayed et al. 2015).
This review article is devoted to these S. leeuwenhoekii strains, summarises the current literature on the specialised metabolites produced by them and provides new insights into the biosynthetic gene clusters for the chaxalactins and lasso peptides of strain C34T.
An overview of Streptomyces leeuwenhoekii species: habitat and phylogeny
The Chaxa Lagoon, located in Los Flamencos National Reserve at 2300 metres above sea level in the Salar de Atacama near Toconao in northern Chile, is a hyper-arid and hypersaline lake and possesses the highest content of sodium, sulphate, magnesium, bicarbonate, chlorine, nitrate and arsenic ions when compared with other lagoons, ponds and springs found in Salar de Huasco, Salar de Ascotán and Salar de Atacama (Dorador et al. 2009). In October 2004, a soil sample collected from the Chaxa Lagoon area, with low moisture content (0.007%) and scarce total organic matter (0.03%), was subjected to selective isolation of actinomycetes, resulting in the isolation of strains of the genus Streptomyces (Okoro et al. 2009).
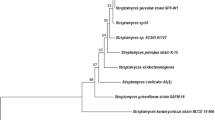
The majority of these isolates, including strains C34, C38, C58 and C79, formed a distinct subclade in the phylogenetic tree of 16S rRNA genes of the genus Streptomyces, sharing 99.5–99.9% nucleotide sequence identity (Busarakam 2014; Okoro et al. 2009). The S. leeuwenhoekii strains grouped together in a clade well-supported by maximum likelihood and maximum parsimony tree-making algorithms (Fig. 1), sharing 99.5% sequence identity with the 16S rRNA genes of Streptomyces mexicanus and Streptomyces warraensis, their closest neighbours. Multi-locus sequence analysis (Rong and Huang 2012) based on the partial sequences of five housekeeping genes (atpD, gyrB, rpoB, recA and trpB) confirmed that strains C34, C38, C58 and C79 were not phylogenetically closely related to any Streptomyces type strains and formed a new centre of taxonomic variation in the genus Streptomyces, a result that was further underpinned by phenotypic and morphological tests (Busarakam 2014). Thus the name S. leeuwenhoekii was proposed, with strain C34 (subsequently referred to as C34T) as the type strain and strains C38, C58 and C79 as additional members. Strain C34T produces an extensively branched substrate mycelium with aerial hyphae that differentiate into smooth-surfaced spores often arranged in a spiral fashion (Fig. 2) (Busarakam et al. 2014).

Maximum likelihood (ML) phylogenetic tree based on the 16S rRNA gene sequences of S. leeuwenhoekii strains C34T, C38, C58 and C79 and closely related type-strains of the genus Streptomyces. The numbers above the branches are bootstrap values greater than 60% for ML (left) and maximum parsimony (right). Sequences of type strains closely related to S. leeuwenhoekii C34T were retrieved from EZBioCloud webserver https://www.ezbiocloud.net (Yoon et al. 2017). The phylogenetic tree was generated using the GGDC webserver (Meier-Kolthoff et al. 2013) available at http://ggdc.dsmz.de/

Scanning electron micrograph of S. leeuwenhoekii strain C34T. Scale bar at top left represents five microns
The gifted S. leeuwenhoekii species, an untapped source of specialised metabolites
Below, we review both the specialised metabolites known to be made by these organisms, as well as their biosynthetic potential as revealed by genome sequencing.
S. leeuwenhoekii strain C34T
The ansamycins are type I polyketide macrolides often with potent anti-bacterial activity and include the clinically used rifamycin (Floss et al. 2011). Twenty one Streptomyces strains isolated from the Atacama Desert, including strain C34T but not the other S. leeuwenhoekii strains, were screened for the presence of the gene encoding 3-amino-5-hydroxybenzoic acid (AHBA) synthase; this enzyme is required for the biosynthesis of AHBA, the precursor molecule for the ansamycins, and its gene is thus a potential key indicator of an ansamycin biosynthesis gene cluster. Only C34T gave a PCR amplicon of the predicted size and DNA sequencing confirmed that it did indeed correspond to an AHBA synthase gene. Metabolite profiling of this strain led to the isolation of four new naphthalenic ansamycin-type polyketides subsequently named chaxamycin A–D after Chaxa Lagoon. All four chaxamycins inhibit Staphylococcus aureus (Rateb et al. 2011a), with chaxamycin D being most active (minimal inhibitory concentration [MIC] 0.05–0.13 µg ml−1). Chaxamycin D also inhibited a number of methicillin-resistant isolates (MRSA, with the exception of the epidemic MRSA 16 strain) with a MIC of 0.25–0.6 µg ml−1. Chaxamycins A–C showed moderate inhibition (46, 45 and 41%, respectively) of the ATPase activity of the human Hsp90 (Rateb et al. 2011a), indicating potential application in cancer chemotherapy (Maloney and Workman 2002).
Based on the well-founded “one strain many compounds” (OSMAC) principle, whereby the metabolite profile of a microorganism can change dramatically when the strain is grown under different nutritional conditions and abiotic parameters, further metabolic profiling of S. leeuwenhoekii C34T was carried out using eight different culture media. Indeed, cultivation of S. leeuwenhoekii C34T in a defined medium led to the isolation of a new group of 22-membered macrolactone polyketides, chaxalactins A–C, in addition to the known siderophore deferroxamine E (Rateb et al. 2011b). Use of the International Streptomyces Project (ISP) medium No. 3 led to the production of the known hygromycin A and 5″-dihydrohygromycin A, whose synthesis was not detected in any other medium. Differential production of chaxamycin A–D was observed in all media tested; for example, chaxamycins C and D were only produced in modified ISP 2 medium, while chaxamycin B was detected in malt-extract peptone, modified Fries media 1 and 2 and also in ISP 2; production of chaxamycin A was detected in all eight media tested, making this molecule a good chemical marker for the strain (Rateb et al. 2011b).
Production of a compound with similar mass to charge ratio (m/z) to that of 14-deoxychaxalactin B (or chaxalactin A) was detected in Streptomyces sp. IB 2014/I/78-8, a distantly related Streptomyces strain to S. leeuwenhoekii C34T and isolated from a Siberian cave ecosystem (Axenov-Gibanov et al. 2016). This finding suggests that these strains might share a highly similar chaxalactin PKS, which is not surprising given the number of examples of similar biosynthetic gene clusters occurring in distinct actinomycetes often isolated from different geographical locations. For instance, the rifamycin biosynthesis gene cluster is present in Amycolatopsis mediterranei S699, a mutant of a strain isolated in France (Kim et al. 1992; Margalith and Beretta 1960) and in Salinispora arenicola CNS-205, isolated from Palau (Gontang et al. 2007; Schultz et al. 2008; Wilson et al. 2010).
Generation of a high-quality genome sequence and genome mining
Given the encouraging results obtained using the OSMAC approach with S. leeuwenhoekii C34T, we set out to explore the full metabolic potential of the strain by analysing its genome sequence, searching for additional biosynthetic gene clusters with potential metabolite novelty (Gomez-Escribano et al. 2015).
A draft genome sequence of S. leeuwenhoekii C34T, obtained from Illumina GA IIx 100 bp paired-end reads and consisting of 658 contigs and a total genome size of 7.86 Mb, was already available (Busarakam et al. 2014). However, initial inspection of this sequence using Artemis (Rutherford et al. 2000), and particularly the GC Frame Plot option contained within it (Bibb et al. 1984), revealed many likely mis-assemblies in contigs containing modular type I polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS) genes. The short-size of the quality reads obtained from high mol% GC DNA (streptomycetes generally possess a GC content of around 73 mol% G+C) using Illumina sequencing (generally less than 250 nt) hampers the correct assembly of the repetitive regions characteristic of these genes (Castro et al. 2015; Gomez-Escribano et al. 2015, 2016). We thus set out to establish a high-quality genome sequence of S. leeuwenhoekii C34T by combining two next generation sequencing technologies: the second-generation Illumina MiSeq renowned for very accurate short nucleotide-reads and the third-generation Single Molecule, Real-Time (SMRT) sequencing platform from Pacific Biosciences (PacBio) that gives long, but less accurate, nucleotide-reads (in our experience with Streptomyces DNA, around 5 kb on average after quality filtering) (Gomez-Escribano et al. 2015).
PacBio sequencing yielded one large contig of 7.9 Mb that was presumed to be the chromosome of S. leeuwenhoekii C34T, and two additional contigs; one of 94.7 kb that contained direct repeats of 8.4 kb present at both ends suggesting that this sequence was a circular plasmid that the assembly programme had failed to deduce, and which was referred to subsequently as pSLE1; and a 9.6 kb contig that appeared to consist of assembled error-containing reads that matched a segment of the 7.9 Mb chromosomal contig and which was discarded from further analysis. Illumina MiSeq sequencing yielded 279 contigs assembled into 175 scaffolds, and surprisingly revealed a second plasmid of 132 kb, contained on two contigs, that had not been present in the assembled PacBio sequence. These two contigs were merged using the PacBio corrected reads, and this sequence was referred to subsequently as the linear plasmid pSLE2 (Gomez-Escribano et al. 2015).
The chromosomal contig generated by PacBio then served as a reference sequence to map the Illumina contigs and to correct the less accurate PacBio sequence (confirmed using Artemis and GC Frame Plot), giving rise to a total of 2976 corrections and indicating an error rate in the PacBio sequence of 0.038%. Most of these differences were omissions in the PacBio sequence of a C or G in homopolymeric runs of three or more Cs or Gs. Further examination of the sequence and contig database revealed an Illumina contig of 5 kb that overlapped one end of the chromosomal sequence, extending its length. This gave rise to a final S. leeuwenhoekii C34T chromosome sequence of 7,903,895 nucleotides (72.76 mol% G+C) that includes just one copy of the 5′-Terminal Inverted Repeat (TIR) that extends for 388 kb; thus the true size of the chromosome is likely to be approximately 8.29 Mb (Gomez-Escribano et al. 2015).
Genome-mining of the final sequence using antiSMASH version 2.0 (Blin et al. 2013) revealed the presence of 35 biosynthetic gene clusters (Gomez-Escribano et al. 2015), several of which are described in further detail below.
The chaxamycin biosynthesis gene cluster
Genome mining led to the identification of a stretch of genomic DNA of 80.2 kb that appeared to encode chaxamycin biosynthesis. Twenty seven clustered genes were found: eight appear to be involved in AHBA biosynthesis, five encode type I PKS subunits, two encode transcriptional regulators and the others are responsible for various tailoring enzymes. Comparison of the putative chaxamycin biosynthesis gene cluster with others devoted to producing ansamycins, such as that for rifamycin in A. mediterranei S699 (AF040570.3), naphthomycin in Streptomyces sp. strain CS (GQ452266.1), and saliniketal in Sal. arenicola CNS-205 (CP000850.1), allowed the identification of its likely boundaries. Cloning of a genomic segment of 145 kb that contained the 80.2 kb region in Streptomyces coelicolor M1152 (which does not produce ansamycins) resulted in the production of all four chaxamycin species in the culture supernatant, confirming that all of the genes specifically required for chaxamycin biosynthesis were present in the 145 kb fragment. Bioinformatic analyses of the genes present in the 80.2 kb region allowed the identification of sle10350–sle10300 (cxmA–E) that encode the polyketide synthase subunits, sle10290 (cxmF) that encodes for a homologue of an amide synthase involved in polyketide release and ansa-ring formation, sle10200 that encodes for a homologue of rif-orf19 that is involved in the formation of the naphthalene ring in rifamycin, sle10150 (cxm24) that encodes for the only putative methyltransferase found in the cluster that could be responsible for C-methylation of the C-3 of the AHBA moiety in the naphthalene ring of all chaxamycin species (Fig. 3), and sle10160 (cxm23) that encodes for a cytochrome P450 monooxygenase potentially involved in the formation of the hydroxyfuran ring of chaxamycin D (Castro et al. 2015).

Chemical structures of molecules produced by members of the phyletic clade Streptomyces leeuwenhoekii. Chaxamycin A–D (compounds 1–4) and chaxalactin A–C (5–7) are produced by strain C34T; atacamycins A–C (8–10) are produced by strain C38; chaxapeptin (11) is produced by strain C58
Deletion of the gene that codes for the AHBA synthase (cxmK) in S. leeuwenhoekii C34T gave rise to a S. leeuwenhoekii mutant (M1653) that was unable to produce chaxamycins in liquid culture. Constitutive expression in trans of cxmK in strain M1653 did not restore chaxamycin production, a result attributed to a polar effect of the deletion on the expression of cxmL downstream of cxmK. However, chaxamycin production was restored in the mutant when a liquid culture of M1653 was supplemented with AHBA, which confirmed the identification of the chaxamycin biosynthesis gene cluster in the native strain (Castro et al. 2015). This led us to conjecture that supplementing M1653 cultures with analogues of AHBA might lead to the production of novel chaxamycins, however preliminary results have yielded only intermediates in chaxamycin biosynthesis (Castro et al., unpublished results).
Chaxamycins and rifamycins are structurally related antibiotics. Rifamycin B is able to inhibit bacterial growth by binding to the β-subunit of the DNA-dependent RNA polymerase encoded by rpoB. Only one copy of this gene occurs in the genome of A. mediterranei and is located at one end of the rifamycin biosynthesis gene cluster (964 nt downstream of rif-orf36). The encoded RpoB (Ames_0654) contains five amino acids in the rif I region known to confer rifampicin resistance to A. mediterranei and to several other organisms that harbour some of these five residues (Campbell et al. 2001): glutamine (Q432), threonine (T434), isoleucine (I437), aspartate (D438) and asparagine (N447) (Fig. 4). Consistent with this, cloning of ames_0654 into the rifampicin-sensitive Mycobacterium smegmatis conferred rifampicin resistance on the strain. Moreover, when point mutations (D430Q, S436D, and S445N) were introduced separately into rpoB of the rifampicin-sensitive Mycobacterium tuberculosis and the three mutant alleles cloned individually into M. smegmatis, each of them conferred rifampicin resistance on the heterologous host (Floss and Yu 2005). In S. leeuwenhoekii, the gene that encodes for RpoB (Sle29840, 77% amino acid sequence identity to Ames_0654) lies 1.29 Mb away from the chaxamycin gene cluster, and its product does not have the amino acid residues required for resistance to rifamycin at the expected positions, instead possessing the same amino acids as S. coelicolor M145 RpoB, which is sensitive to rifamycin (Fig. 4). Thus, it is conceivable that despite the structural similarity to rifamycin, the chaxamycins may not exert their antibiotic activity by binding to RpoB.

Alignment of the amino acid sequences of the RNA polymerase β-subunits (RpoB) of A. mediterranei S699, S. coelicolor M145 and S. leeuwenhoekii C34T illustrating the amino acid residues that confer resistance to rifampicin to A. mediterranei S699
The chaxalactin biosynthesis gene cluster
Biosynthesis of the 22-membered macrolactone chaxalactins A–C is believed to be carried out by a type I PKS distinct from that for the chaxamycins (Castro 2015). Chaxalactin A (Fig. 3) is made from 12 building blocks incorporated by a type I PKS composed of one loading module that primes the biosynthesis of the nascent polyketide chain by incorporating a propionate molecule and 11 extender modules that incorporate additional building blocks into the growing chain. Eight of these extender modules were predicted to incorporate acetate units derived from malonyl-CoA, and another three modules were predicted to incorporate propionate units. The structure of chaxalactin A also revealed that the hydroxyl-group from the malonyl-CoA unit introduced by module 7 at position C-9 is fully reduced, indicating that at least three cis-acting PKS domains (ketoreductase (KR), dehydratase (DH) and enoylreductase (ER)) should be present in module 8 of the chaxalactin PKS. In addition, the cyclisation of the final polyketide chain to form the macrolide ring observed in the structure of chaxalactin A is likely catalysed by a TE domain that is usually located at the C-terminus of the last module of the PKS (Staunton and Weissman 2001). Thus, we searched the S. leeuwenhoekii C34T genome sequence for genes that could encode a type I PKS composed of 12 modules, and found five clustered genes encoding PKS subunits: sle61410, 61430, 61440, 61450 and 61460 (Table 1). The protein domains predicted from the amino acid sequences of these genes gave the following architecture: one loading module, 11 extender modules and a TE domain at the C-terminus of the last PKS subunit, entirely consistent with the modules required for chaxalactin A biosynthesis. Inspection of the rest of the genome sequence failed to reveal any equally suitable type I PKS gene clusters and thus the PKS subunits encoded in the genes mentioned above were named CxlA–E, respectively. In addition, module 8 present in CxlD included the KR, DH and ER domains predicted to be required for the full reduction of the hydroxyl group at position C-9.
Analysis of the domains present in each module of the putative chaxalactin PKS was then carried out to predict their functionality. All 12 acyl carrier protein (ACP) domains possess a serine residue required for attachment of the 4′-phosphopantetheine prosthetic group needed to shuttle extender units and polyketide intermediates from one module to the one downstream. Each of the 12 modules possesses one acyl transferase (AT) domain, which specifically recognises the extender unit that will be linked to the growing polyketide chain. Eight AT domains were predicted to recognise malonyl-CoA as a substrate since they contained the motifs GHS(I/V)G and HAFH, while four AT domains contained the motifs GHSQG and YASH expected for recognition of (2S)-methylmalonyl-CoA, including that of the loading module. Moreover, the order of these AT domains along the linear PKS assembly line is precisely that predicted for chaxalactin A. All AT domains were also predicted to be active because of the presence of the catalytic serine and histidine residues (shown in bold above) in the first and second motifs, respectively (Keatinge-Clay 2012). The linkage of an extender unit to the growing polyketide chain is catalysed by an extending ketosynthase (KS) domain by means of decarboxylation and Claisen condensation (Dewick 2009). The chaxalactin PKS possesses 12 KS domains, 11 of which are located in modules 1–11; all of these 11 are predicted to be functional based on the presence of a cysteine and two histidine residues in the catalytic triad present in motifs TACSSS, EAHGTG and KSNIGHT (Fernández-Moreno et al. 1992; Keatinge-Clay 2012). The remaining KS domain lies in the loading module with the reactive cysteine residue of the TACSSS motif mutated to glutamine and consequently it is unable to perform any Claisen condensation but it could still initiate the elongation of the polyketide chain by catalysing the decarboxylation of acyl-CoA starting molecules (Dewick 2009). This loss of function mutation is common in the KS domains of loading modules of type I PKSs which are consequently referred to as KSQ domains (Keatinge-Clay 2012).
Eleven KR domains were found in the chaxalactin PKS distributed in modules 1–11 (only the loading module lacked a KR domain). KR domains are involved in the reduction of the β-keto group of the polyketide intermediate using NADPH as a cofactor, leaving a β-hydroxyl group as a result (Keatinge-Clay 2007, 2012). Reductase-active KR domains can be inferred by the presence of three diagnostic regions for binding NADPH: a GxGxxG motif, a swap linker and a WGxW motif (Garg et al. 2014). Also the presence of the catalytic triad lysine, serine and tyrosine should also be present in active KR domains (Reid et al. 2003). All of the KR domains found in the chaxalactin PKS possess all of the described motifs and are expected to be active, consistent with the structures of chaxalactins A–C.
After the reduction of the β-keto group of the polyketide intermediate by a KR domain, a DH domain catalyses the dehydration of a β-hydroxyl group of the same polyketide intermediate, leaving an unsaturation in the α,β position. DH domains are present in modules 1, 3, 4, 7, 8, 9, 10 and 11 of the chaxalactin PKS, which are predicted to be active based on the presence of histidine and aspartate residues in motifs HxxxGxxxxP and D(A/V)(V/A)(A/L)(Q/H), respectively (Valenzano et al. 2010). The structure of chaxalactin A contains unsaturations at positions C-23–C-22 (E), C-19–C-18 (E), C-17–C-16 (Z), C-11–C-10 (E), C-7–C-6 (E), C-5–C-4 (E), C-3–C-2 (E), and a full reduction at C-9–C-8. This suggests that dehydration reactions occur on the polyketide intermediates catalysed by active DH domains. Module 8 possesses three cis-acting modifying domains: KR, DH and ER. ER domains reduce the α,β double bond that results from the reduction and dehydration of the β-ketone group by a KR domain and a DH domain, respectively. The ER domain of module 8 has a conserved NADPH-binding motif, HxAx(G/T)GV(G/A)(M/S)A (Keatinge-Clay 2012) and thus is expected to be functional.
The final step in the biosynthesis of chaxalactin A requires the release and cyclisation of the chaxalactin dodecaketide intermediate, catalysed by a TE domain, which in the chaxalactin PKS is fused to the C-terminus of module 11. TE domains harbour a catalytic triad composed of histidine, aspartate and serine residues (Tsai et al. 2001). This catalytic triad is present in the chaxalactin TE domain which is therefore predicted to be functional.
Chaxalactins B and C are thought to be derived from chaxalactin A (Fig. 5) (Castro 2015). The structure of chaxalactin B indicates that it is a product of a tailoring reaction that involves a hydroxylation at position C-14 of chaxalactin A. Subsequently, a C-15 O-methylation reaction at the O-3 position of chaxalactin B would give rise to chaxalactin C (Figs. 3, 5). Thus we might expect that genes encoding for a hydroxylase and a methyltransferase should be located near the chaxalactin PKS of S. leeuwenhoekii C34T. Indeed, upstream of the putative chaxalactin PKS genes, sle61400 encodes for a cytochrome P450 that seems to be transcriptionally coupled to cxlA. Cytochromes P450 are usually involved in hydroxylation reactions (Podust and Sherman 2012) and it is plausible that sle61400 is part of the cluster. Thus, the product of that gene was named CxlF ( Fig. 5; Table 1). In addition, sle61390 encodes for a methyltransferase and is located upstream of CxlF and sle61390 may also be part of the gene cluster. Thus the product of sle61390 was named CxlG.

Proposed biosynthetic pathway for chaxalactin A–C based on genome mining analysis of S. leeuwenhoekii C34T. Red asterisks indicate additional genes that might also be involved in chaxalactin biosynthesis (Table 1)
Other genes that may be part of the chaxalactin biosynthesis cluster include sle61490 and sle61500 located downstream of cxlE and that encode for Rieske non-haem iron oxygenases, also involved in hydroxylation reactions (Que Jr and Tolman 2008). sle61510, located downstream of sle61500, is also predicted to encode a methyltransferase and may also be part of the biosynthetic gene cluster (Fig. 5; Table 1).
Genes for three transport systems were also found near the chaxalactin PKS genes: sle61320, encoding a drug/metabolite transporter (DMT) permease, and sle61350 and sle61360, encoding two ABC transporters. sle61310, located 5390 nt upstream of cxlF and encoding a TetR regulatory protein homologue, lies adjacent to sle61320, and may be responsible for regulation of the latter. Protein domain analysis of Sle61320 indicates that it encodes for a DMT permease with two EamA protein domains with 10 transmembrane helices. EamA domains have been found to be involved in the export of nucleotide sugars (Västermark et al. 2011), hence sle61320 might not be responsible for chaxalactin transport. A similar caveat exists for sle61350 and sle61360, which appear to encode sugar ABC transporter permeases.
Thus the core of the chaxalactin biosynthesis gene cluster (i.e. those genes that encode the minimum set of enzymes required for chaxalactin A–C biosynthesis) lies between genes sle61390–sle61460 spanning a region of 62.7 kb (nt coordinates 7,158,590–7,221,280) (Fig. 5).
Three lasso peptide biosynthesis gene clusters
Lasso peptides are ribosomally synthesised and post-translationally modified peptides having a characteristic structural “lariat-knot” motif that confers a variety of different bioactivities. They are synthesised by relatively small biosynthetic gene clusters which contain genes encoding a precursor peptide and enzymes that process the precursor to yield the mature form of the lasso peptide (Maksimov et al. 2012; Pan et al. 2012).
The S. leeuwenhoekii C34T genome sequence revealed three gene clusters, referred to as lp1, lp2 and lp3, respectively, encoding proteins typically involved in lasso peptide biosynthesis: genes for the precursor peptide, lasso peptide cyclase, a protein of unknown function and a lasso peptide protease. Only lp1 and lp2 contained genes coding for putative exporter proteins, while lp3 was encoded by the plasmid pSLE2 (Gomez-Escribano et al. 2015). An identical nucleotide sequence corresponding to the precursor peptide gene (sle29750) of lp1 was found in the genome of Streptomyces cyanogriseus (accession CP010849, region 4,603,762–4,603,872 nt) and the amino acid sequence of the predicted mature form of the lasso peptide derived from lp1 is identical to that predicted for an uncharacterised protein encoded by TUE45_07344 (accession CUW32593) of Streptomyces reticuli and SAMN05421773_10837 (accession SFC97848) of Streptomyces aidingensins. Highly similar precursor peptides were also identified in sequence data from 11 further Streptomyces species and four other actinomycetes, indicating the likely widespread occurrence of homologues of the lp1 gene cluster. However, no lasso peptides are known to be made by these species. The predicted mature form of the lasso peptide encoded in lp3 is identical to chaxapeptin synthesised by S. leeuwenhoekii C58 (Elsayed et al. 2015). Although lp3 lacks genes for a putative exporter, lasso peptide 3 was readily detected in the supernatant and mycelial extract of S. leeuwenhoekii C34T grown in TSB/YEME medium. Cloning of lp3 in S. coelicolor M1152 also led to the synthesis of lasso peptide 3. Thus the export of this lasso peptide is presumably mediated by a non-specific efflux pump present in many streptomycetes (Gomez-Escribano et al., unpublished results). The amino acid sequence of the predicted mature form of the lasso peptide of lp2 appears thus far to be novel and could not be detected in the draft genome sequence of strain C58 (Gomez-Escribano et al. 2015; Razmilic 2017); lp2 is currently under study (Gomez-Escribano et al., unpublished results).
Gene clusters for other specialised metabolites
Genes (sle01610–sle01870) for the biosynthesis of hygromycin A (Palaniappan et al. 2006) were found in the genome of S. leeuwenhoekii C34T located within the TIR, suggesting that this gene cluster could be duplicated at the other end of the chromosome. Gene clusters for desferrioxamine E (sle44550 to sle44600) and ectoine (sle52690 to sle52700) biosynthesis were also identified using antiSMASH version 2.0 (Blin et al. 2013; Gomez-Escribano et al. 2015; Razmilic 2017).
A putative biosynthetic gene cluster that spans a region of 64 kb encodes a hybrid trans-AT PKS/NRPS, post-synthesis modification enzymes (including a halogenase), transporters and regulators (sle09280–sle09570; nucleotide 1,083,651–1,147,687), and could potentially synthesise a novel specialised metabolite. This gene cluster is similar to that for leinamycin biosynthesis in Streptomyces atroolivaceus, which consists of 28 genes (Hara et al. 1990). All of the proteins encoded in the leinamycin gene cluster except LnmC (a non-characterised protein) and LnmS (ABC transporter component) have similar counterparts (≥ 40% amino acid sequence identity) encoded by the hybrid trans-AT PKS/NRPS cluster of strain C34T. LnmB (a homologue of Cxm21 of the chaxamycin biosynthesis pathway) and a cytochrome P450 (LnmA or LnmZ) are thought to be involved in hydroxylation of C-8 and C-4′, and oxygenation of S-1′ of leinamycin (Tang et al. 2004). On the other hand, there are five genes in the hybrid trans-AT PKS/NRPS cluster that do not have homologues in the leinamycin biosynthesis gene cluster: sle09560 (TetR transcriptional regulator), sle09540 (methyltransferase), sle09470 (halogenase), sle09390 (β-ketoacyl-acyl synthase) and sle09330 (glyoxalase/bleomycin resistance protein/dioxygenase). Despite intensive efforts to detect the product of the C34T gene cluster, we have not been able to detect a chlorinated compound in the culture supernatants of either C34T or S. coelicolor M1152 harbouring a plasmid with the entire 64 kb region (Razmilic 2017).
Genome scale model (GSM) as a tool for metabolic engineering
A GSM of S. leeuwenhoekii C34T was developed as a tool to aid metabolic engineering for the improved biosynthesis of specialised metabolites such as the chaxamycins and chaxalactins. The model was built using the high-quality genome sequence, the derived putative metabolic pathways of strain C34T, and the Gene-Protein-Reaction (GPR) information for streptomycetes retrieved from the Kyoto Encyclopaedia of Genes and Genomes (KEGG) database. The model, iVR1007, consisted of 1722 reactions, 1463 metabolites and 1007 genes that showed 83.7% accuracy in correctly predicting experimental data for growth of S. leeuwenhoekii C34T on different carbon, nitrogen and phosphorous sources. The model was used to predict metabolic engineering targets for enhancing the biosynthesis of chaxamycins, chaxalactins and other specialised metabolites through in silico simulations. The predicted knock-out targets, sle03600 (encoding homoserine O-acetyltransferase) and sle39090 (encoding trehalose-phosphate synthase), were associated with improving precursor supply towards chaxalactin and chaxamycin biosynthesis, respectively. Conversely, overexpression of sle41020 (encoding a UTP-glucose-1-phosphate uridylyltransferase) is predicted to increase flux to chaxamycin production, while overexpression of the following genes is predicted to increase flux to both the chaxamycins and the chaxalactins: sle28060, sle28760, sle22410 or sle22940 (encoding methyl-malonyl-CoA mutase, enhancing the flux to (2S)-methylmalonyl-CoA), and sle47660 and either sle27560, 44630, 39830 or 59710 (genes encoding acetyl-CoA carboxylase predicted to increase the intracellular pool levels of malonyl-CoA). Both (2S)-methylmalonyl-CoA and malonyl-CoA are precursors for both chaxamycin and chaxalactin biosynthesis and therefore it was not surprising that the same targets were found for both specialised metabolites (Razmilic et al., this Special Issue). The predictions generated with the GSM are currently being experimentally tested in S. leeuwenhoekii C34T (Razmilic et al., unpublished results).
S. leeuwenhoekii strain C58
The lasso peptide, chaxapeptin
Mining the draft genome sequence of strain C58 (GCA_001047315) led to the identification of a genomic region of 2.6 kb containing four genes encoding proteins similar to those involved in the biosynthesis of the lasso peptide lariatin. cptA encodes a 44 amino acid precursor peptide of which the 15 C-terminal residues were predicted to constitute the core peptide (the segment of the precursor peptide retained in the final product); cptC encodes a homologue of a peptide cyclase; cptB1 encodes a protein of unknown function and cptB2 encodes a lasso peptide protease. Subsequent growth of S. leeuwenhoekii C58 in ISP 2 (and modified ISP 2) liquid medium led to the isolation of chaxapeptin (Fig. 3), which is similar to another lasso peptide, sungsanpin, produced by a marine streptomycete (Elsayed et al. 2015). Chaxapeptin displayed significant inhibitory activity in a cell invasion assay with human lung cancer cell line A549.
Comparison of S. leeuwenhoekii C34T and S. leeuwenhoekii C58 genomes
The draft genome sequence of S. leeuwenhoekii C58 was determined using Illumina MiSeq technology which yielded 502 contigs with a total genome size of 6.87 Mbp (Genbank accession number: GCA_001047315.1). Seventy eight percent of the predicted proteins of strain C58 have close homologues in strain C34T with identity in the range of 97–100% (amino acid sequence coverage > 90%), while 1.5% of the proteins had homologues in strain C34T with identities of 70–97% (amino acid sequence coverage > 90%).
A search for the chaxamycin biosynthesis genes of strain C34T (Castro et al. 2015) in S. leeuwenhoekii C58 revealed that out of 27 genes, 19 were found with identity values at the protein level in the range of 99–100% (sequence coverage > 90%), including all of the AHBA biosynthesis enzymes, the amide synthase CxmF and Cxm19. Only partial alignments for proteins CxmY, Cxm1 and for all five PKS units were observed, presumably reflecting the draft nature of the C58 genome sequence. Thus, it seems plausible that strain C58 might also be an ansamycin-type polyketide producer.
Similarly, only incomplete lp1 and lp2 biosynthesis gene clusters were detected in the C58 sequence, lacking genes encoding crucial proteins such as the precursor peptide, while few homologues of the chaxalactin biosynthesis machinery of S. leeuwenhoekii C34T (Castro 2015) were convincingly identified (only partial sequences of the polyketide biosynthesis enzymes CxlB and CxlE were detected with 99% amino acid sequence identity but with low sequence coverage (5–59%)). Hence whether the chaxalactin biosynthesis gene cluster occurs in strain C58 remains to be resolved.
Other biosynthetic gene clusters found in S. leeuwenhoekii C34T such as desferrioxamine E and ectoine are present in S. leeuwenhoekii C58 with derived protein sequence identities higher than 98% (99% of amino acid sequence coverage), but, perhaps surprisingly, the hygromycin A biosynthesis gene cluster appears to be completely absent from strain C58, potentially reflecting its location in the TIR of strain C34 (TIRs vary considerably between Streptomyces strains of the same species (Choulet et al. 2006; Weaver et al. 2004), suggesting that they are subject to relatively frequent DNA loss and acquisition).
S. leeuwenhoekii strain C38
The atacamycins
Atacamycins A–C are three new 22-membered macrolactone polyketides that are produced by S. leeuwenhoekii C38 Nachtigall et al. 2011). All atacamycins showed weak anti-bacterial activity towards the phytopathogenic strain Ralstonia solanacearum DSM 9544. Atacamycin B exhibited the strongest activity against phosphodiesterase (PDE-4B2) with an IC50 value of 1.38 mM, whereas atacamycin A and C displayed an IC50 of 2.28 and 4.07 mM, respectively. The atacamycins were also tested for anti-proliferative activity against a panel of 42 different human tumour cell lines, of which atacamycin A was the most active against cell lines of colon cancer (CXF DiFi), breast cancer (MAXF 401NL) and uterine cancer (UXF 1138L) with IC50 values ranging from 2.66 to 5.93 mM (Nachtigall et al. 2011). Based on these data, the atacamycins have been proposed as drug candidates for the treatment of inflammatory diseases, including chronic obstructive pulmonary disease (Diamant and Spina 2011; Nachtigall et al. 2011).
S. leeuwenhoekii strain C79
A potential source of novel antibacterial agents
Strains C79 and C38 displayed different metabolite profiles when compared to other streptomycetes isolated from the Atacama Desert (Nachtigall et al. 2011). To our knowledge, there are no published data on the compounds produced by strain C79; while four new specialised metabolites with antibacterial activity have been detected experimentally, their structures have not yet been determined (Fiedler et al., unpublished results; Idris (2016)).
Summary
The S. leeuwenhoekii strains discussed in this review have proven to be a prolific source of new chemical entities with anti-microbial and anti-cancer activities against a variety of pathogens and cancer cell-lines, respectively. Despite sharing high levels of identity between their 16S rRNA gene sequences (higher than 99.7%), they are able to produce different metabolites. These results support the hypothesis that extreme environments do indeed harbour rare diversity and producers of new chemistry.
The high quality genome sequence of S. leeuwenhoekii strain C34T, obtained by using two next-generation sequencing technologies, proved to be essential for the correct assembly and identification of repetitive regions of biosynthetic gene clusters such as those found in PKS and NRPS gene clusters. Moreover, the genome sequence has allowed the generation of a GSM to study the metabolism of this strain and to identify metabolic engineering targets for improved production of specialised metabolites. This model can also be used as a starting point for the generation of GSMs for the other strains belonging to the S. leeuwenhoekii clade with the intent of understanding their metabolisms and exploiting their biosynthetic potentials.
Comparison of the genomes of strains C34T and C58 revealed that they are highly similar, however the quality of the C58 sequence would need to be improved before the repertoires of specialised metabolites made by the two strains could be confidently compared.
S. leeuwenhoekii strain C34T is currently being analysed using phenotypic microarrays (Biolog Inc.) to link its phenotypic properties with its genome with the aim of deepening our knowledge and understanding of how streptomycetes isolated from the hyper-arid Atacama Desert can survive and propagate in such an extreme environment.
References
Axenov-Gibanov DV, Voytsekhovskaya IV, Tokovenko BT, Protasov ES, Gamaiunov SV, Rebets YV, Luzhetskyy AN, Timofeyev MA (2016) Actinobacteria isolated from an underground lake and moonmilk speleothem from the biggest conglomeratic karstic cave in Siberia as sources of novel biologically active compounds. PLoS ONE 11:e0149216. https://doi.org/10.1371/journal.pone.0149216
Bibb MJ, Findlay PR, Johnson MW (1984) The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene 30:157–166. https://doi.org/10.1016/0378-1119(84)90116-1
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T (2013) antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41:W204–W212. https://doi.org/10.1093/nar/gkt449
Bull AT, Asenjo JA, Goodfellow M, Gómez-Silva B (2016) The Atacama desert: technical resources and the growing importance of novel microbial diversity. Annu Rev Microbiol 70:215–234. https://doi.org/10.1146/annurev-micro-102215-095236
Bull AT, Idris H, Sanderson R, Asenjo J, Andrews B, Goodfellow M (2018) High altitude, hyper-arid soils of the Central-Andes harbor mega-diverse communities of actinobacteria. Extremophiles 22:47–57. https://doi.org/10.1007/s00792-017-0976-5
Busarakam K, Bull AT, Girard G, Labeda DP, van Wezel GP, Goodfellow M (2014) Streptomyces leeuwenhoekii sp. nov., the producer of chaxalactins and chaxamycins, forms a distinct branch in Streptomyces gene trees. Antonie Van Leeuwenhoek 105:849–861. https://doi.org/10.1007/s10482-014-0139-y
Busarakam K (2014) Novel actinobacterial diversity in arid Atacama Desert soils as a source of new drug leads. Doctoral thesis, Newcastle University
Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA (2001) Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104:901–912
Castro JF, Razmilic V, Gomez-Escribano JP, Andrews B, Asenjo JA, Bibb MJ (2015) Identification and heterologous expression of the chaxamycin biosynthesis gene cluster from Streptomyces leeuwenhoekii. Appl Environ Microbiol 81:5820–5831. https://doi.org/10.1128/AEM.01039-15
Castro JF (2015) Identification of the chaxamycin and chaxalactin biosynthesis genes through genome mining of Streptomyces leeuwenhoekii C34 and heterologous production of chaxamycins in Streptomyces coelicolor M1152. Doctoral thesis, University of Chile
Choulet F, Gallois A, Aigle B, Mangenot S, Gerbaud C, Truong C, Francou F-X, Borges F, Fourrier C, Guérineau M, Decaris B, Barbe V, Pernodet J-L, Leblond P (2006) Intraspecific variability of the terminal inverted repeats of the linear chromosome of Streptomyces ambofaciens. J Bacteriol 188:6599–6610. https://doi.org/10.1128/jb.00734-06
Dewick PM (2009) Medicinal natural products: a biosynthetic approach, 3rd edn. Wiley, Chichester
Diamant Z, Spina D (2011) PDE4-inhibitors: a novel, targeted therapy for obstructive airways disease. Pulm Pharmacol Ther 24:353–360. https://doi.org/10.1016/j.pupt.2010.12.011
Dorador C, Meneses D, Urtuvia V, Demergasso C, Vila I, Witzel K-P, Imhoff JF (2009) Diversity of Bacteroidetes in high-altitude saline evaporitic basins in northern Chile. J Geophys Res Biogeosci. https://doi.org/10.1029/2008jg000837
Elsayed SS, Trusch F, Deng H, Raab A, Prokes I, Busarakam K, Asenjo JA, Andrews BA, van West P, Bull AT, Goodfellow M, Yi Y, Ebel R, Jaspars M, Rateb ME (2015) Chaxapeptin, a lasso peptide from extremotolerant Streptomyces leeuwenhoekii strain C58 from the hyperarid Atacama desert. J Org Chem 80:10252–10260. https://doi.org/10.1021/acs.joc.5b01878
Fernández-Moreno MA, Martinez E, Boto L, Hopwood DA, Malpartida F (1992) Nucleotide sequence and deduced functions of a set of cotranscribed genes of Streptomyces coelicolor A3(2) including the polyketide synthase for the antibiotic actinorhodin. J Biol Chem 267:19278–19290
Floss HG, Yu T-W (2005) Rifamycin - Mode of action, resistance, and biosynthesis. Chem Rev 105:621–632. https://doi.org/10.1021/cr030112j
Floss HG, Yu T-W, Arakawa K (2011) The biosynthesis of 3-amino-5-hydroxybenzoic acid (AHBA), the precursor of mC7N units in ansamycin and mitomycin antibiotics: a review. J Antibiot 64:35–44
Garg A, Xie X, Keatinge-Clay A, Khosla C, Cane DE (2014) Elucidation of the cryptic epimerase activity of redox-inactive ketoreductase domains from modular polyketide synthases by tandem equilibrium isotope exchange. JACS 136:10190–10193. https://doi.org/10.1021/ja5056998
Gomez-Escribano JP, Alt S, Bibb M (2016) Next generation sequencing of actinobacteria for the discovery of novel natural products. Mar Drugs 14:78. https://doi.org/10.3390/md14040078
Gomez-Escribano JP, Castro JF, Razmilic V, Chandra G, Andrews B, Asenjo JA, Bibb MJ (2015) The Streptomyces leeuwenhoekii genome: de novo sequencing and assembly in single contigs of the chromosome, circular plasmid pSLE1 and linear plasmid pSLE2. BMC Genom 16:485. https://doi.org/10.1186/s12864-015-1652-8
Gontang EA, Fenical W, Jensen PR (2007) Phylogenetic diversity of Gram-positive bacteria cultured from marine sediments. Appl Environ Microbiol 73:3272–3282. https://doi.org/10.1128/aem.02811-06
Hara M, Saitoh Y, Nakano H (1990) DNA strand scission by the novel antitumor antibiotic leinamycin. Biochemistry 29:5676–5681. https://doi.org/10.1021/bi00476a005
Idris H (2016) Actinobacterial diversity in Atacama Desert habitats as a road map to biodiscovery. Doctoral thesis, Newcastle University
Keatinge-Clay AT (2007) A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem Biol 14:898–908. https://doi.org/10.1016/j.chembiol.2007.07.009
Keatinge-Clay AT (2012) The structures of type I polyketide synthases. Nat Prod Rep 29:1050–1073. https://doi.org/10.1039/C2NP20019H
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces Genetics. John Innes Foundation, Norwich
Kim CG, Kirschning A, Bergon P, Ahn Y, Wang JJ, Shibuya M, Floss HG (1992) Formation of 3-amino-5-hydroxybenzoic acid, the precursor of mC7N units in ansamycin antibiotics, by a new variant of the shikimate pathway. JACS 114:4941–4943. https://doi.org/10.1021/ja00038a090
Maksimov MO, Pelczer I, Link AJ (2012) Precursor-centric genome-mining approach for lasso peptide discovery. Proc Natl Acad Sci USA 109:15223–15228. https://doi.org/10.1073/pnas.1208978109
Maloney A, Workman P (2002) HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther 2:3–24. https://doi.org/10.1517/14712598.2.1.3
Margalith P, Beretta G (1960) Rifomycin. XI. Taxonomic study on Streptomyces mediterranei nov. sp. Mycopathol Mycol Appl 13:321–330. https://doi.org/10.1007/BF02089930
Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:60. https://doi.org/10.1186/1471-2105-14-60
Nachtigall J, Kulik A, Helaly S, Bull AT, Goodfellow M, Asenjo JA, Maier A, Wiese J, Imhoff JF, Süssmuth RD, Fiedler H-P (2011) Atacamycins A-C, 22-membered antitumor macrolactones produced by Streptomyces sp. C38*. J Antibiot 64:775–780
Okoro CK, Brown R, Jones AL, Andrews BA, Asenjo JA, Goodfellow M, Bull AT (2009) Diversity of culturable actinomycetes in hyper-arid soils of the Atacama Desert, Chile. Antonie Van Leeuwenhoek 95:121–133. https://doi.org/10.1007/s10482-008-9295-2
Palaniappan N, Ayers S, Gupta S, el Habib S, Reynolds KA (2006) Production of hygromycin A analogs in Streptomyces hygroscopicus NRRL 2388 through identification and manipulation of the biosynthetic gene cluster. Chem Biol 13:753–764. https://doi.org/10.1016/j.chembiol.2006.05.013
Pan SJ, Rajniak J, Cheung WL, Link AJ (2012) Construction of a single polypeptide that matures and exports the lasso peptide microcin J25. ChemBioChem 13:367–370. https://doi.org/10.1002/cbic.201100596
Podust LM, Sherman DH (2012) Diversity of P450 enzymes in the biosynthesis of natural products. Nat Prod Rep 29:1251–1266. https://doi.org/10.1039/c2np20020a
Que L Jr, Tolman WB (2008) Biologically inspired oxidation catalysis. Nature 455:333–340. https://doi.org/10.1038/nature07371
Rateb ME, Houssen WE, Arnold M, Abdelrahman MH, Deng H, Harrison WTA, Okoro CK, Asenjo JA, Andrews BA, Ferguson G, Bull AT, Goodfellow M, Ebel R, Jaspars M (2011a) Chaxamycins A–D, bioactive ansamycins from a hyper-arid desert Streptomyces sp. J Nat Prod 74:1491–1499. https://doi.org/10.1021/np200320u
Rateb ME, Houssen WE, Harrison WTA, Deng H, Okoro CK, Asenjo JA, Andrews BA, Bull AT, Goodfellow M, Ebel R, Jaspars M (2011b) Diverse metabolic profiles of a Streptomyces strain isolated from a hyper-arid environment. J Nat Prod 74:1965–1971. https://doi.org/10.1021/np200470u
Razmilic V (2017) Metabolism analysis of Streptomyces leeuwenhoekii C34 with a genome scale model and identification of biosynthetic genes of specialised metabolites by genome mining. Doctoral thesis, University of Chile
Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R (2003) A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry 42:72–79. https://doi.org/10.1021/bi0268706
Rong X, Huang Y (2012) Taxonomic evaluation of the Streptomyces hygroscopicus clade using multilocus sequence analysis and DNA-DNA hybridization, validating the MLSA scheme for systematics of the whole genus. Syst Appl Microbiol 35:7–18. https://doi.org/10.1016/j.syapm.2011.10.004
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, Barrell B (2000) Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. https://doi.org/10.1093/bioinformatics/16.10.944
Schultz AW, Oh D-C, Carney JR, Williamson RT, Udwary DW, Jensen PR, Gould SJ, Fenical W, Moore BS (2008) Biosynthesis and structures of cyclomarins and cyclomarazines, prenylated cyclic peptides of marine actinobacterial origin. JACS 130:4507–4516. https://doi.org/10.1021/ja711188x
Staunton J, Weissman KJ (2001) Polyketide biosynthesis: a millennium review. Nat Prod Reports 18:380–416. https://doi.org/10.1039/A909079G
Tang G-L, Cheng Y-Q, Shen B (2004) Leinamycin biosynthesis revealing unprecedented architectural complexity for a hybrid polyketide synthase and nonribosomal peptide synthetase. Chem Biol 11:33–45. https://doi.org/10.1016/j.chembiol.2003.12.014
Tsai S-C, Miercke LJW, Krucinski J, Gokhale R, Chen JC-H, Foster PG, Cane DE, Khosla C, Stroud RM (2001) Crystal structure of the macrocycle-forming thioesterase domain of the erythromycin polyketide synthase: versatility from a unique substrate channel. Proc Natl Acad Sci USA 98:14808–14813. https://doi.org/10.1073/pnas.011399198
Valenzano CR, You Y-O, Garg A, Keatinge-Clay A, Khosla C, Cane DE (2010) Stereospecificity of the dehydratase domain of the erythromycin polyketide synthase. JACS 132:14697–14699. https://doi.org/10.1021/ja107344h
Västermark Å, Almén MS, Simmen MW, Fredriksson R, Schiöth HB (2011) Functional specialization in nucleotide sugar transporters occurred through differentiation of the gene cluster EamA (DUF6) before the radiation of Viridiplantae. BMC Evol Biol 11:123. https://doi.org/10.1186/1471-2148-11-123
Weaver D, Karoonuthaisiri N, Tsai H-H, Huang C-H, Ho M-L, Gai S, Patel KG, Huang J, Cohen SN, Hopwood DA, Chen CW, Kao CM (2004) Genome plasticity in Streptomyces: identification of 1 Mb TIRs in the S. coelicolor A3(2) chromosome. Mol Microbiol 51:1535–1550
Wilson MC, Gulder TAM, Mahmud T, Moore BS (2010) Shared biosynthesis of the saliniketals and rifamycins in Salinispora arenicola is controlled by the sare1259-encoded cytochrome P450. JACS 132:12757–12765. https://doi.org/10.1021/ja105891a
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617. https://doi.org/10.1099/ijsem.0.001755
Acknowledgements
The authors acknowledge funding from the Basal Programme of Conicyt (Chile) to the Centre for Biotechnology and Bioengineering (CeBiB Project FB0001), from the Biotechnological and Biological Sciences Research Council (BBSRC, United Kingdom) Institute Strategic Programme Grant “Understanding and Exploiting Plant and Microbial Secondary Metabolism” (BB/J004561/1) and from the RCUK-CONICYT International Research Project Bioprospecting the Atacama Desert: the discovery and enhancement of novel therapeutic drugs from actinomycetes (Newton Fund JIC CA586).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Castro, J.F., Razmilic, V., Gomez-Escribano, J.P. et al. The ‘gifted’ actinomycete Streptomyces leeuwenhoekii. Antonie van Leeuwenhoek 111, 1433–1448 (2018). https://doi.org/10.1007/s10482-018-1034-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-018-1034-8