
Abstract
There are two vascular networks in mammals that coordinately function as the main supply and drainage systems of the body. The blood vasculature carries oxygen, nutrients, circulating cells, and soluble factors to and from every tissue. The lymphatic vasculature maintains interstitial fluid homeostasis, transports hematopoietic cells for immune surveillance, and absorbs fat from the gastrointestinal tract. These vascular systems consist of highly organized networks of specialized vessels including arteries, veins, capillaries, and lymphatic vessels that exhibit different structures and cellular composition enabling distinct functions. All vessels are composed of an inner layer of endothelial cells that are in direct contact with the circulating fluid; therefore, they are the first responders to circulating factors. However, endothelial cells are not homogenous; rather, they are a heterogenous population of specialized cells perfectly designed for the physiological demands of the vessel they constitute. This review provides an overview of the current knowledge of the specification of arterial, venous, capillary, and lymphatic endothelial cell identities during vascular development. We also discuss how the dysregulation of these processes can lead to vascular malformations, and therapeutic approaches that have been developed for their treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The vascular system is comprised of both the blood and the lymphatic circulation, which function cooperatively to maintain tissue survival, growth, function, and homeostasis. Endothelial cells (ECs) line the innermost layer of all of these vessels and play an important role in sensing the circulating environment and responding to extrinsic signals. ECs exhibit a high degree of heterogeneity in gene/protein expression and structure depending on the vessel type in which they reside (i.e., arterial, venous, capillary or lymphatic), and these acquired differences enable distinct functions, as well as tissue-specific functions.
The processes of EC specification and maturation are vastly different among all of these EC types—from the first stages of specialization to the formation of tissue-specific characteristics. Impairments in any of these developmental pathways result in vascular malformations that can be lethal. This review will highlight important regulatory pathways that promote both blood and lymphatic EC development and specification and describe vascular malformations that occur when they are dysregulated. We will also provide an overview of current therapies for these vascular disorders that have been enabled by our understanding of normal developmental pathways.
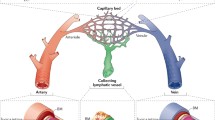
Blood endothelial cell development
The blood vasculature is a closed circulatory system that consists of arterial, venous and capillary networks connected to the heart. The contractile property of the heart propels nutrient- and oxygen-rich blood to all tissues via the arterial system. Capillary networks between arteries and veins enable their distribution into tissues, as well as removal of metabolic waste products. Low-oxygen and low-nutrient blood is then circulated back to the heart and lungs via the venous system. Each vessel type is lined with ECs that not only exhibit a high degree of heterogeneity among different sections of the vascular tree, but also exhibit tissue-specific characteristics, especially at the capillary level. Some capillary ECs express a high level of tight junctions that restrict the passage of nutrients, soluble factors and cells in blood circulation into tissues, such as the brain and retina. In contrast, other capillary ECs have fenestrations that allow for extensive filtration of factors and cells in tissues such as the liver and kidney. Thus, each type of vessel within the blood circulation plays a crucial, yet distinct, role in maintaining tissue homeostasis.
ECs that line blood vessels are surrounded by mural cells [smooth muscle cells (SMCs) and pericytes] to a varying degree, depending on where they are in the circulatory network. For example, large arteries have a thick vessel wall made up of multiple layers of SMCs, whereas arterioles have few mural cells in their vessel wall. An exception to this is the smallest capillaries, which are only made up of a single layer of ECs that are, at times, surrounded by pericyte processes.
Due to their close proximity to circulating factors in the blood, vascular ECs are important for the coordination of vessel responses to changes in nutrients, oxygen and other factors, such as hormones. ECs are the first vascular cells to be differentiated during development and they play a key role in the formation of their vessel wall and a complete vascular network [1, 2]. All blood ECs are thought to be derived from mesodermal progenitors, but they acquire heterogeneous characteristics as they develop and mature into arterial, venous and capillary ECs (Fig. 1). This portion of the review will focus on the signaling mechanisms regulating early blood EC differentiation and their later specification into distinct subtypes.

Blood EC Specification in Health and Disease. (Black) Primordial endothelial cells (ECs) are specified from mesoderm-derived cells and form primitive vascular plexi. Expansion and maturation of these plexi through vasculogenesis and angiogenesis forms the adult vascular network. Several developmental pathways play a significant role in initial EC specification and later in determination of arterial-venous fates. (Red) Changes to key signaling components in these specification pathways are associated with the development of human arteriovenous malformations, including Cutaneo-mucosal venous malformations (VMCM); capillary malformation with arteriovenous malformations (CM-AVM); cerebral cavernous malformations (CCM); and hereditary hemorrhagic telangiectasia (HHT)
Vascular endothelial cell differentiation
Vascular ECs arise from multipotent progenitors in the embryonic and extraembryonic mesoderm [3]. Newly differentiated ECs initially form a primitive vascular plexus in the extraembryonic yolk sac and later the primitive vascular plexi in embryonic tissues that give rise to the cardinal vein (CV) and dorsal aorta, as well as all forming blood vessels in developing organs. This early developmental stage that includes de novo EC differentiation and the formation of primitive vascular plexi is referred to as vasculogenesis. Vasculogenesis is initiated in the mouse at around embryonic day (E)7.0–7.5 with the expression of ETV2, a member of the E26 transformation-specific transcription factors, in mesodermal progenitors that become angioblasts, the precursors to ECs [4, 5]. This early expression of ETV2 drives the differentiation of ECs by promoting the expression of genes including vascular endothelial growth factor receptor 2 (Vegfr2), Cdh5 (encodes VE-cadherin) and Tie2 (angiopoietin receptor), which are important for the formation and maintenance of ECs [3, 6]. Other important signaling factors in the early formation of angioblasts are fibroblast growth factor 2 (FGF2) and bone morphogenic protein 4 (BMP4), which promote the acquisition of EC identity [7]. Activation of these signaling pathways is followed by the induction of vascular endothelial growth factor A (VEGF-A)-VEGFR2 signaling, which promotes EC survival, proliferation and migration, thereby expanding the primitive vascular plexus.
Some reports have suggested that arterial and venous ECs are not derived from the same population of angioblasts, but rather each develops from a distinct pool of precursors. Kohli et al. used the zebrafish model to demonstrate that the arterial and venous ECs that give rise to the major axial vessels in the fish are derived from distinct progenitor populations [8]. They show that angioblasts residing in the midline of the body give rise to the arterial ECs which develop into the dorsal aorta, while a second population of angioblasts in the lateral axis of zebrafish gives rise to venous ECs. They postulated that this difference in expression pattern was due to higher VEGF and sonic hedgehog (SHH) signaling in the midline compared to the lateral axis of the fish, which have been found to promote arterial EC development [8, 9]. Whatever the source of EC progenitors may be, the initial steps of EC differentiation, especially in the formation of the primitive vascular plexi remains largely undebated, although further investigation is needed to more fully understand the regulation of this process.
After the primitive vascular plexi are formed, blood vessels grow, expand and remodel into arterial-venous networks throughout the yolk sac and embryo proper via a process known as angiogenesis. It is important to note that not all blood vessel expansion occurs via vasculogenesis followed by angiogenesis. In some tissues, such as the brain and retina, newly differentiated ECs predominantly exhibit sprouting angiogenesis during vascular network formation, highlighting the heterogeneity in growth patterns of newly formed ECs [10,11,12,13]. Angiogenic-driven vascular expansion relies on the specification of tip cells, which are followed by several trailing stalk cells. The tip cells exhibit a migratory phenotype, while stalk cells actively proliferate to form new vessels [10, 14]; these phenotypic differences are promoted by distinct signaling mechanisms.
Tip and stalk cell specification
As the developing embryo grows, tissue hypoxia promotes angiogenesis of new vessels to unvascularized tissues through the upregulation of VEGF-A, resulting in the induction of vascular EC sprouting [15]. VEGF-A binds to the VEGFR2 receptor on tip cells to promote cell survival and migration [16]. Loss of VEGF-A-VEGFR2 signaling axis impairs vascular development and results in early embryonic lethality [17, 18]. An important signaling factor in the maintenance of tip cell identity is the NOTCH ligand, delta-like protein 4 (DLL4) [19]. DLL4 is preferentially expressed in tip cells and is activated downstream of VEGF-A-VEGFR2 signaling. DLL4 binding to NOTCH receptors on the adjoining stalk cells promotes the expression of VEGFR1, a decoy receptor that sequesters VEGF-A, and inhibits VEGFR2 expression, thereby inhibiting migration and maintaining stalk cell identity [16, 20]. Additional pathways important for tip-stalk cell specification include TGF-β (transforming growth factor β) and BMP9/10 signaling. Binding of TGF-β and BMP9/10 to the TGF-β type-1 receptors ALK5 and ALK1, respectively, promote stalk cell identity through SMAD2/3- and SMAD1/5/8-dependent signaling [21]. Concomitantly, VEGF-A-dependent activation of Neuropilin-1 (NRP-1) in the tip cells inhibits SMAD2/3 and SMAD1/5/8 signaling ultimately repressing the stalk cell phenotype and maintaining tip cell identity [21]. Maintenance of tip and stalk cell identity is further complicated as the vascular plexus grows and matures. Induction of EC branching from already formed vessels is, in part, regulated by BMP-NOTCH signaling cross-talk, whereby BMP signaling promotes EC branching and is directly inhibited by NOTCH [22,23,24]. In zebrafish, BMP6 and BMP2 signaling induce EC branching through a SMAD1/5/8-dependent mechanism [22]. This is counteracted by NOTCH-dependent activation of the inhibitory SMAD6 to carefully regulate the extent of vessel branching, thereby maintaining proper vascular organization during development [22, 23].
Arterial-venous specification
As the embryo continues to develop, the primordial ECs respond to a myriad of signals to specify into arterial or venous phenotypes. Arterial and venous ECs can be distinguished, in part, by the expression of the ligand-receptor partners E-phrinB2 and Ephrin type B receptor 4 (EPHB4) in arteries and veins, respectively [25]. EphrinB2-EPHB4 binding repels arterial and venous ECs from one another maintaining vessel identity in the developing vasculature [25]. ECs start to acquire an arterial or venous phenotype by responding to a complex signaling network which determines EC identity depending on the pathway that is dominantly activated [26]. As blood begins to flow through the developing circulatory network and ECs are exposed to mechanical shear stress, EC specification is further reinforced [27].
Multiple signaling pathways contribute to early arterial-venous specification including VEGF-A, WNT and SHH. VEGF-dependent determination of EC fate is reliant upon the relative concentration of VEGF-A, whereby high and low VEGF-A concentrations promote arterial and venous specification, respectively [28]. In arterial-fated ECs, VEGF-A-VEGFR2 drives arterial specification through activation of NOTCH- and ERK1/2-dependent signaling [29,30,31]. VEGF-A-VEGFR2 signaling activates the transcription factors SOX7, SOX17 and SOX18 to upregulate NOTCH-1 expression [30]. In turn, NOTCH activation and translocation into the nucleus drives the expression of arterial genes [32]. Studies in zebrafish revealed that BMP-dependent signaling potentiates NOTCH-dependent arterial specification [33]. BMP4 activation of the BMP regulatory proteins, BMPER and TWSGI, enhance NOTCH-dependent upregulation of EphB2 [33, 34]. In parallel to NOTCH signaling, VEGF-VEGFR2 activation drives ERK1/2 (MAP kinase signaling)-dependent upregulation of arterial genes [29].
NOTCH expression is also induced through WNT-dependent signaling downstream of VEGFR2 activation [31]. Baseline WNT-β-catenin signaling induces the expression of DLL4, which promotes NOTCH activation in adjacent cells to enable arterial fate specification. Overactivation of this pathway in mice results in defects in arterial-venous specification and the formation of arteriovenous (AV) shunts [31]. SHH-dependent activation is also involved in upregulating NOTCH signaling in ECs. In zebrafish, arterial specification has been shown to be initiated by a SHH-VEGF-A-NOTCH signaling axis [9]. Overexpression of Shh in zebrafish leads to ectopic formation of arterial ECs, while inhibition of the SHH pathway leads to the loss of arterial identity in the developing vasculature [9]. The importance of NOTCH signaling is highlighted by the loss of arterial cell identity, accompanied by EC hyperproliferation and disrupted vascular remodeling, with the loss of NOTCH signaling [35, 36].
The initiation of blood flow through the developing vasculature further specifies arterial ECs [37]. In chick embryos, the start of blood flow in the vessels of the yolk sac upregulates arterial-specific genes including EphB2 [37]. Arterial specification in response to flow is regulated in part by NOTCH signaling [38]. Arterial flow-mediated activation of NOTCH leads to an upregulation of mechanosensitive gap junction protein Connexin 37 (Cx37) and subsequent activation of the cell cycle inhibitor p27; p27-mediated endothelial cell cycle arrest further enables the upregulation of arterial genes [38]. The exact mechanism by which Cx37 regulates p27 and cell cycle arrest is not yet clear, and the role of endothelial cell cycle control in this process is further discussed below.
Arterial specification is inhibited to promote and maintain venous EC fate. Expression of the venous-enriched transcription factor, Nuclear Receptor Subfamily 2 Group F Member 2 (NR2F2/COUP-TFII), inhibits arterial specification through the downregulation of NOTCH signaling [39]. Similarly, ERK1/2-dependent arterial specification is antagonized by PI3K-AKT activation that drives venous EC specification [29]. PI3K-AKT-dependent inhibition of ERK1/2 promotes COUP-TFII expression in developing venous ECs [29, 40]. PI3K is activated in response to VEGF-A which, as mentioned previously, can also promote arterial specification [29, 40]. Thus, arterial-venous identity in response to VEGF-A signaling is determined by the strength of the signal and the downstream pathways activated, revealing the complexity of these developmental pathways [28]. To make matters more complex, studies in zebrafish and mice have shown that arterial ECs can be derived from venous tip cells during development [41,42,43]. Thus, we have much more to learn about the relationships among EC types, and the cross-talk of signaling pathways, in remodeling vascular plexi.
Another pathway that can promote divergent outcomes depending on the receptors and ligands involved is the TGF superfamily-dependent signaling. The identity of the TGF superfamily receptor activated directs whether primitive blood ECs acquire a venous or arterial phenotype. For example, in zebrafish, BMP2/4-ALK3 signaling axis activates SMAD1/5 to promote venous fate by upregulating venous-enriched genes (Ephb4 and Nr2f2) [44]. Other recent studies further support the important role of BMP2-dependent signaling in promoting venous EC identity through the activation of ALK2/3 and BMP receptor II [26, 44].
In contrast, TGF-β-dependent signaling enables arterial cell fate through careful regulation of angiogenic and vascular maturation signals. The TGF superfamily type I receptors discovered to play a role in arterial development include ALK1, ALK5 and Endoglin (ENG), with ALK1 and ALK5 signaling regulating a biphasic response in ECs [45]. Mutations in ALK1 or ENG result in perturbation in arterial-venous specification and ultimate formation of AV malformations [46, 47]. Alk1-, Eng- and Alk5-deficient mice display severe vascular malformations resulting from the inability of the vasculature to undergo arterial-venous maturation [48,49,50,51]. In Eng-deficient mice, specifically, the development of AV malformations is due, in part, to the loss of ENG-dependent inhibition of COUP-TFII expression, resulting in attenuated arterial specification and increased venous EC formation [52]. Similarly, ALK1 activation has been shown to promote arterial specification during development and mice lacking Alk1 are embryonic lethal due to severe vascular malformations [48]. Activation of ALK1 and ENG by TGF-β promotes endothelial cell migration and proliferation through SMAD1/5 signaling, enabling the expansion of developing arteries [53,54,55]. ALK1-ENG pro-angiogenic signaling is antagonized by ALK5 activity. TGF-β activation of ALK5-SMAD2/3 inhibits angiogenesis and promotes vessel maturation [53, 56, 57]. The interplay between ALK1 and ALK5 signaling is further complicated; that is, in vitro studies revealed that ALK5 signaling promotes ALK1 expression and is required for optimal ALK1 signaling [53]. Other studies in mice and cultured ECs argue that ALK1-dependent signaling may be more important in vessel maturation rather than angiogenesis [58,59,60]. The complexity of the signaling pathways and their relative contributions to proper arterial-venous specification continue to be an important area of investigation. However, it is clear that perturbation to any of these pathways severely impacts vascular development resulting in potentially lethal arterial-venous malformations [61,62,63].
Cell cycle regulation of arterial-venous specification
One of the key questions in vascular development is how do similar extrinsic signals cause differential responses in developing ECs. There is growing evidence for a role of cell cycle state in determining the propensity for fate decisions during development. For example, in embryonic stem cells, different cell cycle states create distinct windows of opportunities that enable cells to differentially respond to factors that promote their differentiation toward specific lineages [64, 65]. Recent studies of vascular development similarly suggest that endothelial cell cycle state determines their propensity for arterial-venous specification [38, 39].
Although remodeling ECs are proliferative, their growth is suppressed as arterial-venous specification ensues, suggesting a role for cell cycle arrest in the acquisition of arterial-venous EC identity [38, 43, 66]. In support of this idea, our lab has recently shown that arterial shear stress activates a NOTCH-Cx37-p27 signaling axis to promote cell cycle arrest in G1 phase, which enables the upregulation of arterial genes [38]. Conversely, in venous ECs, COUP-TFII expression promotes cell cycle progression and reduces arterial EC gene expression [39]. Recent work from our group, using FUCCI cell cycle reporter mice [67], revealed that ECs in remodeling veins and arteries are in different states of G1; early G1 vs. late G1, respectively [68]. In addition, these distinct cell cycle states enable BMP vs. TGF-β signaling to upregulate venous vs. arterial genes, respectively [68]. Together, these data highlight the importance of cell cycle regulation in EC fate decisions, although much more work in needed to understand the molecular underpinnings of this process.
Capillary Endothelial Cell Development
Connecting arterial and venous circulation is a vast network of capillaries that serve as the site of gas and nutrient exchange between tissues and the circulatory system. Developing capillaries are composed of a single layer of ECs connected by a thin basement membrane. As capillary networks mature, ECs recruit mural cell precursors via PDGF-B signaling [1, 69]. Upon contact with the capillary ECs, TGF-β is activated and promotes the differentiation of pericytes, which aid in maintaining capillary vessel integrity [1, 70,71,72,73,74].
There is a growing appreciation for the high degree of heterogeneity among EC phenotypes, not only in arteries and veins, but also in capillaries. The structure of capillaries is organ-specific, with morphologies that are perfectly suited for the needs of the tissue [10]. Although the structural heterogeneity among capillary ECs is well described, the transcriptional regulation required to promote these specialized characteristics remains unclear. In early embryonic development, capillaries form the primitive vascular network in the extra- and intra-embryonic tissues [75]. These primitive vascular plexi later remodel and mature into circulatory networks containing arteries, veins and mature capillaries. During the maturation process, some of the primitive capillary networks are pruned away, while new capillaries are formed through angiogenic sprouting of existing blood vessels [75]. Angiogenic sprouting is driven by the migration of endothelial tip cells followed by the proliferative stalk cells to form new capillary tubes, which will migrate until they reach other capillaries and coalesce to form new vessels [16].
As mentioned above, ECs formed from venous tip cells migrate through the remodeling capillary plexi to contribute to forming arterial vessels [41]. Due, in part, to this venous to artery migration, brain capillary ECs share gene expression patterns of both venous and arterial ECs [76]. An analysis of the developing coronary vasculature showed that ECs from the developing coronary plexus are most transcriptionally similar to adult venous and capillary ECs, suggesting that the developing adult coronary capillaries are derived from venous ECs and may only begin to express arterial markers once they make contact with arterial branches [39]. Other recent single-cell RNA sequencing analysis of murine brain capillary ECs also reveals a continuum of gene expression among capillary, arterial and venous ECs [76]. However, distinct capillary EC markers and the specification events that determine capillary EC fate remain to be elucidated.
Arterial-venous malformations
Dysregulation of EC specification leads to vascular malformations, which can be debilitating and even fatal. There are different types of vascular malformations, depending on which process in arterial-capillary-venous development is disrupted (Fig. 1, Table 1). In this section, we provide an overview of different types of blood vascular malformations, what is known about their underlying genetic defects, and what treatments are currently available for affected patients. We also propose new research pathways linking endothelial cell cycle state and fate in the development of vascular malformations.
Hereditary Hemorrhagic Telangiectasia
Hereditary Hemorrhagic Telangiectasia (HHT, OMIM 187300), also known as Osler-Rendu-Weber syndrome, is a genetic vascular disorder affecting 1 in 5,000 individuals worldwide [77]. This disease is characterized by multiple vascular defects including epistaxis, telangiectasias and AV shunts in various organs including brain, lungs and intestines, which are prone to rupture, causing life-threatening hemorrhage [77].
The HHT-causing mutations affect genes encoding different components of the BMP9/10 pathway; namely, ENG encoding the membrane glycoprotein ENDOGLIN in HHT1, ACVRL1 encoding the membrane receptor ALK1 in HHT2, SMAD4 encoding the intracellular signaling molecule SMAD4 in Juvenile polyposis (JP)-HHT, and GDF2 encoding BMP9 ligand in HHT5 [78,79,80,81,82]. To date, all identified mutations result in haploinsufficiency of these genes. In ECs, decreased activity of the BMP9/10 signaling leads to over-activation of the pro-angiogenic factors VEGF-A and angiopoietin-2 (ANGPT-2), triggering EC hyperproliferation, as well as alterations in their permeability and migration, ultimately leading to vascular malformations [78, 83,84,85].
Mouse models of HHT have provided significant insights regarding the functions of the BMP9/10 signaling pathway defects in the development of the vascular anomalies related to HHT. Heterozygous mutations of Eng or Alk1 genes give rise to vascular lesions forming at low frequencies and later in life, making them inconvenient models for further study [86, 87]. Constitutive inactivation of Eng, Alk1 and Smad4 genes in mice leads to embryonic lethality due to multiple cardiovascular defects, again rendering the study of molecular mechanisms difficult [58, 88, 89]. However, postnatal tamoxifen-inducible, EC-specific homozygous deletion of any of these genes induces HHT-like vascular malformations, including excessive angiogenesis, enlarged veins and AV shunts in the neonatal retinal vascularization model [84, 85, 90, 91].
The dysregulation of arterial-venous identity of ECs forming AV shunts has been shown in these different experimental models. The inducible EC-specific deletion of Eng, Alk1 or Smad4 leads to EC hyperproliferation, which is associated with downregulation of several arterial-enriched genes, such as Efnb2, Jag1 and Unc5b, as well as upregulation of venous-enriched genes Ephb4 and Nrp2 [84, 85, 90]. These modifications are also accompanied by increased SMC coverage, or muscularization, of veins and AV shunts, while arteries undergo loss of SMC coverage or de-muscularization.
The molecular mechanism(s) leading to the dysregulation of endothelial identity in AV malformations (AVM) has not been unraveled. However, the modifications of the environment surrounding the ECs induced by AVM, such as defective flow shear stress or impaired nutrient and oxygen supply, could play an important role in this regulation. In fact, impaired-systemic blood flow in mice produces defects in arterial-venous specification and induces AVM during embryonic development [92]. In the context of HHT, fluid shear stress has been shown to potentiate BMP9 signaling through ALK1 and SMAD4 and, thus, enabling the repression of EC proliferation, as well as pro-angiogenic signals [85, 93]. Furthermore, as mentioned above, it has been shown that arterial shear stress, via the NOTCH-Cx37-p27 axis, regulates the cell cycle state of ECs and enables their arterial specification [38]. Nevertheless, the demonstration that fluid shear alterations or endothelial cell cycle state modifications in AVM could be responsible for the loss of EC identity are lacking and represent important research pathways to identify new targets for HHT treatment.
To date, the therapeutic options available for HHT patients are intended to reduce the symptoms of the disease (epistaxis). However, new clinical and preclinical studies are emerging to counter-balance the pro-angiogenic axis over-activated in HHT and, ultimately, to correct telangiectasias and AV shunts into a normal vasculature. The humanized monoclonal anti-VEGF-A antibody, bevacizumab [94], currently in phase III of clinical trial, has significantly reduced bleedings and liver and cardiac anomalies in HHT1, HHT2 and JP-HHT patients [95]. Anti-angiogenic therapies using tyrosine kinase inhibitors targeting VEGF-A signaling such as Nintedanib [96] or Pazopanib [97] have shown encouraging results in treatment of HHT-related bleeding and are now candidates for clinical trials [78]. In mouse models of HHT (Smad4ECiKO and Alk1ECiKO), anti-ANGPT-2 antibodies and PI3K inhibitors have demonstrated robust effects on correcting AVM [83, 84]. Finally, instead of targeting the pro-angiogenic signal, recent therapeutic strategies attempt to restore the BMP9-ALK1-SMAD signaling axis which is defective in HHT. In support of this approach, it has shown that the re-expression of Alk1 gene in Alk1-deficient mice restores BMP9 signaling in ECs and rescues vascular malformations linked to HHT [98]. In a preclinical model (BMP9/10 immunodepletion), Tacrolimus and Sirolimus have proven their efficiency in resolving AVM by promoting the re-activation of the BMP9 signaling pathway [99].
Venous malformations
Venous anomalies are among the most frequent cause of mortality due to vascular defects with an incidence estimated at 1/5,000 – 1/10,000 [100]. The two major categories of venous anomalies are cutaneo-mucosal venous malformations (VMCM, OMIM 600195), representing 95% of cases, and glomuvenous malformations (GVM, OMIM 138000), accounting for the majority of the remaining 5%. Contrary to VMCM, which are linked to EC dysfunction, GVM are due to impaired vascular SMC differentiation and will not be the focus of this review [101].
VMCM are congenital lesions of distorted venous-like vessels. These lesions are soft, compressible, light-to-dark blue, mainly located in the skin and mucosa, but can infiltrate underlying tissues, muscles and joints [102]. Histologically, VMCM are characterized by enlarged vessels, irregular lumens, monolayer of ECs and irregular SMC coverage [103]. Chronic activation of the EC TIE2-PI3K signaling pathway is considered the major cause of VMCM [104].
Genetic and molecular studies have shown that VMCM are caused by gain-of-function mutations in the EC-specific tyrosine kinase receptor TIE2 (TEK) gene [103] or in PIK3CA gene, encoding the p110α catalytic subunit of PI3K [105]. Even if the downstream mechanism of TIE2/PI3K over-activation is not completely understood, several cellular and molecular dysregulations have been identified in VMVC pathogenesis, including defective EC-SMC interactions and venous identity [106,107,108]. In a mouse model that mimics human VMCM, the endothelial expression of Pik3caH1047R, a constitutively active mutant of the p110α, results in EC hyperproliferation, reduction in mural cell coverage of blood vessels, and modification in arterial-venous identity [106]. The venous markers COUP-TFII and EPHB4, and the arterial marker Ephrin-B2 are reduced in EC-Pik3caH1047R postnatal retinas and lungs compared to those in Pik3caWT mice. However, in VMCM patient biopsies, EC proliferation is not a noticeable characteristic [103, 109]. These divergences could be due to the different models and time points of the studies. As already suggested, VMCM could have an initial phase of EC proliferation, where they are more responsive to proliferative cues, followed by a more quiescent state in established lesions [104].
A recurrent hallmark of VMCM is defective SMC coverage in venous lesions [103, 105]. As a consequence of TIE2/PI3K over-activation, the transcription factor FOXO1 and its target gene encoding for PDGF-B, a SMC chemo-attractant, have been shown to be negatively regulated in Pik3caH1047R mice and biopsies from TIE2 mutation-positive VMCM lesions [106, 107]. To date, it is not known whether decreased PDGF-B levels leads to defective SMC coverage in VMCM lesions. However, understanding the consequences of impaired EC-SMC interactions on EC identity in venous malformations could enhance our understanding of how these lesions are formed. Specification of arterial and venous ECs occurs in conjunction with suppression of EC cycle progression [7, 38, 39]. FOXO1 activation plays a role in the regulation of cell cycle progression by promoting cell cycle arrest via the cyclin-dependent kinase inhibitor p27kip1 [110, 111]. Nevertheless, the dysregulation of endothelial cell cycle regulators, such as p27, and its impact on venous identity in VMCM induced by over-activation of PI3K-AKT-FOXO1 axis still need to be investigated.
There is currently no available treatment for VMCM patients. However, in a few preclinical studies, specific inhibitors of TIE2 or PIK3CA over-activation have been tested to prevent or revert VMCM. TIE2 kinase inhibitor or PIK3CA inhibitor (Alpelisib) have demonstrated very modest effects on treating VMCM [112, 113]. The most advanced potential pharmacotherapy for VMCM currently is Sirolimus (also known as Rapamycin). This compound inhibits TIE2- and PIK3CA-mutated venous malformations in vitro and in vivo and, in mouse models, it diminishes lesion growth, normalizes SMC coverage and decreases EC proliferation [106, 112].
Capillary malformations
Capillary malformations (CM, OMIM 163000), also called “port wine stains”, are the most common type of cutaneous vascular malformation, affecting 0.3% to 0.5% of the population [114]. CM consist of dilated capillary-like vessels and lesions that are sporadically flat, red to purple in color, and found most frequently in the head and neck [115]. CM are divided into two categories: CM with AV malformations (CM-AVM, OMIM 608354); and cerebral cavernous malformations (CCM, OMIM 116860) [100].
Capillary malformations with arterial-venous malformations
CM-AVM is an autosomal dominant disorder caused by heterozygous inactivating mutations in the RASA1 (RAS p21 protein activator 1) gene (CM-AVM1) characterized by the presence of capillary malformations, arteriovenous malformations and fistulas, and occasionally vascular overgrowth [116, 117]. RASA1 encodes the RAS-GTPase-activating protein, p120Ras-GAP, that negatively regulates the RAS/MAPK signaling pathway [117, 118]. Among different functions, Ras is an activator of VEGF-A-mediated angiogenesis by promoting the phosphorylation and stabilization of the hypoxia-inducible factor-1 alpha (HIF-1α) transcription factor, which upregulates VEGF-A [119, 120]. As a negative modulator of RAS activity, p120Ras-GAP plays a role in balancing EC signaling by downregulating proliferation, and potentially migration and polarity [121, 122].
There is no ideal vertebrate genetic model of CM-AVM currently available [123, 124]. Rasa1 heterozygous mutant mice are viable, but the homozygous deletion of Rasa1 induces embryonic lethality at E10.5 due to defective vascular development [124]. Overexpression of microRNA-132, a negative regulator of p120Ras-GAP protein expression, promotes neovascularization in a murine tumor model, whereas anti-microRNA-132 inhibits this effect and prevents pathological retinal angiogenesis [121, 125]. CM-like vascular lesions have been reported in those models, but not CM-AVM. Nevertheless, a study using Morpholinos against Rasa1 in zebrafish showed that reduced Rasa1 gene expression leads to impaired circulation and arterial-venous misconnections [123]. Interestingly, this study provides a molecular connection between RASA1 and EPHB4, which is well known for its function in venous specification during embryonic development and is considered a venous marker in the adult vasculature [126, 127]. However, whether dysregulation of EC identity contributes to capillary malformations is still unknown. EPHB4 variants were also reported to cause capillary malformation–arteriovenous malformation 2 (CM-AVM2) [128]. CM-AVM2 mimics RASA1-related CM-AVM1 and HHT and could therefore be considered part of the clinical spectrum of HHT and other vascular malformation syndromes [129].
Cerebral cavernous malformations
CCM occur in approximately 0.5% of the population and can be both sporadic (80%) or autosomal dominant inherited (20%) [130]. The lesions are mostly localized in the brain, but also in the spinal cord and retina [131]. Histologically, CCM consist of dilated vessels, known as cavernomas, that are often assembled into clusters (i.e., mulberry lesions) [130]. These abnormal vessels are formed by a defective layer of ECs that lacks tight junctions and is prone to blood leakage and vascular rupture [131]. Patients can develop symptoms such as headaches, seizures, neurological problems and cerebral hemorrhages, although some are asymptomatic [131, 132].
The inherited forms of CCM are caused by a loss of function mutation in any of the 3 autosomal genes, CCM1/KRIT1, CCM2/OSM or CCM3/PDCD10 [130]. In sporadic forms of CCM, somatic mutations of CCM genes have been observed in cerebral cavernomas [133]. CCM1-3 mutations in ECs are mainly responsible for the vascular defects observed in patients with CCM. Endothelial-specific deletion of Ccm1, Ccm2, or Ccm3 genes in mice induces CCM-like vascular defects in the central nervous system [134, 135], whereas SMC- or neuro-specific mutations of Ccm genes do not induce cavernomas [136]. Loss of function of any of the Ccm genes leads to increased TGF-β/BMP signaling, EC junction defects and endothelial-to-mesenchymal transition (EndMT), which collectively contribute to cavernoma formation [134, 135, 137].
The CCM proteins interact with each other, with CCM2 acting as a linker between CCM1 and CCM3 [138]. The complex binds to VE-cadherin at EC-EC junctions, through β-CATENIN [139]. In the absence of CCM1 or CCM2, the activation of RAP1, a small GTPase known to stabilize the cortical actin cytoskeleton, is impaired [140], and CDC42 is inhibited [141]. This, in turn, promotes disorganization of EC junctions and increases vascular permeability, leading to hemorrhage [142]. Interestingly, endothelial-specific gene deletion of Cdc42 leads to capillary and venous malformations in postnatal mouse retina, similar to CCM defects, due to impaired EC polarized migration [143].
Enhanced TGF-β/BMP signaling after deletion of any of the CCM genes is a common dysregulation causing cavernomas. Two different mechanisms have been identified as responsible for this increased signaling: higher and sustained response to TGF-β by Ccm-deficient ECs; and production of endogenous BMP ligands, such as BMP2 and BMP6 [134, 144, 145]. Increased TGF-β/BMP signaling, through an upregulation of the transcription factor KLF4, promotes EndMT, which contributes to the loss of EC identity in cavernomas [134, 145, 146]. EndMT is characterized by disorganization of EC junctions [134], loss of apical-basal polarity [147], and expression of stem/mesenchymal cell markers [134]. Furthermore, CCM1, via the DLL4-NOTCH pathway, promotes mRNA expression of the cell cycle inhibitors p21 (CIP1) and p27 (KIP1), which inhibit EC proliferation and migration in vitro [148]. Therefore, loss of function of any CCM gene could disrupt endothelial cell cycle control via inhibition of p21 or p27. Since endothelial cell cycle control, via a NOTCH-Cx37-p27 axis, is necessary for the acquisition of arterial EC cell identity, it is possible that dysregulation of this axis contributes to CCM [27, 38]. Thus, further investigation of this pathway could provide additional understanding of the molecular mechanisms leading to CCM and reveal new targets for their treatment.
No pharmacological therapy is currently available for CCM. A partial surgical resection of the vascular lesions is the only therapeutic option at present. However, this strategy is not feasible in critical regions of the brain and does not prevent lesion resurgence. The ideal therapy would be to stabilize the preexisting lesions, inhibit their progression and block the formation of new malformations [149]. Using in vivo and in vitro experimental models of CCM, several pharmacological agents have been tested in preclinical studies. Inhibitors of RhoA (Simvastatin) or ROCK (Fasudil) may be good candidates [150]. Both drugs significantly decrease chronic hemorrhage in vascular lesions in murine model of CCM1 and CCM2. However, Fasudil is more efficient than Simvastatin in improving survival and blunting the development of mature lesions [150]. Exisulib, a β-catenin signaling inhibitor, limits the formation of brain vascular cavernomas in mice with CCM3 ablation in ECs [135]. This drug is currently used clinically to treat different pathologies and may be repurposed for CCM therapy. Similarly, other compounds such as ANGPT-2 neutralizing antibodies [151], multiple kinases inhibitor (Sorafenib) [148] or TGF-β receptors inhibitors [134], have shown efficacy in reducing CCM lesions in preclinical studies, but have not been tested in clinical trials yet.
Lymphatic endothelial cell development
The lymphatic circulation is a specialized vascular network responsible for maintaining tissue fluid homeostasis, immune cell transport and lipid absorption [152]. It is comprised of blunt-ended capillaries, collecting lymphatic vessels and lymph nodes which transport fluid, or lymph, unidirectionally from tissue capillaries into the venous circulation through the subclavian vein [152]. This unidirectional flow of lymph is aided by the presence of intraluminal lymphatic valves within the collecting lymphatic vessels [153]. These lymphatic valves, much like venous valves, help propel lymph against the force of gravity, while preventing retrograde transport. Pathologies which affect the development of the lymphatic circulation result in poor lymphatic flow and severe tissue edema [154,155,156]. Although the end pathologies resulting from aberrant lymphatic development are well understood, there is some debate about the processes that regulate lymphatic development, specifically the source of progenitor cells that give rise to lymphatic ECs (LECs).
LECs form the inner layer of lymphatic vessels and display a high degree of structural and functional heterogeneity throughout the lymphatic network that is evident from the beginning of development. LECs are specified shortly after blood ECs develop from the embryonic mesoderm. In 1902, Florence Sabin was the first to identify venous ECs as a source of LEC progenitors in developing lymphatic vessels in pig embryos [157]. These studies were later supported by lineage tracing of PROX1+ cells (master regulator of LECs described below) in mice and zebrafish, which revealed that venous-derived LEC development was conserved throughout different animal species [158,159,160]. As animal models and lineage-tracing techniques improved, additional sources of lymphatic progenitor cells were identified. In 2015, Stanczuk et al. identified cKit-expressing cells as a novel, non-venous source of LECs in the developing mesenteric lymphatics of the mouse embryo. We will discuss lymphatic vessel development from both progenitor sources [161].
Specification of LECs and their growth pattern differ depending on the source of LEC progenitors and the tissue in which they are developing. Most venous-derived lymphatic vessels form via a process known as lymphangiogenesis, in which newly specified LECs sprout from the endothelium of the CV in organized branches [162, 163]. Conversely, most non-venous-derived lymphatic vessels form via a process known as lymphvasculogenesis [164]. In this process, newly specified LECs form clusters of cells that coalesce into lymphatic vascular plexi before being remodeling into a mature lymphatic network [164]. Regardless of the source of progenitors, all LECs share distinct genetic markers that are unique to the lymphatic circulation.
Cellular markers and their role in LEC development have been reviewed previously [165,166,167,168,169]. Briefly, the unique markers that are used to identify LECs include the transmembrane O-glycoprotein podoplanin (PDPN, gp38, or T1α), the transmembrane receptors that bind vascular endothelial growth factor C (VEGF-C), vascular endothelial growth factor receptor 3 (VEGFR3 or FLT4) and NRP-2, the lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1), and the transcription factors forkhead box C2 (FOXC2) and prospero homeobox 1 (PROX1). Arguably the most important of these genes is the transcription factor PROX1, whose expression commits progenitor ECs into an LEC fate [170, 171]. Embryonic loss of Prox1 expression impairs lymphatic development [163]. Furthermore, Prox1-deficient mice develop severe edema in utero and are embryonic lethal, emphasizing the importance of PROX1 in LEC specification and lymphatic development [163].
Although PROX1 expression is required for the fate commitment of all LECs, their response to growth factors during lymphatic vessel formation appears to be tissue-specific [161, 164, 172]. For example, VEGF-C, the prominent VEGF isoform responsible for promoting lymphatic expansion, affects lymphatic development in different tissues at different developmental time points. That is, Vegf-C overexpression promotes excess lymphatic development in the respiratory tract only during embryogenesis, while promoting dermal lymphatic growth throughout development and into adulthood [173,174,175]. These data highlight the importance of studying lymphatic development in a tissue- and time-specific context. The following sections will review the currently available data about lymphatic endothelial progenitor cell origins and tissue-specific lymphatic vessels into which they develop.
Venous-derived LECs
Venous-derived LECs are specified from the venous ECs in the dorsolateral region of the embryonic CV in mice as early as E9.5 (Fig. 2). Increased expression of PROX1 in this subset of venous ECs drives LEC fate specification [158,159,160, 176, 177]. Increased PROX1 expression is regulated by the transcription factors partners COUP-TFII and SOX18 [178, 179]. COUP-TFII is expressed by venous ECs and, alone, does not drive PROX1 expression. However, when associated with SOX18, it promotes PROX1 expression, and LEC specification and expansion. Loss of either Coup-tfII or Sox18 suppresses LEC specification in the CV [179, 180]. Unlike COUP-TFII, SOX18 expression is restricted to the dorsal region of the CV, which helps guide the polarized specification of LECs during lymphatic development. SOX18 expression is required for lymphatic specification, as evidence by the development of edema and embryonic lethality in Sox18-deficient mice [179]. Concomitantly, increased PROX1 expression enhances SOX18 expression, thereby promoting LEC specification and lymphatic development. This feedback mechanism assures the commitment of LECs from venous ECs, thereby promoting the formation of lymphatic vessels.

Lymphatic EC Specification and Lymphatic Valve Development. (Black) Lymphatic ECs (LECs) are specified from venous and non-venous progenitor cells. LECs form the embryonic lymphatic circulation through lymphangiogenesis and lymphvasculogenesis. Collecting lymphatic vessels have intraluminal valves which are formed from LECs with a high expression of PROX1 (PROX1high) and involves an intricate network of signaling pathways and transcription factors. (Red) Pathways disrupted in human lymphatic development that contribute to lymphatic malformations and lymphedema
Work in the zebrafish model has identified that this LEC specification event happens through the asymmetric division of venous EC progenitors which results in the formation of one venous EC daughter cell and one LEC daughter cell [181]. The newly formed LECs retain expression of COUP-TFII and SOX18, which continue to promote PROX1 expression in the new LEC population [181]. Specified LECs begin to migrate dorsally out of the CV via lymphangiogenesis. Much like angiogenesis during blood vessel formation, lymphangiogenesis involves the sprouting of specified ECs and directionally driven migration. Migrating LECs travel dorsally as a single unit connected by VE-cadherin junctions along the embryo to form the jugular lymphatic sacs and the primordial thoracic ducts [177, 182]. The integrity of these junctions is crucial for proper lymphatic formation and prevention of edema during lymphatic vascular development [182]. Once these structures are formed, LECs begin to migrate once again forming the longitudinal thoracic vessels that will ultimately develop the cervical and thoracic dermal lymphatics and part of the cardiac lymphatic system [158, 176].
Recent reports have identified retinoic acid (RA) as an important signaling molecule regulating the polarized specification of LECs within the CV. Bowles et al. showed that RALDH2 expression, the RA synthesizing enzyme, is higher in the ventrolateral region of the CV, thereby maintaining venous EC identity [183]. Conversely, increased expression of CYB26B1, the enzyme responsible for degrading RA, is co-expressed in the dorsal CV coinciding with LECs expressing PROX1 in E9.5 mouse embryos [183]. In addition, LEC specification and lymphatic vascular growth is impaired in Cyb26b1−/− mice [183]. Together, these data support the concept that carefully regulated levels of RA play an important role in the localized regional specification of LECs from venous ECs.
Spatiotemporal regulation of LEC specification from the CV may also be regulated by BMP signaling [169, 184,185,186]. Using BMP reporter mice, Beets et al. confirmed active BMP signaling in LECs and ECs from the CV in developing embryos [184]. BMP-dependent signaling can have differential effects on LEC specification depending on the identity of the ligand-receptor activated. In zebrafish, BMP2-SMAD1/5/8 signaling inhibits LEC specification. Bmp2-overexpressing zebrafish have impaired thoracic duct development due to attenuated LEC specification [185]. Similarly, Bmp9−/− mice have dilated lymphatic vessels at E15.5, suggesting that BMP9-dependent signaling inhibits overexpansion of LECs during development [186]. In cell culture experiments, BMP9 signaling decreases PROX1 and LYVE1 expression, although this effect may be concentration-dependent [186, 187]. Taken together, BMP2/9 signaling inhibits LEC specification during early development in order to maintain localized lymphatic specification from the dorsolateral region of the CV.
After LECs have been specified in the CV, they migrate dorsally out of the vein to form the first lymphatic vessels. Guided LEC migration is predominantly controlled by VEGF-C-VEGFR3 signaling. As LECs become specified, they begin to express VEGFR3 under the transcriptional regulation of PROX1 [188]. As previously mentioned, VEGFR3 is highly enriched in LECs and its expression is used to distinguish between lymphatic and blood ECs. Increased expression of VEGFR3 at E10.5 enables LECs to respond to VEGF-C signaling to promote LEC migration out of the CV [189, 190]. In Vegf-c-deficient mice, PROX1+ LECs fail to migrate out of the CV resulting in severe edema [162]. Conversely, overexpression of Vegf-c results in ectopic lymphatic growth [191].
A recent report put forth the hypothesis that LEC fate is predetermined at early stages of development prior to CV formation [192]. Stone and Stainier argue that, unlike blood ECs which are derived from the lateral mesoderm, LECs are specified from the paraxial mesoderm and migrate to form the dorsal CV before finally differentiating into LECs [192]. Another group showed a similar pattern in the developing zebrafish. Nicenboim et al. present evidence that LECs are not derived from blood ECs in the posterior CV (PCV, the CV analog in zebrafish), but rather arise from a subset of mesodermal precursor cells [193]. These cells are located in the ventral region of the PCV and begin to migrate dorsally as they specify into LECs. It is these groups of cells from which lymphatic sprouts begin to form [193]. These data highlight the continued debate on the origin of LECs and provide further support for the existing heterogeneity of LECs and plasticity in progenitor ECs.
Non-venous-derived LECs
In 1910, Huntington and McClure were the first to propose a non-venous origin of LEC progenitor cells [194]. They showed that mesoderm-derived endothelial precursor cells specify into LECs independent of the venous endothelium. This theory was largely dismissed for the venous-derived theory until recently, where advancements in lineage-tracing studies provide further support for tissue-specific, non-venous origins of LECs. Using these models, non-venous progenitor cells were found to be derived from multiple cell types including mesenchyme [195,196,197], the paraxial mesoderm [192], hematopoietic cells [198], hemogenic progenitor cells [161, 172], and the skin capillary plexus [199]. The discovery of these non-venous lymphatic progenitor cells revealed a complexity in the diversity of cell progenitors and the LECs that make up the lymphatic circulation. Furthermore, the specific source of LEC progenitor cells seems to ultimately determine the organ- or tissue-specific lymphatic vessels that are formed.
Tissue-specific dependence of lymphatic development is best highlighted by the development of the dermal lymphatics. The dermal lymphatics are derived from both venous and non-venous progenitor cells, each contributing to different regions of the dermal lymphatic network. Deletion of Prox1 in cells expressing EC-enriched Tie2, disrupts development of only the cervical and thoracic dermal lymphatic vessels [164]. These mice show significant subcutaneous edema; however, the growth and specification of the lumbar lymphatic vessels are unaffected, accounting for approximately 30% of LECs that were derived from non-venous origins (Tie2− derived cells) [164]. LECs derived from the Tie2-expressing cells sprout from the developing lymph sacs via lymphangiogenesis. Conversely, the LECs derived from the Tie2-negative population appear as clusters which coalesce and grow into mature lymphatics through lymphvasculogenesis.
Similarly, cardiac and facial lymphatics are derived from both venous and non-venous cell origins. In cardiac development, venous-derived LECs form the dorsal lymphatic vessels. These LECs sprout from the sinus venosus to form the dorsal network [200]. The ventral lymphatic vessels, however, are derived from arterial sub-mesothelial cells that develop from the second heart field [200]. In zebrafish, facial lymphatics develops in three individual steps. The first set of facial lymphatic vessels develops through lymphangiogenesis and sprouts from the dorsal common CV. After the first facial lymphatics form, a subset of angioblasts that resides next to the ventral aorta begins to express PROX1 and proliferates to form the ventral aorta lymphangioblasts that connect to the already developed facial lymphatic vessels [195]. The formation of the ventral facial lymphatic vessels from angioblast progenitors may have evolved out of necessity for a non-venous progenitor pool. The ventral facial area of the developing zebrafish lacks venous circulation and likely necessitated a new source of LECs to be developed in order to have a mature lymphatic network.
Some studies have suggested that non-venous LECs may, in part, be derived from hematopoietic progenitor cells [201,202,203]. This theory is based on the evidence that hematopoietic cell lineage (Vav+) cells also express lymphatic-enriched genes, including PDPN, LYVE1 and VEGFR3. A study using the cornea lymphangiogenesis model in irradiated mice, showed that bone marrow-derived LECs progenitor cells can exit the blood vasculature and infiltrate sites of lymphavasculogenesis [201]. The authors used EGFP-labeled bone marrow cells to trace immune cell infiltration into the cornea and determined that a subset of these EGFP-expressing cells also express lymphatic-enriched genes such as Lyve1 and Vegfr3 [201]. However, the potential of hematopoietic cells as a source of lymphatic progenitor cells continues to be debated.
Hemogenic ECs have also been identified as a potential source of LEC progenitor cells in the developing mesenteric lymphatics. Stanczuk et al. used lineage tracing of cells expressing cKit, which is highly enriched in hemogenic ECs, to assess their contribution in mesenteric lymphatic development [161]. Much like in the dermal lymphatics, mesenteric LECs are derived from both venous and non-venous cell populations. The venous population sprouts from the lymph sac at the mesenteric root and grows outward toward the intestine. These cells are PROX1+ and appear as early as E12.5. By E13, the LECs have grown sufficiently to form large collecting lymphatics. The non-venous cells, which express cKit, appear in clusters around the mesenteric vessels, and form a primitive plexus that joins the sprouting LECs from the lymph sac to form the mature mesenteric lymphatics. These cKit-expressing cells also express PROX1 and NRP-2, suggesting they are lymphatic progenitor cells. Collectively, these studies highlight the importance of understanding the cell and tissue heterogeneity of developing lymphatics in order to better understand the mechanisms that regulate lymphatic vascular development in both health and disease.
Regulation of lymphatic valve formation
Collecting lymphatic vessels have intraluminal bicuspid valves to help prevent retrograde transport of lymph. Lymphatic valves are formed by a layer of LECs surrounding a fibronectin splice isoform EIIIA (FN-EIIIA)-, collagen IV- and laminin α5-rich core [204, 205]. The LECs that make up the valves are specified through a series of genetic programing that promotes LEC migration into the lumen of lymphatic vessels and organization into a functional valve [166, 204,205,206,207]. Mutations in these genetic programs are associated with human lymphedemas (Fig. 2, Table 1) [156, 166, 207].
Lymphatic valve formation occurs in four distinct stages: initiation, circularization, condensation and leaflet elongation [207]. Initiation occurs as a select few LECs upregulate lymphatic markers to specify the valve-forming region. This process is mainly driven by the transcription factor FOXC2 [206]. FOXC2 interaction with other transcription factors, including GATA2 and NFATc1, upregulates the expression of the lymphatic-enriched transcription factor PROX1 [206, 208]. High expression of PROX1 (PROX1high) specifies LECs into valve-forming cells. Deletion of Foxc2 expression in mice reveals aberrant lymphatic valve formation leading to severe edema [209]. Activation of FOXC2 is mediated by shear stress in sites of disturbed flow, mainly due to vessel branching, and promotes valve formation [205]. Specifically, oscillatory shear stress upregulates FOXC2 activating the calcineurin/NFATc1 pathway to form lymphatic valves. FOXC2-dependent activation of NFATc1 is, in part, dependent on mechanosensitive gap junction channel proteins, Cx37 and Cx45 [205]. Loss of Cx37 or Cx43 in mice results in impaired lymphatic valve formation [210]. Further evidence for the role of shear stress in the developing lymphatic valve is seen in the development of lymphedema in Piezo1-deficient mice [211, 212]. PIEZO1 is a mechanosensitive ion channel whose activation has been linked to the development of lymphatic valves. Mice lacking Piezo1 specifically in LECs fail to form lymphatic valves [211, 212]. Conversely, overexpression of Piezo1 leads to increased number of lymphatic valves in the developing mice resulting from increased lymphatic valve gene expression [211]. Together, these findings highlight the importance of mechanical stress on LECs due to increased lymph flow to direct lymphatic valve development.
Similar to early LEC specification from progenitor cells, VEGF-C signaling plays an important role in the upregulation of PROX1 in the cells located in the valve-forming region [213]. VEGF-C-VEGFR3 signaling promotes the upregulation of PROX1 in valve-forming LECs through the activation of the transcriptional co-activators YAP and TAZ [213]. However, the mechanisms by which VEGF-C signaling promotes valve formation in restricted localized areas within the lymphatic vasculature remains unknown. Localized upregulation of PROX1 in valve-forming regions may be, in part, regulated by NOTCH signaling [214]. LEC-specific deletion of Notch1 in mice results in the expansion of PROX1high cells within the valve-forming region and misalignment of valve LECs in the developing valve leaflets [214]. NOTCH-1-dependent valve malformation is caused, in part, by decreased FN-EIIIA and integrin α9 in the valve-forming regions, revealing NOTCH-1 signaling as important, not only for valve LEC specification, but also for migration of these cells into a functional valve structure. BMP9-dependent signaling may also play an important role in lymphatic valve development. Bmp9−/− mice have dilated collecting vessels and lymphatic valve malformations in the mesenteric lymphatics [215]. The collecting lymphatics from these mice maintain expression of LYVE1, which is downregulated during lymphatic maturation, suggesting that BMP9 signaling contributes to lymphatic maturation during late lymphatic development [215, 216].
As early valve-forming LECs are specified around the valve-forming regions (a.k.a. the circularization step), they begin to move and condense into a ring surrounding the lumen. At this time, cells begin to deposit the extracellular matrix that will form the core of the valve leaflets [204, 205]. This extracellular matrix is made up of FN-EIIIA, collagen IV and laminin α5 [204, 205]. Expression of integrin α9 by valve-forming LECs promotes cell migration through the newly deposited matrix to form the classic bicuspid valve [204]. Migration of these LECs is also driven by the activation of planar cell polarity pathways [217]. Specifically, activation of CELSR1 promotes LEC migration by destabilizing VE-cadherin interactions, while enhancing adherens junction assembly [217]. Careful regulation of all these pathways helps generate functional lymphatic valves, allowing for proper tissue fluid homeostasis in the developing embryo and into adulthood.
Lymphedema and lymphatic malformations
Dysregulation of LEC specification leads to lymphedema and lymphatic vascular malformations, which can be debilitating and even fatal (Fig. 2, Table 1). We provide an overview of these disorders, what is known about their underlying genetic defects, and what treatments are currently available for affected patients.
Lymphedema
The most frequent lymphatic anomaly is lymphedema (LE). LE is characterized by diffuse, localized or extended swelling due to inefficient uptake of interstitial fluid and reduced lymphatic drainage, mostly in the extremities [218]. There are two types of LE: primary LE, which are genetic disorders; and secondary LE, which develop due to extrinsic factors such as surgery or infection of lymphatic vessels.
Primary congenital lymphedema
Inherited primary LE is classified as early-onset congenital LE (Nonne-Milroy lymphedema or Type I lymphedema, OMIM 153100), peripubertal (Meige disease or Type II lymphedema, OMIM 153200), or late-onset congenital LE (after 35 years of age, also named lymphedema tarda) [100, 219]. Around twenty gene mutations have been identified in different forms of LE [220]. The incidence of primary lymphedema is low, affecting 1 in 100,000 people worldwide [221]. Most of the genes involved encode for proteins in the VEGF-C/VEGFR3-signaling pathway [220].
The first mutations were discovered in FLT4, the gene encoding VEGFR3. Autosomal dominant and recessive missense mutations in the tyrosine kinase domain of this receptor leads to type I lymphedema [220, 222]. These mutations inhibit VEGFR3 phosphorylation and prevent its downstream signaling [222]. The homozygous Flt4 knockout mice die around E9.5 due to irregular and unorganized vessels, edema and cardiovascular failure [223]. The Chy mice, a model of Type I lymphedema, have heterozygous inactivating mutations in Vegfr3 and develop dysfunctional hypoplastic lymphatic vessels and swelling of the limbs [224]. Collagen and calcium-binding EGF domain-containing protein 1 (CCBE1) binds to the extracellular matrix to potentiate VEGF-C effects via VEGFR3 [225]. In humans, homozygous and heterozygous mutations that impair CCBE1 function cause the Hennekan lymphangiectasia-lymphedema syndrome (OMIM 235510), which includes generalized lymphatic anomalies, including LE, visceral lymphangiectasias and mental retardation [226]. In zebrafish, it was shown that CCBE1 is required for lymphangioblast budding and angiogenic sprouting from venous endothelium and its gene mutation leads to the full of fluid (fof) mutant phenotype [227]. PTPN14 is a tyrosine-phosphatase that regulates VEGFR3 activation after VEGF-C binding. A loss of function mutation of PTPN14 leads to LE (OMIM 608911) due to VEGFR3-signaling hyperactivation [228].
Downstream of the VEGF-C-VEGFR3-signaling pathway, several transcription factors are activated and regulate numerous targets genes [220]. For example, truncated and missense mutations of FOXC2 are found in patients with hereditary type II lymphedema (late-onset LE) [229]. Foxc2 homozygous deletion in mice results in defective lymphatic patterning and arrested lymphatic valve development. Foxc2 heterozygous and endothelial-specific deletion in mice leads to lymphatic hyperplasia and impaired valve function in collecting lymphatic vessels [209, 230]. A rare form of LE with variable onset, Hypotrichosis-Lymphedema-Telengiectasia (OMIM 607823), has been associated with recessive and dominant mutations in the transcription factor SOX18 [231, 232]. SOX18 regulates PROX1, a transcription factor essential for lymphangiogenesis which, in turn, positively regulates FLT4 gene expression. The observed lymphatic defects may be due to competitive transcription factor binding [231].
GATA2 is another transcription factor that regulates PROX1 and FOXC2 expression. Loss-of-function mutations in GATA2 have been reported in patients with primary LE and myelodysplasia (Emberger syndrome, OMIM 614038) [208]. GATA2 in expressed by ECs, hematopoietic stem and progenitor cells, and lymphatic valve-forming cells. Gata2-homozygous deletion in mice leads to embryonic lethally at mid-gestation due to anemia and reduced myeloid-erythroid progenitor cells. However, no vascular defects have been detected in this model, which may be due to redundancy among GATA family members [233, 234].
Impaired lymphatic valve development/function could contribute to LE, as mutations in GJC1 encoding Cx43 and GJC2 encoding Cx47, which are expressed in lymphatic valves, have been identified in some patients [235, 236]. The related mutations are amino acid substitutions that alter connexin functions. Substitutions of highly conserved amino acids in GJC2 (Cx47) cause LE in all four extremities [235], whereas loss-of-function mutations cause Hypomyelinating leukodystrophy 2 (OMIM 608804), in which LE does not occur. The amino acid substitutions may have gain-of function effects, since Gjc2 homozygous deletion in mice does not result in lymphatic defects [237]. Various mutations in GJA1 (Cx43) are known to cause oculodentodigital dysplasia (OMIM 164200), and at least one has been linked to primary LE [236]. FOXC2 transcription factors also control the expression of several other proteins involved in lymphangiogenesis, such as Cx37, which is involved in lymphatic valve formation [210, 238]. Indeed, Cx37 expression is drastically reduced in mesenteric lymphatic vessels of mice lacking Foxc2, suggesting that Gja4 (encodes Cx37) may be regulated by FOXC2 [210]. However, the mechanism underlying Cx37-, Cx43- or Cx47-related LE is still unclear and could be related to abnormal lymphatic vessel and/or valve development, or defective cell–cell communication in lymphatic vessels leading to impaired coordination of pulsatile lymphatic flow [239]. Interestingly, Cx37 is a potent inhibitor of cell cycle progression, and it is therefore possible that Cx37 plays a similar cell cycle arrest role via p27 to enable specification toward lymphatic EC fates [240].
Lymphatic malformations
Lymphatic malformations (LM) consist of masses of abnormal dilated lymphatic channels not connected to the lymphatic system but filled with fluid, most commonly located in the head and neck [100, 220]. LMs are sporadic and their etiopathogenesis is unknown. LM can be part of a syndrome, such as Turner syndrome (due to monosomy X), or overgrowth syndrome, such as Klippel-Trenaunay syndrome (capillary-lymphatico-venous malformation, OMIM 149000) that is caused by mutations in the PI3K/AKT pathway [220].
Clinical treatments for lymphatic vascular malformations
The current therapy for lymphedema consists of decongestive physiotherapy to reduce edema and maintain the health of the skin and surrounding structures. Manual lymphatic drainage, as well as skin care and exercise, and the use of compression bandages are the gold standards. Occasionally surgery is performed [241, 242]. These treatments can only result in symptomatic improvements, but they do not cure the underlying dysfunction. Medical treatment of complicated LM with Sirolimus has showed substantial clinical benefits, though further research is needed to determine the efficacy of this medication for diverse subsets of lymphatic malformations [242, 243]. Furthermore, preclinical studies in mice have shown that Sirolimus treatment combined with anti-VEGF-C therapy can promote the regression of LM in Pik3ca-mutant model [244].
Summary
EC specification is a critical step in vascular development. Perturbations in the signaling pathways that determine blood and lymphatic EC identity result in vascular malformations. A combination of factors, including growth factor signaling, transcriptional regulation, and mechanotransduction, determine EC fate in the developing embryo. Furthermore, there is a growing appreciation for the role of cell cycle regulation in EC fate determination. Impairment of cell cycle control results in aberrant EC growth and fate determination that may ultimately lead to vascular malformations. Further research is still needed to elucidate the mechanisms of EC specification, and how their dysregulation leads to vascular malformations. Gaining further insights will help to improve treatment for such disorders.
References
Hirschi KK, Rohovsky SA, D’Amore PA (1998) PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol 141(3):805–814. https://doi.org/10.1083/jcb.141.3.805
Hirschi KK, Rohovsky SA, Beck LH, Smith SR, D’Amore PA (1999) Endothelial cells modulate the proliferation of mural cell precursors via platelet-derived growth factor-BB and heterotypic cell contact. Circ Res 84(3):298–305. https://doi.org/10.1161/01.res.84.3.298
Proulx K, Lu A, Sumanas S (2010) Cranial vasculature in zebrafish forms by angioblast cluster-derived angiogenesis. Dev Biol 348(1):34–46. https://doi.org/10.1016/j.ydbio.2010.08.036
Lee D, Park C, Lee H, Lugus JJ, Kim SH, Arentson E, Chung YS, Gomez G, Kyba M, Lin S, Janknecht R, Lim DS, Choi K (2008) ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell 2(5):497–507. https://doi.org/10.1016/j.stem.2008.03.008
Ferdous A, Caprioli A, Iacovino M, Martin CM, Morris J, Richardson JA, Latif S, Hammer RE, Harvey RP, Olson EN, Kyba M, Garry DJ (2009) Nkx2-5 transactivates the Ets-related protein 71 gene and specifies an endothelial/endocardial fate in the developing embryo. Proc Natl Acad Sci USA 106(3):814–819. https://doi.org/10.1073/pnas.0807583106
Castro PR, Barbosa AS, Pereira JM, Ranfley H, Felipetto M, Gonçalves CAX, Paiva IR, Berg BB, Barcelos LS (2018) Cellular and molecular heterogeneity associated with vessel formation processes. Biomed Res Int 2018:6740408. https://doi.org/10.1155/2018/6740408
Marcelo KL, Goldie LC, Hirschi KK (2013) Regulation of endothelial cell differentiation and specification. Circ Res 112(9):1272–1287. https://doi.org/10.1161/CIRCRESAHA.113.300506
Kohli V, Schumacher JA, Desai SP, Rehn K, Sumanas S (2013) Arterial and venous progenitors of the major axial vessels originate at distinct locations. Dev Cell 25(2):196–206. https://doi.org/10.1016/j.devcel.2013.03.017
Lawson ND, Vogel AM, Weinstein BM (2002) sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell 3(1):127–136. https://doi.org/10.1016/s1534-5807(02)00198-3
Potente M, Mäkinen T (2017) Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol 18(8):477–494. https://doi.org/10.1038/nrm.2017.36
Stone J, Itin A, Alon T, Pe’er J, Gnessin H, Chan-Ling T, Keshet E (1995) Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci 15(7 Pt 1):4738–4747. https://doi.org/10.1523/jneurosci.15-07-04738.1995
Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E (1995) Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1(10):1024–1028. https://doi.org/10.1038/nm1095-1024
Raab S, Beck H, Gaumann A, Yüce A, Gerber HP, Plate K, Hammes HP, Ferrara N, Breier G (2004) Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb Haemost 91(3):595–605. https://doi.org/10.1160/th03-09-0582
Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161(6):1163–1177. https://doi.org/10.1083/jcb.200302047
Simons M, Gordon E, Claesson-Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17(10):611–625. https://doi.org/10.1038/nrm.2016.87
Blanco R, Gerhardt H (2013) VEGF and Notch in tip and stalk cell selection. Cold Spring Harb Perspect Med 3(1):a006569. https://doi.org/10.1101/cshperspect.a006569
Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380(6573):435–439. https://doi.org/10.1038/380435a0
Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC (1995) Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376(6535):62–66. https://doi.org/10.1038/376062a0
Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ (2007) Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA 104(9):3219–3224. https://doi.org/10.1073/pnas.0611206104
Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K (2015) VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci USA 112(3):761–766. https://doi.org/10.1073/pnas.1423278112
Aspalter IM, Gordon E, Dubrac A, Ragab A, Narloch J, Vizán P, Geudens I, Collins RT, Franco CA, Abrahams CL, Thurston G, Fruttiger M, Rosewell I, Eichmann A, Gerhardt H (2015) Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nat Commun 6:7264. https://doi.org/10.1038/ncomms8264
Mouillesseaux KP, Wiley DS, Saunders LM, Wylie LA, Kushner EJ, Chong DC, Citrin KM, Barber AT, Park Y, Kim JD, Samsa LA, Kim J, Liu J, Jin SW, Bautch VL (2016) Notch regulates BMP responsiveness and lateral branching in vessel networks via SMAD6. Nat Commun 7:13247. https://doi.org/10.1038/ncomms13247
Itoh F, Itoh S, Goumans MJ, Valdimarsdottir G, Iso T, Dotto GP, Hamamori Y, Kedes L, Kato M, ten Dijke PP (2004) Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J 23(3):541–551. https://doi.org/10.1038/sj.emboj.7600065
Mack JJ, Iruela-Arispe ML (2018) NOTCH regulation of the endothelial cell phenotype. Curr Opin Hematol 25(3):212–218. https://doi.org/10.1097/moh.0000000000000425
Wang HU, Chen ZF, Anderson DJ (1998) Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93(5):741–753. https://doi.org/10.1016/s0092-8674(00)81436-1
Wiley DM, Kim JD, Hao J, Hong CC, Bautch VL, Jin SW (2011) Distinct signalling pathways regulate sprouting angiogenesis from the dorsal aorta and the axial vein. Nat Cell Biol 13(6):686–692. https://doi.org/10.1038/ncb2232
Fang J, Hirschi K (2019) Molecular regulation of arteriovenous endothelial cell specification. F1000Res. https://doi.org/10.12688/f1000research.16701.1
Casie Chetty S, Rost MS, Enriquez JR, Schumacher JA, Baltrunaite K, Rossi A, Stainier DY, Sumanas S (2017) Vegf signaling promotes vascular endothelial differentiation by modulating etv2 expression. Dev Biol 424(2):147–161. https://doi.org/10.1016/j.ydbio.2017.03.005
Hong CC, Peterson QP, Hong JY, Peterson RT (2006) Artery/vein specification is governed by opposing phosphatidylinositol-3 kinase and MAP kinase/ERK signaling. Curr Biol 16(13):1366–1372. https://doi.org/10.1016/j.cub.2006.05.046
Chiang IK, Fritzsche M, Pichol-Thievend C, Neal A, Holmes K, Lagendijk A, Overman J, D’Angelo D, Omini A, Hermkens D, Lesieur E, Liu K, Ratnayaka I, Corada M, Bou-Gharios G, Carroll J, Dejana E, Schulte-Merker S, Hogan B, Beltrame M, De Val S, Francois M (2017) SoxF factors induce Notch1 expression via direct transcriptional regulation during early arterial development. Development 144(14):2629–2639. https://doi.org/10.1242/dev.146241
Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, Taketo MM, Iruela-Arispe ML, Adams RH, Dejana E (2010) The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev Cell 18(6):938–949. https://doi.org/10.1016/j.devcel.2010.05.006
Iso T, Maeno T, Oike Y, Yamazaki M, Doi H, Arai M, Kurabayashi M (2006) Dll4-selective Notch signaling induces ephrinB2 gene expression in endothelial cells. Biochem Biophys Res Commun 341(3):708–714. https://doi.org/10.1016/j.bbrc.2006.01.020
Esser JS, Steiner RE, Deckler M, Schmitt H, Engert B, Link S, Charlet A, Patterson C, Bode C, Zhou Q, Moser M (2018) Extracellular bone morphogenetic protein modulator BMPER and twisted gastrulation homolog 1 preserve arterial-venous specification in zebrafish blood vessel development and regulate Notch signaling in endothelial cells. Febs J 285(8):1419–1436. https://doi.org/10.1111/febs.14414
Osmanagic-Myers S, Rezniczek GA (2018) Arteriovenous specification: BMPER and TWSG1 determine endothelial cell fate via activation of synergistic BMP and Notch signaling. Febs J 285(8):1399–1402. https://doi.org/10.1111/febs.14439
Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, Weinstein BM (2001) Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128(19):3675–3683
Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T (2000) Notch signaling is essential for vascular morphogenesis in mice. Genes Dev 14(11):1343–1352
le Noble F, Moyon D, Pardanaud L, Yuan L, Djonov V, Matthijsen R, Bréant C, Fleury V, Eichmann A (2004) Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development 131(2):361–375. https://doi.org/10.1242/dev.00929
Fang JS, Coon BG, Gillis N, Chen Z, Qiu J, Chittenden TW, Burt JM, Schwartz MA, Hirschi KK (2017) Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nat Commun 8(1):2149. https://doi.org/10.1038/s41467-017-01742-7
Su T, Stanley G, Sinha R, D’Amato G, Das S, Rhee S, Chang AH, Poduri A, Raftrey B, Dinh TT, Roper WA, Li G, Quinn KE, Caron KM, Wu S, Miquerol L, Butcher EC, Weissman I, Quake S, Red-Horse K (2018) Single-cell analysis of early progenitor cells that build coronary arteries. Nature 559(7714):356–362. https://doi.org/10.1038/s41586-018-0288-7
Chu M, Li T, Shen B, Cao X, Zhong H, Zhang L, Zhou F, Ma W, Jiang H, Xie P, Liu Z, Dong N, Xu Y, Zhao Y, Xu G, Lu P, Luo J, Wu Q, Alitalo K, Koh GY, Adams RH, He Y (2016) Angiopoietin receptor Tie2 is required for vein specification and maintenance via regulating COUP-TFII. Elife. https://doi.org/10.7554/eLife.21032
Xu C, Hasan SS, Schmidt I, Rocha SF, Pitulescu ME, Bussmann J, Meyen D, Raz E, Adams RH, Siekmann AF (2014) Arteries are formed by vein-derived endothelial tip cells. Nat Commun 5:5758. https://doi.org/10.1038/ncomms6758
Hasan SS, Tsaryk R, Lange M, Wisniewski L, Moore JC, Lawson ND, Wojciechowska K, Schnittler H, Siekmann AF (2017) Endothelial Notch signalling limits angiogenesis via control of artery formation. Nat Cell Biol 19(8):928–940. https://doi.org/10.1038/ncb3574
Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF, Langen UH, Stehling M, Nagasawa T, Ferrara N, Borggrefe T, Adams RH (2017) Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol 19(8):915–927. https://doi.org/10.1038/ncb3555
Neal A, Nornes S, Payne S, Wallace MD, Fritzsche M, Louphrasitthiphol P, Wilkinson RN, Chouliaras KM, Liu K, Plant K, Sholapurkar R, Ratnayaka I, Herzog W, Bond G, Chico T, Bou-Gharios G, De Val S (2019) Venous identity requires BMP signalling through ALK3. Nat Commun 10(1):453. https://doi.org/10.1038/s41467-019-08315-w
Goumans MJ, Liu Z, ten Dijke P (2009) TGF-beta signaling in vascular biology and dysfunction. Cell Res 19(1):116–127. https://doi.org/10.1038/cr.2008.326
Fish JE, Wythe JD (2015) The molecular regulation of arteriovenous specification and maintenance. Dev Dyn 244(3):391–409. https://doi.org/10.1002/dvdy.24252
Lesca G, Burnichon N, Raux G, Tosi M, Pinson S, Marion MJ, Babin E, Gilbert-Dussardier B, Rivière S, Goizet C, Faivre L, Plauchu H, Frébourg T, Calender A, Giraud S (2006) Distribution of ENG and ACVRL1 (ALK1) mutations in French HHT patients. Hum Mutat 27(6):598. https://doi.org/10.1002/humu.9421
Urness LD, Sorensen LK, Li DY (2000) Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet 26(3):328–331. https://doi.org/10.1038/81634
Sorensen LK, Brooke BS, Li DY, Urness LD (2003) Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFbeta coreceptor. Dev Biol 261(1):235–250. https://doi.org/10.1016/s0012-1606(03)00158-1
Carvalho RL, Itoh F, Goumans MJ, Lebrin F, Kato M, Takahashi S, Ema M, Itoh S, van Rooijen M, Bertolino P, Ten Dijke P, Mummery CL (2007) Compensatory signalling induced in the yolk sac vasculature by deletion of TGFbeta receptors in mice. J Cell Sci 120(Pt 24):4269–4277. https://doi.org/10.1242/jcs.013169
Larsson J, Goumans MJ, Sjöstrand LJ, van Rooijen MA, Ward D, Levéen P, Xu X, ten Dijke P, Mummery CL, Karlsson S (2001) Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J 20(7):1663–1673. https://doi.org/10.1093/emboj/20.7.1663
Mancini ML, Terzic A, Conley BA, Oxburgh LH, Nicola T, Vary CP (2009) Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev Dyn 238(10):2479–2493. https://doi.org/10.1002/dvdy.22066
Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P (2003) Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell 12(4):817–828. https://doi.org/10.1016/s1097-2765(03)00386-1
Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P (2004) Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J 23(20):4018–4028. https://doi.org/10.1038/sj.emboj.7600386
Blanco FJ, Santibanez JF, Guerrero-Esteo M, Langa C, Vary CP, Bernabeu C (2005) Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J Cell Physiol 204(2):574–584. https://doi.org/10.1002/jcp.20311
Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P (2002) Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J 21(7):1743–1753. https://doi.org/10.1093/emboj/21.7.1743
Itoh F, Itoh S, Carvalho RL, Adachi T, Ema M, Goumans MJ, Larsson J, Karlsson S, Takahashi S, Mummery CL, Dijke PT, Kato M (2009) Poor vessel formation in embryos from knock-in mice expressing ALK5 with L45 loop mutation defective in Smad activation. Lab Invest 89(7):800–810. https://doi.org/10.1038/labinvest.2009.37
Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E (2000) Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA 97(6):2626–2631. https://doi.org/10.1073/pnas.97.6.2626
Lamouille S, Mallet C, Feige JJ, Bailly S (2002) Activin receptor-like kinase 1 is implicated in the maturation phase of angiogenesis. Blood 100(13):4495–4501. https://doi.org/10.1182/blood.V100.13.4495
Ota T, Fujii M, Sugizaki T, Ishii M, Miyazawa K, Aburatani H, Miyazono K (2002) Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-beta in human umbilical vein endothelial cells. J Cell Physiol 193(3):299–318. https://doi.org/10.1002/jcp.10170
Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, Breviario F, Conti V, Briot A, Iruela-Arispe ML, Adams RH, Dejana E (2013) Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun 4:2609. https://doi.org/10.1038/ncomms3609
Larrivée B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A (2012) ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell 22(3):489–500. https://doi.org/10.1016/j.devcel.2012.02.005
Rochon ER, Menon PG, Roman BL (2016) Alk1 controls arterial endothelial cell migration in lumenized vessels. Development 143(14):2593–2602. https://doi.org/10.1242/dev.135392
Dalton S (2015) Linking the cell cycle to cell fate decisions. Trends Cell Biol 25(10):592–600. https://doi.org/10.1016/j.tcb.2015.07.007
Pauklin S, Vallier L (2013) The cell-cycle state of stem cells determines cell fate propensity. Cell 155(1):135–147. https://doi.org/10.1016/j.cell.2013.08.031
Ristanović D, Milosević NT, Stefanović IB, Marić D, Popov I (2009) Cell image area as a tool for neuronal classification. J Neurosci Methods 182(2):272–278. https://doi.org/10.1016/j.jneumeth.2009.06.004
Mort RL, Ford MJ, Sakaue-Sawano A, Lindstrom NO, Casadio A, Douglas AT, Keighren MA, Hohenstein P, Miyawaki A, Jackson IJ (2014) Fucci2a: a bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell Cycle 13(17):2681–2696. https://doi.org/10.4161/15384101.2015.945381
Chavkin NW, Genet G, Poulet M, Genet N, Marziano C, Vasavada H, Nelson EA, Kour A, McDonnell SP, Huba M, Walsh K, Hirschi KK (2020) Endothelial Cell Cycle State Determines Propensity for Arterial-Venous Fate. bioRxiv:2020.2008.2012.246512. https://doi.org/10.1101/2020.08.12.246512
Lindahl P, Johansson BR, Levéen P, Betsholtz C (1997) Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277(5323):242–245. https://doi.org/10.1126/science.277.5323.242
Sato Y, Tsuboi R, Lyons R, Moses H, Rifkin DB (1990) Characterization of the activation of latent TGF-beta by co-cultures of endothelial cells and pericytes or smooth muscle cells: a self-regulating system. J Cell Biol 111(2):757–763. https://doi.org/10.1083/jcb.111.2.757
Flaumenhaft R, Abe M, Sato Y, Miyazono K, Harpel J, Heldin CH, Rifkin DB (1993) Role of the latent TGF-beta binding protein in the activation of latent TGF-beta by co-cultures of endothelial and smooth muscle cells. J Cell Biol 120(4):995–1002. https://doi.org/10.1083/jcb.120.4.995
Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE (2009) Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 114(24):5091–5101. https://doi.org/10.1182/blood-2009-05-222364
Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M, Tsuruo T, Sawada Y, Niwa M, Kataoka Y (2005) Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res 1038(2):208–215. https://doi.org/10.1016/j.brainres.2005.01.027
van Dijk CG, Nieuweboer FE, Pei JY, Xu YJ, Burgisser P, van Mulligen E, el Azzouzi H, Duncker DJ, Verhaar MC, Cheng C (2015) The complex mural cell: pericyte function in health and disease. Int J Cardiol 190:75–89. https://doi.org/10.1016/j.ijcard.2015.03.258
Conway EM, Collen D, Carmeliet P (2001) Molecular mechanisms of blood vessel growth. Cardiovasc Res 49(3):507–521. https://doi.org/10.1016/s0008-6363(00)00281-9
Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554(7693):475–480. https://doi.org/10.1038/nature25739
Shovlin CL (2010) Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev 24(6):203–219. https://doi.org/10.1016/j.blre.2010.07.001
Robert F, Desroches-Castan A, Bailly S, Dupuis-Girod S, Feige JJ (2020) Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis 15(1):4. https://doi.org/10.1186/s13023-019-1281-4
McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J et al (1994) Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 8(4):345–351. https://doi.org/10.1038/ng1294-345
Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA (1996) Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 13(2):189–195. https://doi.org/10.1038/ng0696-189
Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA (2004) A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 363(9412):852–859. https://doi.org/10.1016/s0140-6736(04)15732-2
Wooderchak-Donahue WL, McDonald J, O’Fallon B, Upton PD, Li W, Roman BL, Young S, Plant P, Fülöp GT, Langa C, Morrell NW, Botella LM, Bernabeu C, Stevenson DA, Runo JR, Bayrak-Toydemir P (2013) BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet 93(3):530–537. https://doi.org/10.1016/j.ajhg.2013.07.004
Crist AM, Zhou X, Garai J, Lee AR, Thoele J, Ullmer C, Klein C, Zabaleta J, Meadows SM (2019) Angiopoietin-2 inhibition rescues arteriovenous malformation in a Smad4 hereditary hemorrhagic telangiectasia mouse model. Circulation 139(17):2049–2063. https://doi.org/10.1161/circulationaha.118.036952
Ola R, Dubrac A, Han J, Zhang F, Fang JS, Larrivee B, Lee M, Urarte AA, Kraehling JR, Genet G, Hirschi KK, Sessa WC, Canals FV, Graupera M, Yan M, Young LH, Oh PS, Eichmann A (2016) PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat Commun 7:13650. https://doi.org/10.1038/ncomms13650
Ola R, Kunzel SH, Zhang F, Genet G, Chakraborty R, Pibouin-Fragner L, Martin K, Sessa W, Dubrac A, Eichmann A (2018) SMAD4 prevents flow induced arteriovenous malformations by inhibiting Casein Kinase 2. Circulation 138(21):2379–2394. https://doi.org/10.1161/CIRCULATIONAHA.118.033842
Bourdeau A, Dumont DJ, Letarte M (1999) A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 104(10):1343–1351. https://doi.org/10.1172/JCI8088
Srinivasan S, Hanes MA, Dickens T, Porteous ME, Oh SP, Hale LP, Marchuk DA (2003) A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet 12(5):473–482. https://doi.org/10.1093/hmg/ddg050
Lan Y, Liu B, Yao H, Li F, Weng T, Yang G, Li W, Cheng X, Mao N, Yang X (2007) Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol Cell Biol 27(21):7683–7692. https://doi.org/10.1128/MCB.00577-07
Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, Charlton R, Parums DV, Jowett T, Marchuk DA, Burn J, Diamond AG (2000) Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol 217(1):42–53. https://doi.org/10.1006/dbio.1999.9534
Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, Fruttiger M, Arthur HM (2010) Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res 106(8):1425–1433. https://doi.org/10.1161/CIRCRESAHA.109.211037
Selvam S, Kumar T, Fruttiger M (2018) Retinal vasculature development in health and disease. Prog Retin Eye Res 63:1–19. https://doi.org/10.1016/j.preteyeres.2017.11.001
Hwa JJ, Beckouche N, Huang L, Kram Y, Lindskog H, Wang RA (2017) Abnormal arterial-venous fusions and fate specification in mouse embryos lacking blood flow. Sci Rep 7(1):11965. https://doi.org/10.1038/s41598-017-12353-z
Baeyens N, Larrivee B, Ola R, Hayward-Piatkowskyi B, Dubrac A, Huang B, Ross TD, Coon BG, Min E, Tsarfati M, Tong H, Eichmann A, Schwartz MA (2016) Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J Cell Biol 214(7):807–816. https://doi.org/10.1083/jcb.201603106
Flieger D, Hainke S, Fischbach W (2006) Dramatic improvement in hereditary hemorrhagic telangiectasia after treatment with the vascular endothelial growth factor (VEGF) antagonist bevacizumab. Ann Hematol 85(9):631–632. https://doi.org/10.1007/s00277-006-0147-8
Buscarini E, Botella LM, Geisthoff U, Kjeldsen AD, Mager HJ, Pagella F, Suppressa P, Zarrabeitia R, Dupuis-Girod S, Shovlin CL, Vascern HHT (2019) Safety of thalidomide and bevacizumab in patients with hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis 14(1):28. https://doi.org/10.1186/s13023-018-0982-4
Kovacs-Sipos E, Holzmann D, Scherer T, Soyka MB (2017) Nintedanib as a novel treatment option in hereditary haemorrhagic telangiectasia. BMJ Case Rep. https://doi.org/10.1136/bcr-2017-219393
Faughnan ME, Gossage JR, Chakinala MM, Oh SP, Kasthuri R, Hughes CCW, McWilliams JP, Parambil JG, Vozoris N, Donaldson J, Paul G, Berry P, Sprecher DL (2019) Pazopanib may reduce bleeding in hereditary hemorrhagic telangiectasia. Angiogenesis 22(1):145–155. https://doi.org/10.1007/s10456-018-9646-1
Hwan Kim Y, Vu PN, Choe SW, Jeon CJ, Arthur HM, Vary CPH, Lee YJ, Oh SP (2020) Overexpression of Activin Receptor-Like Kinase 1 in endothelial cells suppresses development of arteriovenous malformations in mouse models of hereditary hemorrhagic telangiectasia. Circ Res 127(9):1122–1137. https://doi.org/10.1161/CIRCRESAHA.119.316267
Ruiz S, Chandakkar P, Zhao H, Papoin J, Chatterjee PK, Christen E, Metz CN, Blanc L, Campagne F, Marambaud P (2017) Tacrolimus rescues the signaling and gene expression signature of endothelial ALK1 loss-of-function and improves HHT vascular pathology. Hum Mol Genet 26(24):4786–4798. https://doi.org/10.1093/hmg/ddx358
Uebelhoer M, Boon LM, Vikkula M (2012) Vascular anomalies: from genetics toward models for therapeutic trials. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a009688
Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibe M, Warman ML, Tan OT, Olsen BR, Vikkula M (2002) Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet 70(4):866–874. https://doi.org/10.1086/339492
Dompmartin A, Vikkula M, Boon LM (2010) Venous malformation: update on aetiopathogenesis, diagnosis and management. Phlebology 25(5):224–235. https://doi.org/10.1258/phleb.2009.009041
Vikkula M, Boon LM, Carraway KL 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR (1996) Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell 87(7):1181–1190. https://doi.org/10.1016/s0092-8674(00)81814-0
Kangas J, Natynki M, Eklund L (2018) Development of molecular therapies for venous malformations. Basic Clin Pharmacol Toxicol 123(Suppl 5):6–19. https://doi.org/10.1111/bcpt.13027
Limaye N, Kangas J, Mendola A, Godfraind C, Schlogel MJ, Helaers R, Eklund L, Boon LM, Vikkula M (2015) Somatic activating PIK3CA mutations cause venous malformation. Am J Hum Genet 97(6):914–921. https://doi.org/10.1016/j.ajhg.2015.11.011
Castillo SD, Tzouanacou E, Zaw-Thin M, Berenjeno IM, Parker VE, Chivite I, Mila-Guasch M, Pearce W, Solomon I, Angulo-Urarte A, Figueiredo AM, Dewhurst RE, Knox RG, Clark GR, Scudamore CL, Badar A, Kalber TL, Foster J, Stuckey DJ, David AL, Phillips WA, Lythgoe MF, Wilson V, Semple RK, Sebire NJ, Kinsler VA, Graupera M, Vanhaesebroeck B (2016) Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aad9982
Uebelhoer M, Natynki M, Kangas J, Mendola A, Nguyen HL, Soblet J, Godfraind C, Boon LM, Eklund L, Limaye N, Vikkula M (2013) Venous malformation-causative TIE2 mutations mediate an AKT-dependent decrease in PDGFB. Hum Mol Genet 22(17):3438–3448. https://doi.org/10.1093/hmg/ddt198
Natynki M, Kangas J, Miinalainen I, Sormunen R, Pietila R, Soblet J, Boon LM, Vikkula M, Limaye N, Eklund L (2015) Common and specific effects of TIE2 mutations causing venous malformations. Hum Mol Genet 24(22):6374–6389. https://doi.org/10.1093/hmg/ddv349
Rossler J, Haubold M, Gilsbach R, Juttner E, Schmitt D, Niemeyer CM, Hein L (2013) beta1-Adrenoceptor mRNA level reveals distinctions between infantile hemangioma and vascular malformations. Pediatr Res 73(4 Pt 1):409–413. https://doi.org/10.1038/pr.2013.16
Medema RH, Kops GJ, Bos JL, Burgering BM (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404(6779):782–787. https://doi.org/10.1038/35008115
Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH (2002) Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol 22(22):7842–7852. https://doi.org/10.1128/mcb.22.22.7842-7852.2002
Boscolo E, Limaye N, Huang L, Kang KT, Soblet J, Uebelhoer M, Mendola A, Natynki M, Seront E, Dupont S, Hammer J, Legrand C, Brugnara C, Eklund L, Vikkula M, Bischoff J, Boon LM (2015) Rapamycin improves TIE2-mutated venous malformation in murine model and human subjects. J Clin Invest 125(9):3491–3504. https://doi.org/10.1172/JCI76004
Castel P, Carmona FJ, Grego-Bessa J, Berger MF, Viale A, Anderson KV, Bague S, Scaltriti M, Antonescu CR, Baselga E, Baselga J (2016) Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaf1164
Jacobs AH, Walton RG (1976) The incidence of birthmarks in the neonate. Pediatrics 58(2):218–222
Garzon MC, Huang JT, Enjolras O, Frieden IJ (2007) Vascular malformations: Part I. J Am Acad Dermatol 56(3):353–370. https://doi.org/10.1016/j.jaad.2006.05.069 (quiz 371–354)
Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, Burrows PE, Clapuyt P, Hammer F, Dubois J, Baselga E, Brancati F, Carder R, Quintal JM, Dallapiccola B, Fischer G, Frieden IJ, Garzon M, Harper J, Johnson-Patel J, Labreze C, Martorell L, Paltiel HJ, Pohl A, Prendiville J, Quere I, Siegel DH, Valente EM, Van Hagen A, Van Hest L, Vaux KK, Vicente A, Weibel L, Chitayat D, Vikkula M (2008) Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat 29(7):959–965. https://doi.org/10.1002/humu.20746
Orme CM, Boyden LM, Choate KA, Antaya RJ, King BA (2013) Capillary malformation–arteriovenous malformation syndrome: review of the literature, proposed diagnostic criteria, and recommendations for management. Pediatr Dermatol 30(4):409–415. https://doi.org/10.1111/pde.12112
Glanzer JG, Liao L, Baker T, McMullen MH, Langan AS, Crandall LZ, Vorce RL (2002) Organization and regulation of the human rasGAP gene. Gene 285(1–2):149–156. https://doi.org/10.1016/s0378-1119(02)00415-8
Meadows KN, Bryant P, Vincent PA, Pumiglia KM (2004) Activated Ras induces a proangiogenic phenotype in primary endothelial cells. Oncogene 23(1):192–200. https://doi.org/10.1038/sj.onc.1206921
Serban D, Leng J, Cheresh D (2008) H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ Res 102(11):1350–1358. https://doi.org/10.1161/CIRCRESAHA.107.169664
Anand S, Majeti BK, Acevedo LM, Murphy EA, Mukthavaram R, Scheppke L, Huang M, Shields DJ, Lindquist JN, Lapinski PE, King PD, Weis SM, Cheresh DA (2010) MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med 16(8):909–914. https://doi.org/10.1038/nm.2186
Kulkarni SV, Gish G, van der Geer P, Henkemeyer M, Pawson T (2000) Role of p120 Ras-GAP in directed cell movement. J Cell Biol 149(2):457–470. https://doi.org/10.1083/jcb.149.2.457
Kawasaki J, Aegerter S, Fevurly RD, Mammoto A, Mammoto T, Sahin M, Mably JD, Fishman SJ, Chan J (2014) RASA1 functions in EPHB4 signaling pathway to suppress endothelial mTORC1 activity. J Clin Invest 124(6):2774–2784. https://doi.org/10.1172/JCI67084
Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K, Shih TS, Jacks T, Pawson T (1995) Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 377(6551):695–701. https://doi.org/10.1038/377695a0
Westenskow PD, Kurihara T, Aguilar E, Scheppke EL, Moreno SK, Wittgrove C, Marchetti V, Michael IP, Anand S, Nagy A, Cheresh D, Friedlander M (2013) Ras pathway inhibition prevents neovascularization by repressing endothelial cell sprouting. J Clin Invest 123(11):4900–4908. https://doi.org/10.1172/JCI70230
Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R (1999) Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev 13(3):295–306. https://doi.org/10.1101/gad.13.3.295
Muto A, Yi T, Harrison KD, Davalos A, Fancher TT, Ziegler KR, Feigel A, Kondo Y, Nishibe T, Sessa WC, Dardik A (2011) Eph-B4 prevents venous adaptive remodeling in the adult arterial environment. J Exp Med 208(3):561–575. https://doi.org/10.1084/jem.20101854
Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, Chung W, Dubois J, Lacour JP, Martorell L, Mazereeuw-Hautier J, Pyeritz RE, Amor DJ, Bisdorff A, Blei F, Bombei H, Dompmartin A, Brooks D, Dupont J, Gonzalez-Ensenat MA, Frieden I, Gerard M, Kvarnung M, Hanson-Kahn AK, Hudgins L, Leaute-Labreze C, McCuaig C, Metry D, Parent P, Paul C, Petit F, Phan A, Quere I, Salhi A, Turner A, Vabres P, Vicente A, Wargon O, Watanabe S, Weibel L, Wilson A, Willing M, Mulliken JB, Boon LM, Vikkula M (2017) Germline loss-of-function mutations in EPHB4 cause a second form of Capillary Malformation-Arteriovenous Malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation 136(11):1037–1048. https://doi.org/10.1161/CIRCULATIONAHA.116.026886
Wooderchak-Donahue WL, Akay G, Whitehead K, Briggs E, Stevenson DA, O’Fallon B, Velinder M, Farrell A, Shen W, Bedoukian E, Skrabann CM, Antaya RJ, Henderson K, Pollak J, Treat J, Day R, Jacher JE, Hannibal M, Bontempo K, Marth G, Bayrak-Toydemir P, McDonald J (2019) Phenotype of CM-AVM2 caused by variants in EPHB4: how much overlap with hereditary hemorrhagic telangiectasia (HHT)? Genet Med 21(9):2007–2014. https://doi.org/10.1038/s41436-019-0443-z
Labauge P, Denier C, Bergametti F, Tournier-Lasserve E (2007) Genetics of cavernous angiomas. Lancet Neurol 6(3):237–244. https://doi.org/10.1016/S1474-4422(07)70053-4
Revencu N, Vikkula M (2006) Cerebral cavernous malformation: new molecular and clinical insights. J Med Genet 43(9):716–721. https://doi.org/10.1136/jmg.2006.041079
Rigamonti D, Hadley MN, Drayer BP, Johnson PC, Hoenig-Rigamonti K, Knight JT, Spetzler RF (1988) Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med 319(6):343–347. https://doi.org/10.1056/NEJM198808113190605
McDonald DA, Shi C, Shenkar R, Gallione CJ, Akers AL, Li S, De Castro N, Berg MJ, Corcoran DL, Awad IA, Marchuk DA (2014) Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet 23(16):4357–4370. https://doi.org/10.1093/hmg/ddu153
Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, Tournier-Lasserve E, Chapon F, Richichi C, Retta SF, Lampugnani MG, Dejana E (2013) EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498(7455):492–496. https://doi.org/10.1038/nature12207
Bravi L, Rudini N, Cuttano R, Giampietro C, Maddaluno L, Ferrarini L, Adams RH, Corada M, Boulday G, Tournier-Lasserve E, Dejana E, Lampugnani MG (2015) Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. Proc Natl Acad Sci USA 112(27):8421–8426. https://doi.org/10.1073/pnas.1501352112
He Y, Zhang H, Yu L, Gunel M, Boggon TJ, Chen H, Min W (2010) Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci Signal 3(116):ra26. https://doi.org/10.1126/scisignal.2000722
Takada S, Hojo M, Tanigaki K, Miyamoto S (2017) Contribution of endothelial-to-mesenchymal transition to the pathogenesis of human cerebral and orbital cavernous malformations. Neurosurgery 81(1):176–183. https://doi.org/10.1093/neuros/nyx078
Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, Marchuk DA (2005) CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet 14(17):2521–2531. https://doi.org/10.1093/hmg/ddi256
Hilder TL, Malone MH, Bencharit S, Colicelli J, Haystead TA, Johnson GL, Wu CC (2007) Proteomic identification of the cerebral cavernous malformation signaling complex. J Proteome Res 6(11):4343–4355. https://doi.org/10.1021/pr0704276
Glading A, Han J, Stockton RA, Ginsberg MH (2007) KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol 179(2):247–254. https://doi.org/10.1083/jcb.200705175
Whitehead KJ, Chan AC, Navankasattusas S, Koh W, London NR, Ling J, Mayo AH, Drakos SG, Jones CA, Zhu W, Marchuk DA, Davis GE, Li DY (2009) The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med 15(2):177–184. https://doi.org/10.1038/nm.1911
Stockton RA, Shenkar R, Awad IA, Ginsberg MH (2010) Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med 207(4):881–896. https://doi.org/10.1084/jem.20091258
Lavina B, Castro M, Niaudet C, Cruys B, Alvarez-Aznar A, Carmeliet P, Bentley K, Brakebusch C, Betsholtz C, Gaengel K (2018) Defective endothelial cell migration in the absence of Cdc42 leads to capillary-venous malformations. Development. https://doi.org/10.1242/dev.161182
Cunha SI, Magnusson PU, Dejana E, Lampugnani MG (2017) Deregulated TGF-beta/BMP signaling in vascular malformations. Circ Res 121(8):981–999. https://doi.org/10.1161/CIRCRESAHA.117.309930
Morikawa M, Koinuma D, Tsutsumi S, Vasilaki E, Kanki Y, Heldin CH, Aburatani H, Miyazono K (2011) ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res 39(20):8712–8727. https://doi.org/10.1093/nar/gkr572
Dejana E, Hirschi KK, Simons M (2017) The molecular basis of endothelial cell plasticity. Nat Commun 8:14361. https://doi.org/10.1038/ncomms14361
Lampugnani MG, Orsenigo F, Rudini N, Maddaluno L, Boulday G, Chapon F, Dejana E (2010) CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J Cell Sci 123(Pt 7):1073–1080. https://doi.org/10.1242/jcs.059329
Wustehube J, Bartol A, Liebler SS, Brutsch R, Zhu Y, Felbor U, Sure U, Augustin HG, Fischer A (2010) Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc Natl Acad Sci USA 107(28):12640–12645. https://doi.org/10.1073/pnas.1000132107
Lampugnani MG, Malinverno M, Dejana E, Rudini N (2017) Endothelial cell disease: emerging knowledge from cerebral cavernous malformations. Curr Opin Hematol 24(3):256–264. https://doi.org/10.1097/MOH.0000000000000338
Shenkar R, Shi C, Austin C, Moore T, Lightle R, Cao Y, Zhang L, Wu M, Zeineddine HA, Girard R, McDonald DA, Rorrer A, Gallione C, Pytel P, Liao JK, Marchuk DA, Awad IA (2017) RhoA kinase inhibition with Fasudil versus Simvastatin in Murine models of cerebral cavernous malformations. Stroke 48(1):187–194. https://doi.org/10.1161/STROKEAHA.116.015013
Zhou HJ, Qin L, Zhang H, Tang W, Ji W, He Y, Liang X, Wang Z, Yuan Q, Vortmeyer A, Toomre D, Fuh G, Yan M, Kluger MS, Wu D, Min W (2016) Erratum: Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat Med 22(12):1502. https://doi.org/10.1038/nm1216-1502c
Schulte-Merker S, Sabine A, Petrova TV (2011) Lymphatic vascular morphogenesis in development, physiology, and disease. J Cell Biol 193(4):607–618. https://doi.org/10.1083/jcb.201012094
Lauweryns JM, Boussauw L (1973) The ultrastructure of lymphatic valves in the adult rabbit lung. Z Zellforsch Mikrosk Anat 143(2):149–168. https://doi.org/10.1007/bf00307476
Gordon K, Schulte D, Brice G, Simpson MA, Roukens MG, van Impel A, Connell F, Kalidas K, Jeffery S, Mortimer PS, Mansour S, Schulte-Merker S, Ostergaard P (2013) Mutation in vascular endothelial growth factor-C, a ligand for vascular endothelial growth factor receptor-3, is associated with autosomal dominant milroy-like primary lymphedema. Circ Res 112(6):956–960. https://doi.org/10.1161/circresaha.113.300350
Balboa-Beltran E, Fernández-Seara MJ, Pérez-Muñuzuri A, Lago R, García-Magán C, Couce ML, Sobrino B, Amigo J, Carracedo A, Barros F (2014) A novel stop mutation in the vascular endothelial growth factor-C gene (VEGFC) results in Milroy-like disease. J Med Genet 51(7):475–478. https://doi.org/10.1136/jmedgenet-2013-102020
Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, Seaver LH, Glover TW (2000) Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet 67(6):1382–1388. https://doi.org/10.1086/316915
Sabin F (1902) On the origin of the lymphatic system from the veins, and the development of the lymph hearts and thoracic duct in the pig. Am J Anat 1:367–389. https://doi.org/10.1002/aja.1000010310
Srinivasan RS, Dillard ME, Lagutin OV, Lin FJ, Tsai S, Tsai MJ, Samokhvalov IM, Oliver G (2007) Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev 21(19):2422–2432. https://doi.org/10.1101/gad.1588407
Küchler AM, Gjini E, Peterson-Maduro J, Cancilla B, Wolburg H, Schulte-Merker S (2006) Development of the zebrafish lymphatic system requires VEGFC signaling. Curr Biol 16(12):1244–1248. https://doi.org/10.1016/j.cub.2006.05.026
Yaniv K, Isogai S, Castranova D, Dye L, Hitomi J, Weinstein BM (2006) Live imaging of lymphatic development in the zebrafish. Nat Med 12(6):711–716. https://doi.org/10.1038/nm1427
Stanczuk L, Martinez-Corral I, Ulvmar MH, Zhang Y, Laviña B, Fruttiger M, Adams RH, Saur D, Betsholtz C, Ortega S, Alitalo K, Graupera M, Mäkinen T (2015) cKit Lineage hemogenic endothelium-derived cells contribute to mesenteric lymphatic vessels. Cell Rep 10(10):1708–1721. https://doi.org/10.1016/j.celrep.2015.02.026
Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K (2004) Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol 5(1):74–80. https://doi.org/10.1038/ni1013
Wigle JT, Oliver G (1999) Prox1 function is required for the development of the murine lymphatic system. Cell 98(6):769–778. https://doi.org/10.1016/s0092-8674(00)81511-1
Martinez-Corral I, Ulvmar MH, Stanczuk L, Tatin F, Kizhatil K, John SW, Alitalo K, Ortega S, Makinen T (2015) Nonvenous origin of dermal lymphatic vasculature. Circ Res 116(10):1649–1654. https://doi.org/10.1161/circresaha.116.306170
Ulvmar MH, Mäkinen T (2016) Heterogeneity in the lymphatic vascular system and its origin. Cardiovasc Res 111(4):310–321. https://doi.org/10.1093/cvr/cvw175
Vittet D (2014) Lymphatic collecting vessel maturation and valve morphogenesis. Microvasc Res 96:31–37. https://doi.org/10.1016/j.mvr.2014.07.001
Yang Y, Oliver G (2014) Development of the mammalian lymphatic vasculature. J Clin Invest 124(3):888–897. https://doi.org/10.1172/jci71609
Koltowska K, Betterman KL, Harvey NL, Hogan BM (2013) Getting out and about: the emergence and morphogenesis of the vertebrate lymphatic vasculature. Development 140(9):1857–1870. https://doi.org/10.1242/dev.089565
Lee YJ (2020) Cell fate determination of lymphatic endothelial cells. Int J Mol Sci. https://doi.org/10.3390/ijms21134790
Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, Gunn MD, Jackson DG, Oliver G (2002) An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J 21(7):1505–1513. https://doi.org/10.1093/emboj/21.7.1505
Johnson NC, Dillard ME, Baluk P, McDonald DM, Harvey NL, Frase SL, Oliver G (2008) Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev 22(23):3282–3291. https://doi.org/10.1101/gad.1727208
Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dubé KN, Bollini S, Matsuzaki F, Carr CA, Riley PR (2015) Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 522(7554):62–67. https://doi.org/10.1038/nature14483
Yao LC, Testini C, Tvorogov D, Anisimov A, Vargas SO, Baluk P, Pytowski B, Claesson-Welsh L, Alitalo K, McDonald DM (2014) Pulmonary lymphangiectasia resulting from vascular endothelial growth factor-C overexpression during a critical period. Circ Res 114(5):806–822. https://doi.org/10.1161/circresaha.114.303119
Saaristo A, Veikkola T, Enholm B, Hytönen M, Arola J, Pajusola K, Turunen P, Jeltsch M, Karkkainen MJ, Kerjaschki D, Bueler H, Ylä-Herttuala S, Alitalo K (2002) Adenoviral VEGF-C overexpression induces blood vessel enlargement, tortuosity, and leakiness but no sprouting angiogenesis in the skin or mucous membranes. Faseb J 16(9):1041–1049. https://doi.org/10.1096/fj.01-1042com
Enholm B, Karpanen T, Jeltsch M, Kubo H, Stenback F, Prevo R, Jackson DG, Yla-Herttuala S, Alitalo K (2001) Adenoviral expression of vascular endothelial growth factor-C induces lymphangiogenesis in the skin. Circ Res 88(6):623–629. https://doi.org/10.1161/01.res.88.6.623
Hägerling R, Pollmann C, Andreas M, Schmidt C, Nurmi H, Adams RH, Alitalo K, Andresen V, Schulte-Merker S, Kiefer F (2013) A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J 32(5):629–644. https://doi.org/10.1038/emboj.2012.340
Yang Y, García-Verdugo JM, Soriano-Navarro M, Srinivasan RS, Scallan JP, Singh MK, Epstein JA, Oliver G (2012) Lymphatic endothelial progenitors bud from the cardinal vein and intersomitic vessels in mammalian embryos. Blood 120(11):2340–2348. https://doi.org/10.1182/blood-2012-05-428607
Lee S, Kang J, Yoo J, Ganesan SK, Cook SC, Aguilar B, Ramu S, Lee J, Hong YK (2009) Prox1 physically and functionally interacts with COUP-TFII to specify lymphatic endothelial cell fate. Blood 113(8):1856–1859. https://doi.org/10.1182/blood-2008-03-145789
Francois M, Caprini A, Hosking B, Orsenigo F, Wilhelm D, Browne C, Paavonen K, Karnezis T, Shayan R, Downes M, Davidson T, Tutt D, Cheah KS, Stacker SA, Muscat GE, Achen MG, Dejana E, Koopman P (2008) Sox18 induces development of the lymphatic vasculature in mice. Nature 456(7222):643–647. https://doi.org/10.1038/nature07391
Srinivasan RS, Geng X, Yang Y, Wang Y, Mukatira S, Studer M, Porto MP, Lagutin O, Oliver G (2010) The nuclear hormone receptor Coup-TFII is required for the initiation and early maintenance of Prox1 expression in lymphatic endothelial cells. Genes Dev 24(7):696–707. https://doi.org/10.1101/gad.1859310
Koltowska K, Lagendijk AK, Pichol-Thievend C, Fischer JC, Francois M, Ober EA, Yap AS, Hogan BM (2015) Vegfc regulates bipotential precursor division and Prox1 expression to promote lymphatic identity in Zebrafish. Cell Rep 13(9):1828–1841. https://doi.org/10.1016/j.celrep.2015.10.055
Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM (2012) Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am J Pathol 180(6):2561–2575. https://doi.org/10.1016/j.ajpath.2012.02.019
Bowles J, Secker G, Nguyen C, Kazenwadel J, Truong V, Frampton E, Curtis C, Skoczylas R, Davidson TL, Miura N, Hong YK, Koopman P, Harvey NL, François M (2014) Control of retinoid levels by CYP26B1 is important for lymphatic vascular development in the mouse embryo. Dev Biol 386(1):25–33. https://doi.org/10.1016/j.ydbio.2013.12.008
Beets K, Staring MW, Criem N, Maas E, Schellinx N, de Sousa Lopes SM, Umans L, Zwijsen A (2016) BMP-SMAD signalling output is highly regionalized in cardiovascular and lymphatic endothelial networks. BMC Dev Biol 16(1):34. https://doi.org/10.1186/s12861-016-0133-x
Dunworth WP, Cardona-Costa J, Bozkulak EC, Kim JD, Meadows S, Fischer JC, Wang Y, Cleaver O, Qyang Y, Ober EA, Jin SW (2014) Bone morphogenetic protein 2 signaling negatively modulates lymphatic development in vertebrate embryos. Circ Res 114(1):56–66. https://doi.org/10.1161/circresaha.114.302452
Yoshimatsu Y, Lee YG, Akatsu Y, Taguchi L, Suzuki HI, Cunha SI, Maruyama K, Suzuki Y, Yamazaki T, Katsura A, Oh SP, Zimmers TA, Lee SJ, Pietras K, Koh GY, Miyazono K, Watabe T (2013) Bone morphogenetic protein-9 inhibits lymphatic vessel formation via activin receptor-like kinase 1 during development and cancer progression. Proc Natl Acad Sci USA 110(47):18940–18945. https://doi.org/10.1073/pnas.1310479110
Subileau M, Merdzhanova G, Ciais D, Collin-Faure V, Feige JJ, Bailly S, Vittet D (2019) Bone Morphogenetic Protein 9 regulates early lymphatic-specified endothelial cell expansion during mouse embryonic stem cell differentiation. Stem Cell Rep 12(1):98–111. https://doi.org/10.1016/j.stemcr.2018.11.024
Srinivasan RS, Escobedo N, Yang Y, Interiano A, Dillard ME, Finkelstein D, Mukatira S, Gil HJ, Nurmi H, Alitalo K, Oliver G (2014) The Prox1-Vegfr3 feedback loop maintains the identity and the number of lymphatic endothelial cell progenitors. Genes Dev 28(19):2175–2187. https://doi.org/10.1101/gad.216226.113
Kukk E, Lymboussaki A, Taira S, Kaipainen A, Jeltsch M, Joukov V, Alitalo K (1996) VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development 122(12):3829–3837
Zhang L, Zhou F, Han W, Shen B, Luo J, Shibuya M, He Y (2010) VEGFR-3 ligand-binding and kinase activity are required for lymphangiogenesis but not for angiogenesis. Cell Res 20(12):1319–1331. https://doi.org/10.1038/cr.2010.116
Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, Rauvala H, Swartz M, Fukumura D, Jain RK, Alitalo K (1997) Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science 276(5317):1423–1425. https://doi.org/10.1126/science.276.5317.1423
Stone OA, Stainier DYR (2019) Paraxial mesoderm is the major source of lymphatic endothelium. Dev Cell 50(2):247-255.e243. https://doi.org/10.1016/j.devcel.2019.04.034
Nicenboim J, Malkinson G, Lupo T, Asaf L, Sela Y, Mayseless O, Gibbs-Bar L, Senderovich N, Hashimshony T, Shin M, Jerafi-Vider A, Avraham-Davidi I, Krupalnik V, Hofi R, Almog G, Astin JW, Golani O, Ben-Dor S, Crosier PS, Herzog W, Lawson ND, Hanna JH, Yanai I, Yaniv K (2015) Lymphatic vessels arise from specialized angioblasts within a venous niche. Nature 522(7554):56–61. https://doi.org/10.1038/nature14425
Huntington GS, McClure CFW (1910) The anatomy and development of the jugular lymph sacs in the domestic cat (Felis domestica). Dev Dyn 10(1):177–312
Eng TC, Chen W, Okuda KS, Misa JP, Padberg Y, Crosier KE, Crosier PS, Hall CJ, Schulte-Merker S, Hogan BM, Astin JW (2019) Zebrafish facial lymphatics develop through sequential addition of venous and non-venous progenitors. EMBO Rep. https://doi.org/10.15252/embr.201847079
Ny A, Koch M, Schneider M, Neven E, Tong RT, Maity S, Fischer C, Plaisance S, Lambrechts D, Héligon C, Terclavers S, Ciesiolka M, Kälin R, Man WY, Senn I, Wyns S, Lupu F, Brändli A, Vleminckx K, Collen D, Dewerchin M, Conway EM, Moons L, Jain RK, Carmeliet P (2005) A genetic Xenopus laevis tadpole model to study lymphangiogenesis. Nat Med 11(9):998–1004. https://doi.org/10.1038/nm1285
Wilting J, Aref Y, Huang R, Tomarev SI, Schweigerer L, Christ B, Valasek P, Papoutsi M (2006) Dual origin of avian lymphatics. Dev Biol 292(1):165–173. https://doi.org/10.1016/j.ydbio.2005.12.043
Maruyama K, Ii M, Cursiefen C, Jackson DG, Keino H, Tomita M, Van Rooijen N, Takenaka H, D’Amore PA, Stein-Streilein J, Losordo DW, Streilein JW (2005) Inflammation-induced lymphangiogenesis in the cornea arises from CD11b-positive macrophages. J Clin Invest 115(9):2363–2372. https://doi.org/10.1172/jci23874
Pichol-Thievend C, Betterman KL, Liu X, Ma W, Skoczylas R, Lesieur E, Bos FL, Schulte D, Schulte-Merker S, Hogan BM, Oliver G, Harvey NL, Francois M (2018) A blood capillary plexus-derived population of progenitor cells contributes to genesis of the dermal lymphatic vasculature during embryonic development. Development. https://doi.org/10.1242/dev.160184
Lioux G, Liu X, Temiño S, Oxendine M, Ayala E, Ortega S, Kelly RG, Oliver G, Torres M (2020) A Second Heart Field-Derived Vasculogenic Niche Contributes to Cardiac Lymphatics. Dev Cell 52(3):350-363.e356. https://doi.org/10.1016/j.devcel.2019.12.006
Religa P, Cao R, Bjorndahl M, Zhou Z, Zhu Z, Cao Y (2005) Presence of bone marrow-derived circulating progenitor endothelial cells in the newly formed lymphatic vessels. Blood 106(13):4184–4190. https://doi.org/10.1182/blood-2005-01-0226
Lee JY, Park C, Cho YP, Lee E, Kim H, Kim P, Yun SH, Yoon YS (2010) Podoplanin-expressing cells derived from bone marrow play a crucial role in postnatal lymphatic neovascularization. Circulation 122(14):1413–1425. https://doi.org/10.1161/circulationaha.110.941468
Buttler K, Lohrberg M, Gross G, Weich HA, Wilting J (2016) Integration of CD45-positive leukocytes into newly forming lymphatics of adult mice. Histochem Cell Biol 145(6):629–636. https://doi.org/10.1007/s00418-015-1399-y
Bazigou E, Xie S, Chen C, Weston A, Miura N, Sorokin L, Adams R, Muro AF, Sheppard D, Makinen T (2009) Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev Cell 17(2):175–186. https://doi.org/10.1016/j.devcel.2009.06.017
Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hagerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, Calmes JM, Adams RH, Makinen T, Kiefer F, Kwak BR, Petrova TV (2012) Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev Cell 22(2):430–445. https://doi.org/10.1016/j.devcel.2011.12.020
Norrmén C, Ivanov KI, Cheng J, Zangger N, Delorenzi M, Jaquet M, Miura N, Puolakkainen P, Horsley V, Hu J, Augustin HG, Ylä-Herttuala S, Alitalo K, Petrova TV (2009) FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J Cell Biol 185(3):439–457. https://doi.org/10.1083/jcb.200901104
Bazigou E, Wilson JT, Moore JE Jr (2014) Primary and secondary lymphatic valve development: molecular, functional and mechanical insights. Microvasc Res 96:38–45. https://doi.org/10.1016/j.mvr.2014.07.008
Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, Hsu AP, Dyack S, Fernandez CV, Chong CE, Babic M, Bardy PG, Shimamura A, Zhang MY, Walsh T, Holland SM, Hickstein DD, Horwitz MS, Hahn CN, Scott HS, Harvey NL (2012) Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood 119(5):1283–1291. https://doi.org/10.1182/blood-2011-08-374363
Petrova TV, Karpanen T, Norrmén C, Mellor R, Tamakoshi T, Finegold D, Ferrell R, Kerjaschki D, Mortimer P, Ylä-Herttuala S, Miura N, Alitalo K (2004) Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med 10(9):974–981. https://doi.org/10.1038/nm1094
Kanady JD, Dellinger MT, Munger SJ, Witte MH, Simon AM (2011) Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev Biol 354(2):253–266. https://doi.org/10.1016/j.ydbio.2011.04.004
Choi D, Park E, Jung E, Cha B, Lee S, Yu J, Kim PM, Lee S, Hong YJ, Koh CJ, Cho CW, Wu Y, Li Jeon N, Wong AK, Shin L, Kumar SR, Bermejo-Moreno I, Srinivasan RS, Cho IT, Hong YK (2019) Piezo1 incorporates mechanical force signals into the genetic program that governs lymphatic valve development and maintenance. JCI Insight. https://doi.org/10.1172/jci.insight.125068
Nonomura K, Lukacs V, Sweet DT, Goddard LM, Kanie A, Whitwam T, Ranade SS, Fujimori T, Kahn ML, Patapoutian A (2018) Mechanically activated ion channel PIEZO1 is required for lymphatic valve formation. Proc Natl Acad Sci USA 115(50):12817–12822. https://doi.org/10.1073/pnas.1817070115
Cha B, Ho YC, Geng X, Mahamud MR, Chen L, Kim Y, Choi D, Kim TH, Randolph GJ, Cao X, Chen H, Srinivasan RS (2020) YAP and TAZ maintain PROX1 expression in the developing lymphatic and lymphovenous valves in response to VEGF-C signaling. Development. https://doi.org/10.1242/dev.195453
Murtomaki A, Uh MK, Kitajewski C, Zhao J, Nagasaki T, Shawber CJ, Kitajewski J (2014) Notch signaling functions in lymphatic valve formation. Development 141(12):2446–2451. https://doi.org/10.1242/dev.101188
Levet S, Ciais D, Merdzhanova G, Mallet C, Zimmers TA, Lee SJ, Navarro FP, Texier I, Feige JJ, Bailly S, Vittet D (2013) Bone morphogenetic protein 9 (BMP9) controls lymphatic vessel maturation and valve formation. Blood 122(4):598–607. https://doi.org/10.1182/blood-2012-12-472142
Mäkinen T, Adams RH, Bailey J, Lu Q, Ziemiecki A, Alitalo K, Klein R, Wilkinson GA (2005) PDZ interaction site in ephrinB2 is required for the remodeling of lymphatic vasculature. Genes Dev 19(3):397–410. https://doi.org/10.1101/gad.330105
Tatin F, Taddei A, Weston A, Fuchs E, Devenport D, Tissir F, Makinen T (2013) Planar cell polarity protein Celsr1 regulates endothelial adherens junctions and directed cell rearrangements during valve morphogenesis. Dev Cell 26(1):31–44. https://doi.org/10.1016/j.devcel.2013.05.015
Dale RF (1985) The inheritance of primary lymphoedema. J Med Genet 22(4):274–278. https://doi.org/10.1136/jmg.22.4.274
Rockson SG (2001) Lymphedema. Am J Med 110(4):288–295. https://doi.org/10.1016/s0002-9343(00)00727-0
Brouillard P, Boon L, Vikkula M (2014) Genetics of lymphatic anomalies. J Clin Invest 124(3):898–904. https://doi.org/10.1172/JCI71614
Smeltzer DM, Stickler GB, Schirger A (1985) Primary lymphedema in children and adolescents: a follow-up study and review. Pediatrics 76(2):206–218
Irrthum A, Karkkainen MJ, Devriendt K, Alitalo K, Vikkula M (2000) Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet 67(2):295–301. https://doi.org/10.1086/303019
Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K (1998) Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 282(5390):946–949. https://doi.org/10.1126/science.282.5390.946
Karkkainen MJ, Saaristo A, Jussila L, Karila KA, Lawrence EC, Pajusola K, Bueler H, Eichmann A, Kauppinen R, Kettunen MI, Yla-Herttuala S, Finegold DN, Ferrell RE, Alitalo K (2001) A model for gene therapy of human hereditary lymphedema. Proc Natl Acad Sci USA 98(22):12677–12682. https://doi.org/10.1073/pnas.221449198
Bos FL, Caunt M, Peterson-Maduro J, Planas-Paz L, Kowalski J, Karpanen T, van Impel A, Tong R, Ernst JA, Korving J, van Es JH, Lammert E, Duckers HJ, Schulte-Merker S (2011) CCBE1 is essential for mammalian lymphatic vascular development and enhances the lymphangiogenic effect of vascular endothelial growth factor-C in vivo. Circ Res 109(5):486–491. https://doi.org/10.1161/CIRCRESAHA.111.250738
Alders M, Hogan BM, Gjini E, Salehi F, Al-Gazali L, Hennekam EA, Holmberg EE, Mannens MM, Mulder MF, Offerhaus GJ, Prescott TE, Schroor EJ, Verheij JB, Witte M, Zwijnenburg PJ, Vikkula M, Schulte-Merker S, Hennekam RC (2009) Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nat Genet 41(12):1272–1274. https://doi.org/10.1038/ng.484
Hogan BM, Bos FL, Bussmann J, Witte M, Chi NC, Duckers HJ, Schulte-Merker S (2009) Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat Genet 41(4):396–398. https://doi.org/10.1038/ng.321
Au AC, Hernandez PA, Lieber E, Nadroo AM, Shen YM, Kelley KA, Gelb BD, Diaz GA (2010) Protein tyrosine phosphatase PTPN14 is a regulator of lymphatic function and choanal development in humans. Am J Hum Genet 87(3):436–444. https://doi.org/10.1016/j.ajhg.2010.08.008
Bell R, Brice G, Child AH, Murday VA, Mansour S, Sandy CJ, Collin JR, Brady AF, Callen DF, Burnand K, Mortimer P, Jeffery S (2001) Analysis of lymphoedema-distichiasis families for FOXC2 mutations reveals small insertions and deletions throughout the gene. Hum Genet 108(6):546–551. https://doi.org/10.1007/s004390100528
Sabine A, Bovay E, Demir CS, Kimura W, Jaquet M, Agalarov Y, Zangger N, Scallan JP, Graber W, Gulpinar E, Kwak BR, Makinen T, Martinez-Corral I, Ortega S, Delorenzi M, Kiefer F, Davis MJ, Djonov V, Miura N, Petrova TV (2015) FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J Clin Invest 125(10):3861–3877. https://doi.org/10.1172/JCI80454
Irrthum A, Devriendt K, Chitayat D, Matthijs G, Glade C, Steijlen PM, Fryns JP, Van Steensel MA, Vikkula M (2003) Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am J Hum Genet 72(6):1470–1478. https://doi.org/10.1086/375614
Pennisi D, Gardner J, Chambers D, Hosking B, Peters J, Muscat G, Abbott C, Koopman P (2000) Mutations in Sox18 underlie cardiovascular and hair follicle defects in ragged mice. Nat Genet 24(4):434–437. https://doi.org/10.1038/74301
Zhou Y, Lim KC, Onodera K, Takahashi S, Ohta J, Minegishi N, Tsai FY, Orkin SH, Yamamoto M, Engel JD (1998) Rescue of the embryonic lethal hematopoietic defect reveals a critical role for GATA-2 in urogenital development. EMBO J 17(22):6689–6700. https://doi.org/10.1093/emboj/17.22.6689
Fujiwara Y, Chang AN, Williams AM, Orkin SH (2004) Functional overlap of GATA-1 and GATA-2 in primitive hematopoietic development. Blood 103(2):583–585. https://doi.org/10.1182/blood-2003-08-2870
Ferrell RE, Baty CJ, Kimak MA, Karlsson JM, Lawrence EC, Franke-Snyder M, Meriney SD, Feingold E, Finegold DN (2010) GJC2 missense mutations cause human lymphedema. Am J Hum Genet 86(6):943–948. https://doi.org/10.1016/j.ajhg.2010.04.010
Brice G, Ostergaard P, Jeffery S, Gordon K, Mortimer PS, Mansour S (2013) A novel mutation in GJA1 causing oculodentodigital syndrome and primary lymphoedema in a three generation family. Clin Genet 84(4):378–381. https://doi.org/10.1111/cge.12158
Tress O, Maglione M, Zlomuzica A, May D, Dicke N, Degen J, Dere E, Kettenmann H, Hartmann D, Willecke K (2011) Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes Pelizaeus-Merzbacher-like disease in humans. PLoS Genet 7(7):e1002146. https://doi.org/10.1371/journal.pgen.1002146
Kanady JD, Simon AM (2011) Lymphatic communication: connexin junction, what’s your function? Lymphology 44(3):95–102
Castorena-Gonzalez JA, Zawieja SD, Li M, Srinivasan RS, Simon AM, de Wit C, de la Torre R, Martinez-Lemus LA, Hennig GW, Davis MJ (2018) Mechanisms of Connexin-Related Lymphedema. Circ Res 123(8):964–985. https://doi.org/10.1161/CIRCRESAHA.117.312576
Burt JM, Nelson TK, Simon AM, Fang JS (2008) Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am J Physiol Cell Physiol 295(5):C1103-1112. https://doi.org/10.1152/ajpcell.299.2008
Kayiran O, De La Cruz C, Tane K, Soran A (2017) Lymphedema: From diagnosis to treatment. Turk J Surg 33(2):51–57. https://doi.org/10.5152/turkjsurg.2017.3870
Elluru RG, Balakrishnan K, Padua HM (2014) Lymphatic malformations: diagnosis and management. Semin Pediatr Surg 23(4):178–185. https://doi.org/10.1053/j.sempedsurg.2014.07.002
Tian R, Liang Y, Zhang W, Wang J, Shan Y, Gao H, Xie C, Li J, Xu M, Gu S (2020) Effectiveness of sirolimus in the treatment of complex lymphatic malformations: Single center report of 56 cases. J Pediatr Surg 55(11):2454–2458. https://doi.org/10.1016/j.jpedsurg.2019.12.021
Martinez-Corral I, Zhang Y, Petkova M, Ortsater H, Sjoberg S, Castillo SD, Brouillard P, Libbrecht L, Saur D, Graupera M, Alitalo K, Boon L, Vikkula M, Makinen T (2020) Blockade of VEGF-C signaling inhibits lymphatic malformations driven by oncogenic PIK3CA mutation. Nat Commun 11(1):2869. https://doi.org/10.1038/s41467-020-16496-y
Funding
NIH R01 HL146056, R01 DK118728, and U2EB017103 to KKH; NIH T32 HL007284 to CM.
Author information
Authors and Affiliations
Contributions
Drs. Corina Marziano, Gael Genet, and Karen K. Hirschi contributed to the conception, production, revision, and final approval of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marziano, C., Genet, G. & Hirschi, K.K. Vascular endothelial cell specification in health and disease. Angiogenesis 24, 213–236 (2021). https://doi.org/10.1007/s10456-021-09785-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-021-09785-7