
Abstract
Endothelial cells (ECs) have been found to be capable of acquiring a mesenchymal phenotype through a process known as endothelial-to-mesenchymal transition (EndMT). First seen in the developing embryo, EndMT can be triggered postnatally under certain pathological conditions. During this process, ECs dedifferentiate into mesenchymal stem-like cells (MSCs) and subsequently give rise to cell types belonging to the mesoderm lineage. As EndMT contributes to a multitude of diseases, pharmacological modulation of the signaling pathways underlying EndMT may prove to be effective as a therapeutic treatment. Additionally, EndMT in ECs could also be exploited to acquire multipotent MSCs, which can be readily re-differentiated into various distinct cell types. In this review, we will consider current models of EndMT, how manipulation of this process might improve treatment of clinically important pathologies and how it could be harnessed to advance regenerative medicine and tissue engineering.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A substantial body of experimental evidence has shown that epithelial cells possess the intrinsic capability to become mesenchymal cells in a process called epithelial-to-mesenchymal transition (EMT) [1, 2]. EMT is a reversible cell differentiation event associated with extensive alterations at the transcriptional, translational, and morphological level. It is an essential physiological mechanism which is indispensable for several stages of embryogenesis [3] as well as wound healing [4], but it can also promote pathological phenomena such as cancer metastasis and fibrosis [1]. In the past few years, endothelial cells (ECs) have also been found to undergo a similar dedifferentiation process known as endothelial-to-mesenchymal transition (EndMT) [5]. Throughout this highly dynamic process, ECs progressively dedifferentiate into mesenchymal stem-like cells (MSCs) and acquire the characteristics of multipotent cells. During EndMT, ECs spawn a wide spectrum of intermediate phenotypes [6]. These changes in differentiation status and cell behavior are illustrative of their inherent plasticity since their ability to transition is reversible (i.e., mesenchymal-to-endothelial transition) and the process can be either full or partial [7].
ECs that undergo EndMT are characterized by a phenotypic switch involving: (i) loss of cellular adhesion due to the downregulation of proteins involved in cell–cell junctions; (ii) cytoskeletal reorganization, which converts tightly compacted cobblestone-like cells into spindle-shaped cells with no apical-basal polarity [5]; (iii) reduced expression of distinctive EC markers, such as vascular endothelial (VE)-cadherin, CD31/PECAM-1, TIE1, TIE2, and von Willebrand Factor (vWF); (iv) increased expression of mesenchymal cell markers, such as fibroblast-specific protein-1 (FSP-1), alpha-smooth muscle actin (α-SMA), vimentin, and N-cadherin [8]. EndMT-derived cells thus exhibit an enhanced migratory potential and increased extracellular matrix (ECM) production, both of which are hallmarks of invasive cells [9, 10].
EndMT was first observed in the developing embryo, where it was shown to occur in subsets of ECs during cardiogenesis and vasculogenesis. ECs in the endocardium undergo EndMT, invade the cardiac jelly and eventually generate the cardiac cushions. Disruption of EndMT at this embryonic stage results in abnormal formation of the cardiac valves and embryonic lethality [11,12,13]. Similarly to EMT, EndMT can be triggered postnatally under certain pathological conditions, such as tissue damage or inflammation, thereby giving rise to fibroblasts and myofibroblasts [14]. Through the combination of genetic labeling of ECs and disease animal models [5], EndMT was demonstrated to contribute to wound healing [12], pulmonary arterial hypertension (PAH) [15], atherosclerosis [16], cardiac and renal fibrosis [12, 17, 18], fibrodysplasia ossificans progressiva (FOP) [19], and cancer progression [10]. Accordingly, most EndMT research has focused on its role in disease and approaches to block this process. By example, recently, researchers have used small molecules to modify the signaling pathways governing EndMT in an attempt to inhibit or reverse its effects [5]. Interestingly, EndMT could also be used in a different manner, wherein ECs may be exploited to derive multipotent MSCs, which can be readily re-differentiated into various distinct cell types [20].
Here, we will review the evidence that EndMT is integral to the development and evolution of certain pathologies and that targeting EndMT represents a potential therapeutic avenue to treat disease. First, we will describe the signaling pathways that stimulate ECs to undergo EndMT, including the inhibitory mechanisms that prevent this mesenchymal transition. Next, we will discuss how EndMT has been targeted in different disease contexts. Finally, the potential for exploiting EndMT in regenerative medicine and tissue engineering will be assessed.
EndMT-promoting mechanisms
The extent to which ECs lose their distinctive characteristics and gain mesenchymal properties is dependent on the tissue and signaling contexts. It is established that numerous different stimuli can promote EndMT. Below, some of the principal pro-EndMT cues are considered.
Signaling pathways
As ECs share a number of characteristics with epithelial cells (e.g. apical-basal polarity, tight cell junctions, absence of migratory features), it is reasonable to assume that EndMT is related to the process of EMT, and is thus modulated by many of the same pathways and effectors [7]. Ultimately, activation of these pathways results in the expression of common transcription factors, such as Snail, Slug, Twist, ZEB1, ZEB2, and Sox2 [5, 17, 20, 21]. These well-characterized transcription factors initiate EndMT, likely by repressing the expression of endothelial genes (e.g. CDH5 and PECAM1) and subsequently activating the expression of mesenchymal genes (e.g. VIM and COL5A1) [22], thereby transforming ECs into a mesenchymal state.
The best-studied mediators of EndMT are the transforming growth factor (TGF)-β and bone morphogenetic protein (BMP) family of growth factors, which signal through both Smad-dependent and Smad-independent pathways [23, 24]. This diverse superfamily of proteins (i.e. TGF-βs, BMPs, activins, and growth differentiation factors (GDFs)) exert pleiotropic effects in most, if not all, tissues and are indispensable for many physiological processes, including inflammation and wound repair [25]. Members of the TGF-β family signal via specific receptor complexes at the cell membrane. An archetypal response is illustrated by TGF-β1, which binds with high affinity to the type II TGF-β receptor (TGF-βRII) resulting in the recruitment and phosphorylation-dependent activation of the type I TGF-β receptor (activin receptor-like kinase (ALK) 5) [5]. The active ALK5 binds and phosphorylates Smad2/3, which interacts with Smad4 to form a transcription complex that translocates to the nucleus and triggers the expression of specific genes [6, 23]. This subset of genes includes those upregulated in EndMT, such as NOTCH1, TWIST1, and SNAI1/2 [22]. In addition, certain TGF-β family members (TGF-β2, BMP2, and BMP4) were found to induce EndMT by signaling through ALK2 [23, 26]. In vivo relevance of this mechanism is illustrated by the EC-derived heterotopic ossification observed in patients with FOP, which is due to an overactive mutant ALK2 [6, 26]. The pivotal role that the TGF-β superfamily plays in the initiation of EndMT has not only been observed in vitro [5, 6, 27, 28], but has also been validated in multiple in vivo mice studies, which showed that the knockdown and knockout of several TGF-β signaling-related genes, such as SMAD2, SMAD3, and TGFBR2, prevented EndMT [29, 30].
TGF-β signaling can induce EndMT either directly, as described above, or indirectly, as exemplified by the Wnt pathway, caveolin-1 (CAV1), and endothelin-1 (ET-1). The Wnt pathway comprises a multigene family of secreted glycoproteins that play important roles during embryogenesis and heart cushion development [31, 32]. Several studies have confirmed the involvement of Wnt proteins in the induction of EndMT via Smad-dependent TGF-β signaling, and canonical (i.e. involving β-catenin) and non-canonical Wnt signaling pathways [33,34,35,36]. Additionally, studies have found that canonical Notch signaling can act in concert with TGF-β to induce EndMT by activating expression of Snail [37,38,39]. It should be noted that Kaposi’s sarcoma-associated herpesvirus was found to cause EndMT via Notch signaling independently of the TGF-β pathway [40]. Caveolin 1 (CAV1) is the major component of caveolae that controls TGF-β signaling by internalizing, trafficking, and degrading TGF-β receptors [41]. Mice lacking CAV1 undergo spontaneous EndMT, which can be augmented by treatment with TGF-β [42]. Finally, recent studies using human ECs have demonstrated that ET-1, an endogenous vasoconstrictor polypeptide, can stimulate EndMT and yield myofibroblasts, either alone or in combination with TGF-β [43,44,45].
Inflammation, metabolic status, and shear stress
Several lines of evidence support the view that inflammation, metabolic status, and shear stress can all strongly influence EndMT. Firstly, proinflammatory molecules such as interleukin (IL)-1β, IL-6‚ interferon (IFN)-γ, and tumor necrosis factor (TNF)-α have been shown to stimulate EndMT by activating expression of Snail and Slug in synergy with TGF-β [46,47,48]. Secondly, matrix metalloproteinases (MMPs) play a role in several physiological processes and contribute to tissue homeostasis and remodeling, and also function during inflammation by regulating various cytokines, chemokines, and ECM proteins [49]. They are known to initiate EMT through cleavage of cell–cell junction proteins, and, more recently, have been shown to be associated with EndMT [22, 50, 51]. Thirdly, recent studies demonstrated that EndMT was induced by hypoxia via activation of Snail, and hypoxia-inducible factor-1 α (HIF-1α) was observed to promote the process during the development of radiation-induced pulmonary fibrosis [52, 53]. Additionally, HIF-1 has been shown to increase the levels of platelet-derived growth factor (PDGF)-β) and TGF-β1 signaling leading to EndMT via downregulation of neprilysin (NEP) [54]. Differential oxygen concentrations drive EndMT in a different manner. Reactive oxygen species (ROS) are a byproduct of oxygen metabolism whose levels fluctuate as a consequence of environmental stresses (e.g temperature changes and UV light). ROS stimulate EndMT, e.g. by inducing TGF-β expression, which in turn leads to the production of ROS via a positive feedback loop [55]. ROS also activate nuclear factor-κΒ (NF-κB) signaling, which drives EndMT in synergy with TGF-β [56]. Furthermore, NADPH oxidase 4 (NOX4), an enzyme responsible for the production of ROS, was found to mediate TGF-β-dependent production of myofibroblast by EndMT [57, 58]. Recently, the Akt/mammalian target of rapamycin (mTOR)/70 kDa ribosomal S6 kinase (p70S6K) signaling pathway was also shown to be involved in TGF-β1-induced EndMT in transplant kidney interstitial fibrosis [59]. Finally, hemodynamic forces have been demonstrated to strongly modulate EndMT [60]. Shear stress, a fundamental force governing homeostasis of ECs, suppresses EndMT via a number of TGF-β signaling-dependent mechanisms [61]. Correspondingly, whereas high shear stress appears to inhibit EndMT [61], disturbed flow is a potent EndMT inducer in vivo as well as in organ-on-a-chip devices. Under these conditions, genetic inhibition of extracellular-signal-regulated kinase (ERK) 5 signaling enhances EndMT, whereas ERK5 overactivation prevents EndMT in cells exposed to disturbed flow or stimulated by TGF-β in static conditions [62]. A different mechanical stress, termed cyclic strain, and caused by a perpendicular stretching force on the vessel wall, has been shown to potentiate EndMT by augmenting both TGF-β and Wnt signaling [63, 64].
microRNAs
MicroRNAs (miRNAs) control EndMT by altering the activity of signaling intermediates leading to changes in signaling amplitude and output. miRNA 125b has been shown to contribute to EndMT progression [65], and it has been demonstrated that miRNA21 mediates TGF-β-induced EndMT by controlling actin remodeling and promoting the secretion of inflammatory cytokines [66]. Several other miRNAs were also found to be positive modulators of EndMT, such as miR-31, which is required for the expression of EndMT markers following TGF-β-treatment [67], and miR-9, a miRNA regulated by TNF-α signaling [68]. Additionally, metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), a long non-coding RNA, was found to modulate TGF-β1-induced EndMT of endothelial progenitor cells (EPCs) through regulation of TGF-βRII and Smad3 via decreased miR-145 expression [69].
EndMT-inhibiting signaling pathways and mechanisms
In addition to stimuli favoring EndMT, there are also a number of different factors involved in the negative regulation of this process.
Signaling pathways
Although TGF-β and BMP are known to induce EndMT under specific conditions, they can also bind ALK1 to activate Smad1/5/8, which induces proliferation at the expense of EndMT [70]. Endoglin, an accessory type III TGF-β receptor, partially regulates the equilibrium between ALK1/ALK5 activation. By stimulating downstream Smad1/5/8 responses it can indirectly inhibit ALK5 signaling, and thus inhibit EndMT [71]. Interestingly, BMP7 appears to be a negative regulator of EndMT [72], presumably through the activation of ALK2 alone (and the associated Smad1/5/8 pathway), in contrast to BMP2 and BMP4 which bind to ALK2 in conjunction with ALK5 and thereby promote EndMT [6, 26].
Another known mechanism of EndMT inhibition is vascular endothelial growth factor A (VEGF-A)-stimulated VEGF receptor (VEGFR)2 signaling [73]. This process, however, is counteracted by VEGF-A sequestration by VEGFR1, thereby preventing its interaction with VEGFR2, and leading to EndMT [74]. Two other layers of regulation of this network are repression of VEGF-A by BMP signaling [75], and attenuation of VEGF-A signaling by mechanical cyclic strain [19].
Other signaling cascades and factors that abrogate EndMT include: (i) activation of the Src signaling pathway by hydrogen sulfide during endoplasmic reticulum (ER) stress [76]; (ii) glucagon-like peptide-1 (GLP-1) suppression of hyperglycemia-induced EndMT via reduced expression of ROS and inhibition of ROS-activated poly(ADP-ribose) polymerase 1 (PARP-1) [77]; (iii) high-density lipoprotein (HDL) inhibition of TGF-β1-induced EndMT [78]; (iv) endothelial heat shock protein beta-1 (HSPB-1)-mediated EndMT inhibition after stimulation with fibrotic cytokines [79]; (v) netrin-1-mediated attenuation of EndMT during renal dysfunction, as demonstrated in a nephrectomy rat model [80]; (vi) expression of ECM protein fibulin-1 via reduced expression of TGF-β2 [81]; (vii) secretion of cytokines and angiogenic factors by macrophages sustains endothelial differentiation of EPCs and consequently restricts EndMT during muscle regeneration [82].
miRNAs
miRNAs have been shown to block EndMT in numerous different tissues. Several miRNAs, such as miR-15a, miR-23b, and miR-199a, have been found to impair EndMT during heart development [83]. TGF-β-induced EndMT was blocked by miR-126 in bone marrow-derived EPCs through direct targeting of the phosphoinositide 3-kinase (PI3K) subunit p85 [84]. miR-155 was found to be a potent inhibitor of TGF-β-induced EndMT via inhibition of RhoA expression [85, 86]. Furthermore, miR-302c was observed to suppress EndMT in hepatocellular carcinoma by negatively regulating the expression of metadherin (MTDH) [87]. Fibroblast growth factor receptor 1 (FGFR1) signaling can also inhibit TGF-β-induced EndMT by promoting the expression of miRNA let-7, a negative regulator of TGF-β signaling [88]. N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP), a peptide substrate of angiotensin-converting enzyme (ACE), contributes to this by upregulating let-7 and restoring FGFR levels [89]. FGF-2, although found to induce EndMT in some types of ECs [90], has also been demonstrated to abrogate TGF-β-induced EndMT through miR-20 [91]. Lastly, miR-630 was shown to inhibit EndMT in heterotopic ossification by targeting Slug [92].
Autophagy
Recently, autophagy has emerged as a potentially important player in controlling EndMT by decreasing TGF-β2-induced EndMT [93]. Activation of autophagy was also shown to reduce expression of Snail by decreasing the phosphorylation levels of Smad3, thus counteracting EndMT [94]. Furthermore, pharmacological inhibition of mTOR resulted in the activation of autophagy and a decrease of EndMT [95], providing evidence of a causal link between mTOR-dependent inhibition of autophagy and EndMT. These findings suggest that targeting autophagy may be a productive way of limiting EndMT.
Therapeutic modulation of EndMT
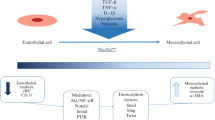
Figure 1 highlights those signaling networks that could be plausible targets for therapeutically inhibiting EndMT as a treatment for several different pathologies. Neutralizing antibodies or chemical inhibitors targeting molecules required for EndMT could be an effective means of impeding the process [5]. Proof-of-principle evidence of this comes from experiments showing that inhibition of ALK5, TGF-βRII, β-glycan, and endoglin prevent embryonic EndMT in the endothelium of mice [96,97,98]. Consistently, reducing expression of EndMT regulators e.g. ALK2, ALK5, or Snail expression, resulted in a comparable block in EndMT in EC cultures [19, 24]. Another study has found that local and circulating ECs are capable of undergoing EndMT in response to musculoskeletal injury, suggesting that targeting early EC recruitment and trafficking could potentially impede pathological EndMT [99].

EndMT as a target for therapeutic intervention. ECs differentiate into MSCs via the process of EndMT, which is regulated by various signaling mechanisms. Numerous compounds can be used to block this differentiation step, thereby disrupting the process and potentially ameliorating the effects of pathological EndMT
To date, several compounds have been tested, with mixed degrees of success, for their ability to inhibit EndMT. Many of these compounds interfere with TGF-β signaling. The dipeptidyl peptidase-4 (DPP-4) inhibitor, linagliptin, could block TGF-β2-induced EndMT by impairing its interaction with integrin β1 [100]. Arginylglycylaspartic acid (RGD) is an Arg-Gly-Asp tripeptide motif that is found in many matrix proteins and is responsible for integrin-dependent cell adhesion to the ECM. One recently developed RGD antagonist, RGD-2, was found to revert TGF-β1-induced EndMT and consequently has the potential to be employed as an anti-fibrotic therapeutic treatment [101]. A specific inhibitor of Smad3 (SIS3) was shown to block EndMT and reduce renal fibrosis [102]. EndMT was also inhibited by the ALK5 inhibitor SB-431542 in cultured ECs [9], and dorsomorphin blocked EndMT of endothelial cultures by inhibiting the kinase activity of a mutant ALK2 in FOP [19]. Celastrol was found to block TGF-β1-induced EndMT and has been promoted as a possible therapy for cardiac fibrosis [103]. TGF-β-induced EndMT was inhibited by kallistatin via upregulation of endothelial nitric oxide synthase (eNOS) and downregulation of EndMT-promoting miR-21 [104]. EndMT was also impaired by the angiotensin II type 1 receptor inhibitor losartan, which blocked TGF-β signaling [105]. Other compounds can disrupt EndMT by inhibiting different signaling pathways and/or intermediates (Table 1).
EndMT in regenerative medicine and in vitro modeling applications
An extensive literature focused on the pathological consequences of EndMT should not overshadow several lines of evidence supporting the idea that EndMT could be harnessed for the purpose of tissue engineering, predicated on the fact that EndMT generates MSCs that can be programmed to differentiate into a wide variety of different cell types. In FOP, heterotopic bone is formed by a gain-of-function mutation in ALK2 [116]. Studies with lineage tracing and biomarker experiments have shown that this mutation causes ECs to undergo EndMT, thereby acquiring properties of MSCs [19, 117]. They have further demonstrated that these cells can be differentiated into osteoblasts, chondrocytes, or adipocytes [19]. The generation of osteoprogenitor cells via EndMT has also been seen in vascular [118], valvular [119], and tumor calcifications [120]. ECs were shown to be differentiated to chondrocytes via EndMT by high glucose levels [121], and ECs lining the vessels of white and brown adipose tissue have been shown to give rise to preadipocytes [122]. A number of studies have demonstrated the ability of ECs from vascular tumors to undergo EndMT in culture and form adipocytes, pericytes and smooth muscle cells (SMCs) [123], whilst related work has shown that EPCs can transform into smooth muscle cells [9]. The ability of ECs to form skeletal myocytes has also been observed during muscle repair [124]. ECs were also found to contribute to the cardiac renewal process [125].
EndMT could thus be manipulated to generate multipotent MSCs from ECs, which can thereafter be transformed into different cell types. Via full or partial reprogramming, where intermediary cell types would suffice, EndMT could potentially be used in the treatment of a variety of diseases (Fig. 2). Bone disorders such as osteoporosis or osteoarthritis could be treated by EndMT-derived osteocytes or chondrocytes [20]. EndMT-mediated (cardio)myogenesis could be employed in the regeneration of cardiomyocytes after myocardial infarction [20]. Moreover, vascular tissue could be regenerated by EndMT via its ability to produce SMCs and pericytes [20]. Manipulating EndMT could offer a potential solution to controlling aberrant angiogenesis since expression of the EndMT-inducing transcription factor Slug was shown to regulate vessel sprouting [126]. Another study proposed that this angiogenic sprouting may represent a partial EndMT. Their results also clearly indicate the importance of the Snail family of transcription factors during angiogenesis [7], and suggest the involvement of EndMT, at least partially, in vasculature formation.

EndMT in tissue engineering and in vitro modeling. EndMT-derived MSCs can be differentiated into various mesenchymal cell types. Once the desired cell type is obtained, they can be used for tissue engineering and subsequent transplantation into the patient. The acquired cells can also be employed in experimental in vitro applications, such as in the construction of a vascularized 3D-organoid model
The appeal of employing EndMT in tissue engineering lies in the fact that the process can take place both in vivo and ex vivo. As pointed out by others, suitable drugs could be applied locally to degenerate tissues to reprogram ECs, which are present in abundance in any vascularized tissue, into the desired mesenchymal cell type [20]. To engineer tissues ex vivo, ECs can be isolated and, under the right 3D-culturing conditions, induced to undergo EndMT to become MSCs, using protocols more simple and cost-effective than those for induced pluripotent stem cells (iPSCs). After differentiating these stem cells into the cell type of interest, they can then be transplanted into the patient [20]. Perhaps not entirely unexpectedly, EndMT could also be beneficial in pathologies where fibrotic cells are actually desired, thus not requiring the additional step of differentiating MSCs to specialized cell types. One such study found that EndMT contributed to the therapeutic effects of bleomycin, a sclerosant used for the treatment of venous malformations (VMs), pointing to a possible role for this process in sclerotherapy [127]. Moreover, as ECM contributes to the mechanical functioning of cardiovascular tissue-engineered grafts, EndMT could aid the formation of cells, from ECs, that are capable of producing and remodeling ECM [128].
EndMT could also be employed for in vitro experimental purposes, such as the culturing and modeling of in vitro organs, which can be used as a substitute for experimental animal models (Fig. 2). One study established such an in vitro model with human embryonic stem cell (hESC)-derived ECs to study the regulation of Notch signaling in the induction of EndMT in cardiogenesis [129]. Another study generated an organoid-based EMT model from intestinal epithelial cells. These cells exhibited an in vivo physiology and, therefore, could be used to study EMT-associated intestinal fibrosis [130]. Such an approach could also be feasible for harvested ECs to study EndMT-related diseases. Although it is possible to grow organoids in vitro, a main restriction of 3D-culture systems is the lack of a vascular network [131]. In light of the fact that EndMT demonstrably plays a role in angiogenesis [126], cultured ECs could potentially be used to create vascular networks through EndMT, contributing to the development of a fully vascularized organoid.
Concluding remarks
EndMT has an established role in many different pathologies. Targeting the signaling pathways responsible for EndMT could, therefore, be an effective means of facilitating wound healing as well as treating EndMT-associated diseases. This is, of course, not an easy undertaking, not least because EndMT is controlled by complex signaling networks and not simple, linear, and discrete signaling modules. This makes the selection of suitable therapeutic targets a far from trivial proposition. For instance, TGF-β could be an obvious candidate, however, it exerts pleiotropical effects in the regulation of a multitude of processes in various tissues and targeting this pathway could lead to major, unwanted side-effects [5]. Deciphering in greater depth, the activating and inhibitory EndMT signaling map could identify unique targets that offer realistic hopes of developing viable therapeutic strategies to modulate EndMT. Alongside a potential role in inhibiting pathological processes, EndMT could be exploited to play a more ‘creative’ role in tissue engineering. EndMT gives rise to multipotent MSCs that can be reprogrammed into various distinct cell types, offering the possibility that this capacity could be harnessed to advance regenerative medicine.
References
Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA (2009) Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 119(6):1438–1449. https://doi.org/10.1172/JCI38019
Kalluri R, Weinberg R (2009) Review series the basics of epithelial-mesenchymal transition. J Clin Invest 119(6):1420–1428. https://doi.org/10.1172/JCI39104.1420
Lim J, Thiery JP (2012) Epithelial-mesenchymal transitions: insights from development. Development 139(19):3471–3486. https://doi.org/10.1242/dev.071209
Kalluri R, Neilson EG (2003) Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 112(12):1776–1784. https://doi.org/10.1172/JCI200320530
Sanchez-Duffhues G, Orlova V, ten Dijke P (2016) In brief: endothelial-to-mesenchymal transition. J Pathol 238(3):378–380. https://doi.org/10.1002/path.4653
Medici D, Kalluri R (2012) Endothelial–mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin Cancer Biol 144(5):724–732. https://doi.org/10.1038/jid.2014.371
Welch-Reardon KM, Wu N, Hughes CCW (2015) A role for partial endothelial–mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol 35(2):303–308. https://doi.org/10.1161/ATVBAHA.114.303220
Frid MG, Kale VA, Stenmark KR (2002) Mature vascular endothelium can give rise to smooth muscle cells via endothelialmesenchymal transdifferentiation: in vitro analysis. Circ Res 90(11):1189–1196. https://doi.org/10.1161/01.RES.0000021432.70309.28
Moonen JRAJ, Krenning G, Brinker MGL, Koerts JA, Van Luyn MJA, Harmsen MC (2010) Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc Res 86(3):506–515. https://doi.org/10.1093/cvr/cvq012
Potenta S, Zeisberg E, Kalluri R (2008) The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer 99(9):1375–1379. https://doi.org/10.1038/sj.bjc.6604662
Arciniegas E, Neves CY, Carrillo LM, Zambrano E, Ramírez R (2005) Endothelial–mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium 12(4):193–200. https://doi.org/10.1080/10623320500227283
Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V (2012) Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition from cardiovascular development to disease. Circulation 125(14):1795–1808. https://doi.org/10.1161/CIRCULATIONAHA.111.040352
Armstrong EJ, Bischoff J (2004) Heart valve development: endothelial cell signaling and differentiation. Circ Res 95(5):459–470. https://doi.org/10.1161/01.RES.0000141146.95728.da
Lin F, Wang N, Zhang TC (2012) The role of endothelial–mesenchymal transition in development and pathological process. IUBMB Life 64(9):717–723. https://doi.org/10.1002/iub.1059
Ranchoux B, Antigny F, Rucker-Martin C et al (2015) Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 131(11):1006–1018. https://doi.org/10.1161/CIRCULATIONAHA.114.008750
Chen PY, Qin L, Baeyens N et al (2015) Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 125(12):4514–4528. https://doi.org/10.1172/JCI82719
Piera-Velazquez S, Mendoza F, Jimenez S (2016) Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J Clin Med 5(4):45. https://doi.org/10.3390/jcm5040045
Kriz W, Kaissling B, Le Hir M (2011) Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest 121(2):468–474. https://doi.org/10.1172/JCI44595
Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16(12):1400–1406. https://doi.org/10.1038/nm.2252
Medici D (2016) Endothelial–mesenchymal transition in regenerative medicine. Stem Cells Int. https://doi.org/10.1155/2016/6962801
Guihard PJ, Yao J, Blazquez-medela AM, Iruela-arispe L (2016) Endothelial–mesenchymal transition in vascular calcification of Ins2 Akita/+ mice. PLoS ONE 11(12):1–12. https://doi.org/10.1371/journal.pone.0167936
Evrard SM, Lecce L, Michelis KC et al (2016) Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. https://doi.org/10.1038/ncomms11853
van Meeteren LA, ten Dijke P (2011) Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res 347(1):177–186. https://doi.org/10.1007/s00441-011-1222-6
Medici D, Potenta S, Kalluri R (2011) Transforming growth factor-β2 promotes Snail-mediated endothelial–mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J 437(3):515–520. https://doi.org/10.1042/BJ20101500
Gonzalez DM, Medici D (2014) Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal 7(344):re8–re8. https://doi.org/10.1126/scisignal.2005189
Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16(12):1400–1406. https://doi.org/10.1038/nm0411-514d
Piera-Velazquez S, Jimenez S (2012) Molecular mechanisms of endothelial to mesenchymal cell transition (EndoMT) in experimentally induced fibrotic diseases. Fibrogenesis Tissue Repair. 5 (Suppl 1 Proceedings of Fibroproliferative disorders: from biochemical analysis to targeted therapiesPetro E Petrides and David Brenner):S7. https://doi.org/10.1186/1755-1536-5-S1-S7
Li Z, Jimenez SA (2011) Protein kinase Cδ and c-Abl kinase are required for transforming growth factor β induction of endothelial–mesenchymal transition in vitro. Arthritis Rheum 63(8):2473–2483. https://doi.org/10.1002/art.30317
Cooley BC, Nevado J, Mellad J et al (2014) TGF-β signaling mediates endothelial to mesenchymal transition (EndMT) during vein graft remodeling. Sci Trans Med 6(227):1–22. https://doi.org/10.1126/scitranslmed.3006927.TGF-
Xavier S, Vasko R, Matsumoto K et al (2015) Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial–mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol 26(4):817–829. https://doi.org/10.1681/ASN.2013101137
Clevers H, Nusse R (2012) Wnt/β-catenin signaling and disease. Cell 149(6):1192–1205. https://doi.org/10.1016/j.cell.2012.05.012
Liebner S, Cattelino A, Gallini R et al (2004) β-catenin is required for endothelial–mesenchymal transformation during heart cushion development in the mouse. J Cell Biol 166(3):359–367. https://doi.org/10.1083/jcb.200403050
Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK (2011) Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech 4(4):469–483. https://doi.org/10.1242/dmm.006510
Beyer C, Schramm A, Akhmetshina A et al (2012) β-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann Rheum Dis 71(5):761–767. https://doi.org/10.1136/annrheumdis-2011-200568
Li L, Chen L, Zang J et al (2015) C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism 64(5):597–610. https://doi.org/10.1016/j.metabol.2015.01.014
Wang S-H, Chang JS, Hsiao J-R et al (2016) Tumour cell-derived WNT5B modulates in vitro lymphangiogenesis via induction of partial endothelial–mesenchymal transition of lymphatic endothelial cells. Oncogene 36(April):1–13. https://doi.org/10.1038/onc.2016.317
Noseda M, McLean G, Niessen K et al (2004) Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res 94(7):910–917. https://doi.org/10.1161/01.RES.0000124300.76171.C9
Chang ACY, Fu Y, Garside VC et al (2011) Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev Cell 21(2):288–300. https://doi.org/10.1016/j.devcel.2011.06.022
Fu Y, Chang A, Chang L et al (2009) Differential regulation of transforming growth factor β signaling pathways by Notch in human endothelial cells. J Biol Chem 284(29):19452–19462. https://doi.org/10.1074/jbc.M109.011833
Gasperini P, Espigol-Frigole G, McCormick PJ et al (2012) Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through notch-dependent signaling. Cancer Res 72(5):1157–1169. https://doi.org/10.1158/0008-5472.CAN-11-3067
Del Galdo F, Lisanti MP, Jimenez SA (2008) Caveolin-1, transforming growth factor-β receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol 20(6):713–719
Li Z, Wermuth PJ, Benn BS, Lisanti MP, Jimenez SA (2013) Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am J Pathol 182(2):325–331. https://doi.org/10.1016/j.ajpath.2012.10.022
Widyantoro B, Emoto N, Nakayama K et al (2010) Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 121(22):2407–2418. https://doi.org/10.1161/CIRCULATIONAHA.110.938217
Wermuth PJ, Li Z, Mendoza FA, Jimenez SA. Stimulation of transforming growth factor-β1-induced endothelial-to-mesenchymal transition and tissue fibrosis by endothelin-1 (ET-1): A novel profibrotic effect of ET-1. PLoS ONE. 2016;11(9). https://doi.org/10.1371/journal.pone.0161988
Cipriani P, Di Benedetto P, Ruscitti P et al (2015) The endothelial–mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-β and may be blocked by Macitentan, a dual endothelin-1 receptor antagonist. J Rheumatol 42(10):1808–1816. https://doi.org/10.3899/jrheum.150088
Rieder F, Kessler SP, West GA et al (2011) Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol 179(5):2660–2673. https://doi.org/10.1016/j.ajpath.2011.07.042
Mahler GJ, Farrar EJ, Butcher JT (2013) Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol 33(1):121–130. https://doi.org/10.1161/ATVBAHA.112.300504
Chrobak I, Lenna S, Stawski L, Trojanowska M (2013) Interferon-γ promotes vascular remodeling in human microvascular endothelial cells by upregulating endothelin (ET)-1 and transforming growth factor (TGF) β2. J Cell Physiol 228(8):1774–1783. https://doi.org/10.1002/jcp.24337
Parks WC, Parks WC, Wilson CL, Wilson CL, López-Boado YS, López-Boado YS (2004) Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. https://doi.org/10.1038/nri1418
Ho WT, Chang JS, Su CC et al (2015) Inhibition of matrix metalloproteinase activity reverses corneal endothelial–mesenchymal transition. Am J Pathol 185(8):2158–2167. https://doi.org/10.1016/j.ajpath.2015.04.005
Zhao Y, Qiao X, Wang L et al (2016) Matrix metalloproteinase 9 induces endothelial–mesenchymal transition via Notch activation in human kidney glomerular endothelial cells. BMC Cell Biol. https://doi.org/10.1186/s12860-016-0101-0
Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM (2015) Snail Is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem 290(27):16553–16664. https://doi.org/10.1074/jbc.M115.636944
Choi SH, Hong ZY, Nam JK et al (2015) A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res 21(16):3716–3726. https://doi.org/10.1158/1078-0432.CCR-14-3193
Song S, Zhang M, Yi Z et al (2016) The role of PDGF-B/TGF-β1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell Signal 28(10):1489–1501. https://doi.org/10.1016/j.cellsig.2016.06.022
Liu R-M, Gaston Pravia KA (2010) Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Radic Biol Med 48(1):1–15. https://doi.org/10.1016/j.freeradbiomed.2009.09.026
Maleszewska M, Moonen J-RAJ, Huijkman N, van de Sluis B, Krenning G, Harmsen MC (2013) IL-1β and TGFβ2 synergistically induce endothelial to mesenchymal transition in an NFκB-dependent manner. Immunobiology 218(4):443–454. https://doi.org/10.1016/j.imbio.2012.05.026
Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J (2010) NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFβ1-induced fibroblast differentiation into myofibroblasts. Thorax 65(8):733–738. https://doi.org/10.1136/thx.2009.113456
Liu RM, Desai LP (2015) Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol 6:565–577. https://doi.org/10.1016/j.redox.2015.09.009
Wang Z, Han Z, Tao J et al. (2017) Role of endothelial-to-mesenchymal transition induced by TGF-β1 in transplant kidney interstitial fibrosis. J Cell Mol Med 21(10): 2359–2369
Hahn C, Schwartz MA (2009) Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol 10(1):53–62. https://doi.org/10.1038/nrm2596
Krenning G, Barauna VG, Krieger JE, Harmsen MC, Moonen JRAJ (2016) Endothelial plasticity: shifting phenotypes through force feedback. Stem Cells Int. https://doi.org/10.1155/2016/9762959
Moonen JRAJ, Lee ES, Schmidt M et al (2015) Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc Res 108(3):377–386. https://doi.org/10.1093/cvr/cvv175
Balachandran K, Alford PW, Wylie-Sears J et al (2011) Cyclic strain induces dual-mode endothelial–mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci 108(50):19943–19948. https://doi.org/10.1073/pnas.1106954108
Mai J, Hu Q, Xie Y et al (2014) Dyssynchronous pacing triggers endothelial–mesenchymal transition through heterogeneity of mechanical stretch in a canine model. Circ J 79(1):201–209. https://doi.org/10.1253/circj.CJ-14-0721
Nagpal V, Rai R, Place AT et al (2016) MiR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation 133(3):291–301. https://doi.org/10.1161/CIRCULATIONAHA.115.018174
Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park DH, Thum T (2012) Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by MicroRNA-21. Arterioscler Thromb Vasc Biol 32(2):361–369. https://doi.org/10.1161/ATVBAHA.111.234286
Katsura A, Suzuki HI, Ueno T et al (2016) MicroRNA-31 is a positive modulator of endothelial–mesenchymal transition and associated secretory phenotype induced by TGF-β Genes Cells 21(1):99–116. https://doi.org/10.1111/gtc.12323
Chakraborty S, Zawieja DC, Davis MJ, Muthuchamy M (2015) MicroRNA signature of inflamed lymphatic endothelium and role of miR-9 in lymphangiogenesis and inflammation. Am J Physiol Cell Physiol 309(10):C680–C692. https://doi.org/10.1152/ajpcell.00122.2015
Xiang Y, Zhang Y, Tang Y, Li Q (2017) MALAT1 modulates TGF-β1-induced endothelial-to-mesenchymal transition through downregulation of miR-145. Cell Physiol Biochem 42(1):357–372. https://doi.org/10.1159/000477479
Goumans MJ, Valdimarsdottir G, Itoh S et al (2003) Activin receptor-like kinase (ALK) 1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol Cell 12:817–828. https://doi.org/10.1016/S1097-2765(03)00386-1
Lebrin F, Goumans M-J, Jonker L et al (2004) Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J 23(20):4018–4028. https://doi.org/10.1038/sj.emboj.7600386
Zeisberg EM, Tarnavski O, Zeisberg M et al (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13(8):952–961. https://doi.org/10.1038/nm1613
Paruchuri S, Yang J, Aikawa E et al (2006) Human pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to vascular endothelial growth factor: a and transforming growth factor-β2. Circ Res 99(8):861–869. https://doi.org/10.1161/01.RES.0000245188.41002.2c
Tao J, Doughman Y, Yang K, Ramirez-Bergeron D, Watanabe M (2013) Epicardial HIF signaling regulates vascular precursor cell invasion into the myocardium. Dev Biol 376(2):136–149. https://doi.org/10.1016/j.ydbio.2013.01.026
Bai Y, Wang J, Morikawa Y, Bonilla-Claudio M, Klysik E, Martin JF (2013) Bmp signaling represses Vegfa to promote outflow tract cushion development. Development 140(16):3395–3402. https://doi.org/10.1242/dev.097360
Ying R, Wang XQ, Yang Y et al (2016) Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci 144:208–217. https://doi.org/10.1016/j.lfs.2015.11.025
Yan F, Zhang G-H, Feng M et al (2015) Glucagon-like peptide 1 protects against hyperglycemic-induced endothelial-to-mesenchymal transition and improves myocardial dysfunction by suppressing poly (ADP-ribose) polymerase 1 activity. Mol Med 21:15–25. https://doi.org/10.2119/molmed.2014.00259
Spillmann F, Miteva K, Pieske B, Tschöpe C, Van Linthout S (2015) High-density lipoproteins reduce endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol 35(8):1774–1777. https://doi.org/10.1161/ATVBAHA.115.305887
Choi SH, Nam JK, Kim BY et al (2016) HSPB1 inhibits the endothelial-to-mesenchymal transition to suppress pulmonary fibrosis and lung tumorigenesis. Cancer Res 76(5):1019–1030. https://doi.org/10.1158/0008-5472.CAN-15-0952
Bai J, Hao J, Zhang X, Cui H, Han J, Cao N (2016) Netrin-1 attenuates the progression of renal dysfunction by blocking endothelial-to-mesenchymal transition in the 5/6 nephrectomy rat model. BMC Nephrol 17(1):47. https://doi.org/10.1186/s12882-016-0260-4
Harikrishnan K, Cooley MA, Sugi Y et al (2015) Fibulin-1 suppresses endothelial to mesenchymal transition in the proximal outflow tract. Mech Dev 136:123–132. https://doi.org/10.1016/j.mod.2014.12.005
Zordan P, Rigamonti E, Freudenberg K et al (2014) Macrophages commit postnatal endothelium-derived progenitors to angiogenesis and restrict endothelial to mesenchymal transition during muscle regeneration. Cell Death Dis 5(1):e1031. https://doi.org/10.1038/cddis.2013.558
Bonet F, Dueñas Á, López-Sánchez C, García-Martínez V, Aránega AE, Franco D (2015) MiR-23b and miR-199a impair epithelial-to-mesenchymal transition during atrioventricular endocardial cushion formation. Dev Dyn 244(10):1259–1275. https://doi.org/10.1002/dvdy.24309
Zhang J, Zhang Z, Zhang DY, Zhu J, Zhang T, Wang C (2013) MicroRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2-PI3K/Akt signalling pathway. PLoS ONE. 8(12). https://doi.org/10.1371/journal.pone.0083294
Kumar S, Kim CW, Simmons RD, Jo H (2014) Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis mechanosensitive athero-miRs. Arterioscler Thromb Vasc Biol 34(10):2206–2216. https://doi.org/10.1161/ATVBAHA.114.303425
Bijkerk R, de Bruin RG, van Solingen C et al (2012) MicroRNA-155 functions as a negative regulator of RhoA signaling in TGF-β-induced endothelial to mesenchymal transition. MicroRNA (Shariqah United Arab Emirates) 1(1):2–10. https://doi.org/10.2174/2211536611201010002
Zhu K, Pan Q, Jia L-Q et al (2014) MiR-302c inhibits tumor growth of hepatocellular carcinoma by suppressing the endothelial–mesenchymal transition of endothelial cells. Sci Rep 4:5524. https://doi.org/10.1038/srep05524
Chen PY, Qin L, Barnes C et al (2012) FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep 2(6):1684–1696. https://doi.org/10.1016/j.celrep.2012.10.021
Nagai T, Kanasaki M, Srivastava SP et al. (2014) N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial–mesenchymal transition. Biomed Res Int. https://doi.org/10.1155/2014/696475
Lee JG, Ko MK, Kay EP (2012) Endothelial mesenchymal transformation mediated by IL-1β-induced FGF-2 in corneal endothelial cells. Exp Eye Res 95(1):35–39. https://doi.org/10.1016/j.exer.2011.08.003
Correia ACP, Moonen J-RAJ, Brinker MGL, Krenning G (2016) FGF2 inhibits endothelial–mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-β signaling. J Cell Sci 129(3):569–579. https://doi.org/10.1242/jcs.176248
Sun Y, Cai J, Yu S, Chen S, Li F, Fan C (2016) MiR-630 inhibits endothelial–mesenchymal transition by targeting slug in traumatic heterotopic ossification. Sci Rep 6:22729. https://doi.org/10.1038/srep22729
Grassi G, Di Caprio G, Santangelo L et al (2015) Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis 6(9):e1880. https://doi.org/10.1038/cddis.2015.249
Wang J, Feng Y, Wang Y, Xiang D, Zhang X, Yuan F (2017) Autophagy regulates endothelial–mesenchymal transition by decreasing the phosphorylation level of Smad3. Biochem Biophys Res Commun 487(3):740–747. https://doi.org/10.1016/j.bbrc.2017.04.130
Marchi S, Trapani E, Corricelli M, Goitre L, Pinton P, Retta SF (2016) Beyond multiple mechanisms and unique drugs: defective autophagy as pivotal player in cerebral cavernous malformation pathogenesis and implications for targeted therapies. Rare Dis 5511(May):00–00. https://doi.org/10.1080/21675511.2016.1142640
Boyer AS, Ayerinskas II, Vincent EB, McKinney LA, Weeks DL, Runyan RB (1999) TGFβ2 and TGFβ3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Biol 208(2):530–545. https://doi.org/10.1006/dbio.1999.9211
Mercado-Pimentel ME, Runyan RB (2007) Multiple transforming growth factor-β isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs 185:146–156. https://doi.org/10.1159/000101315
Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V (2008) Signaling via the Tgf-β type I receptor Alk5 in heart development. Dev Biol 322(1):208–218. https://doi.org/10.1016/j.ydbio.2008.07.038
Agarwal S, Loder S, Cholok D et al (2016) Local and circulating endothelial cells undergo endothelial to mesenchymal transition (EndMT) in response to musculoskeletal injury. Sci Rep 6:32514. https://doi.org/10.1038/srep32514
Shi S, Srivastava SP, Kanasaki M et al (2015) Interactions of DPP-4 and integrin β1 influences endothelial-to-mesenchymal transition. Kidney Int 88(3):479–489. https://doi.org/10.1038/ki.2015.103
Bianchini F, Peppicelli S, Fabbrizzi P et al (2017) Triazole RGD antagonist reverts TGFβ1-induced endothelial-to-mesenchymal transition in endothelial precursor cells. Mol Cell Biochem 424(1–2):99–110. https://doi.org/10.1007/s11010-016-2847-2
Li J, Qu X, Yao J et al. (2010) Blockade of endothelial–mesenchymal transition by a smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. 59. https://doi.org/10.2337/db09-1631
Gong F, Zhao F, Gan X.(2017) Celastrol protects TGF- β 1-induced endothelial–mesenchymal transition. 37(2):185–190. https://doi.org/10.1007/s11596-017-1713-0
Guo Y, Li P, Bledsoe G, Yang ZR, Chao L, Chao J (2015) Kallistatin inhibits TGF-β-induced endothelial–mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp Cell Res 337(1):103–110. https://doi.org/10.1016/j.yexcr.2015.06.021
Wylie-Sears J, Levine RA, Bischoff J (2014) Losartan inhibits endothelial-to-mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor-β-induced phosphorylation of ERK. Biochem Biophys Res Commun 446(4):870–875. https://doi.org/10.1016/j.bbrc.2014.03.014
Chen X, Cai J, Zhou X et al (2015) Protective effect of spironolactone on endothelial-to-mesenchymal transition in HUVECS via notch pathway. Cell Physiol Biochem 36(1):191–200. https://doi.org/10.1159/000374063
Zhou H, Chen X, Chen L et al (2014) Anti-fibrosis effect of scutellarin via inhibition of endothelial–mesenchymal transition on isoprenaline-induced myocardial fibrosis in rats. Molecules 19(10):15611–15623. https://doi.org/10.3390/molecules191015611
Corallo C, Cutolo M, Kahaleh B et al (2016) Bosentan and macitentan prevent the endothelial-to-mesenchymal transition (EndoMT) in systemic sclerosis: in vitro study. Arthritis Res Ther 18(1):228. https://doi.org/10.1186/s13075-016-1122-y
Gao H, Zhang J, Liu T, Shi W (2011) Rapamycin prevents endothelial cell migration by inhibiting the endothelial-to-mesenchymal transition and matrix metalloproteinase-2 and -9: an in vitro study. Mol Vis 17(12):3406–3414.
Zhou X, Chen X, Cai JJ et al (2015) Relaxin inhibits cardiac fibrosis and endothelial–mesenchymal transition via the Notch pathway. Drug Des Devel Ther 9:4599–4611. https://doi.org/10.2147/DDDT.S85399
Bravi L, Rudini N, Cuttano R et al (2015) Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. Proc Natl Acad Sci 112(27):8421–8426. https://doi.org/10.1073/pnas.1501352112
wu M, Tang R, Liu H, Pan M, Liu B (2016) Cinacalcet ameliorates aortic calcification in uremic rats via suppression of endothelial-to-mesenchymal transition. Nat Publ Gr 37(11):1–9. https://doi.org/10.1038/aps.2016.83
Deng Y, Guo Y, Liu P et al (2016) Blocking protein phosphatase 2A signaling prevents endothelial-to-mesenchymal transition and renal fibrosis: a peptide-based drug therapy. Sci Rep 6(1):19821. https://doi.org/10.1038/srep19821
Furihata T, Kawamatsu S, Ito R et al (2015) Hydrocortisone enhances the barrier properties of HBMEC/ciβ, a brain microvascular endothelial cell line, through mesenchymal-to-endothelial transition-like effects. Fluids Barriers CNS 12(1):1–15. https://doi.org/10.1186/s12987-015-0003-0
Qi Q, Mao Y, Tian Y et al (2017) Geniposide inhibited endothelial–mesenchymal transition via the mTOR signaling pathway in a bleomycin-induced scleroderma mouse model. Am J Transl Res 9(3):1025–1036
Shore EM, Xu M, Feldman GJ et al (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38(5):525–527. https://doi.org/10.1038/ng1783
Lounev VY, Ramachandran R, Wosczyna MN et al (2009) Identification of progenitor cells that contribute to heterotopic identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am 91(3):652–663. https://doi.org/10.2106/JBJS.H.01177
Yao J, Guihard PJ, Blazquez-Medela AM et al (2015) Serine protease activation essential for endothelial–mesenchymal transition in vascular calcification. Circ Res 117(9):758–769. https://doi.org/10.1161/CIRCRESAHA.115.306751
Hjortnaes J, Shapero K, Goettsch C et al (2015) Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis 242(1):251–260. https://doi.org/10.1016/j.atherosclerosis.2015.07.008
Dudley AC, Khan ZA, Shih SC et al (2008) Calcification of multipotent prostate tumor endothelium. Cancer Cell 14(3):201–211. https://doi.org/10.1016/j.ccr.2008.06.017
Tang R, Wu M, Gao M, Liu H, Zhang X, Liu B (2012) High glucose mediates endothelial-to-chondrocyte transition in human aortic endothelial cells. Cardiovasc Diabetol 11(1):113. https://doi.org/10.1186/1475-2840-11-113
Tran K-V, Gealekman O, Frontini A et al (2012) The vascular endothelium of the adipose tissue gives rise to both white and brown fat cells. Cell Metab 15(2):222–229. https://doi.org/10.1016/j.cmet.2012.01.008
Huang L, Nakayama H, Klagsbrun M, Mulliken JB, Bischoff J (2015) Glucose transporter 1-positive endothelial cells in infantile hemangioma exhibit features of facultative stem cells. Stem Cells 33(1):133–145. https://doi.org/10.1002/stem.1841
Huang P, Schulz TJ, Beauvais A, Tseng Y-H, Gussoni E (2014) Intramuscular adipogenesis is inhibited by myo-endothelial progenitors with functioning Bmpr1a signalling. Nat Commun 5:4063. https://doi.org/10.1038/ncomms5063
Fioret BA, Heimfeld JD, Paik DT, Hatzopoulos AK (2014) Endothelial cells contribute to generation of adult ventricular myocytes during cardiac homeostasis. Cell Rep 8(1):229–241. https://doi.org/10.1016/j.celrep.2014.06.004
Welch-Reardon KM, Ehsan SM, Wang K et al (2014) Angiogenic sprouting is regulated by endothelial cell expression of Slug. J Cell Sci 127(Pt 9):2017–2028. https://doi.org/10.1242/jcs.143420
Zhang W, Chen G, Ren JG, Zhao YF (2013) Bleomycin induces endothelial mesenchymal transition through activation of mTOR pathway: A possible mechanism contributing to the sclerotherapy of venous malformations. Br J Pharmacol 170(6):1210–1220. https://doi.org/10.1111/bph.12355
Muylaert DEP, de Jong OG, Slaats GGG et al (2015) Environmental influences on endothelial to mesenchymal transition in developing implanted cardiovascular tissue-engineered grafts. Tissue Eng Part B Rev 22(1):58–67. https://doi.org/10.1089/ten.teb.2015.0167
Reichman D, Man L, Park L et al (2016) Notch hyper-activation drives trans-differentiation of hESC-derived endothelium. Stem Cell Res 17(2):391–400. https://doi.org/10.1016/j.scr.2016.09.005
Hahn S, Nam M-O, Noh JH et al (2017) Organoid-based epithelial to mesenchymal transition (OEMT) model: from an intestinal fibrosis perspective. Sci Rep 7(1):2435. https://doi.org/10.1038/s41598-017-02190-5
Ibrahim M, Richardson MK (2017) Beyond organoids: in vitro vasculogenesis and angiogenesis using cells from mammals and zebrafish. Reprod Toxicol. https://doi.org/10.1016/j.reprotox.2017.07.002
Acknowledgements
This work was supported by the Netherlands CardioVascular Research Initiative, the Dutch Heart Foundation, the Dutch Federation of University Medical Centers, the Netherlands Organisation for Health Research and Development, the Royal Netherlands Academy of Sciences (Phaedra and Reconnect consortia), and the Cancer Genomics Centre Netherlands.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Man, S., Sanchez Duffhues, G., ten Dijke, P. et al. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis 22, 3–13 (2019). https://doi.org/10.1007/s10456-018-9639-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-018-9639-0