
Abstract
The adsorption of chloroxylenol and chlorophene on halloysite-carbon composites was investigated in batch and flow systems. The synthesis of halloysite-carbon composites through two different methods was performed with microcrystalline cellulose as carbon precursor. The obtained halloysite-carbon composites were characterized by SEM/EDS analysis, the low-temperature nitrogen adsorption/desorption methods, and infrared spectrometry (FT-IR). The SEM/EDS analysis and FT-IR spectra confirmed the presence of carbon on the surface of the halloysite. On the basis of the measurement results in the batch system, the two composites with the best adsorption properties for both adsorbates were chosen for measuring the flow system (using the inverse liquid chromatography). Removal efficiency was equal to 92.26 and 81.36%. It was obtained for chloroxylenol on HNT-m 800 and HNT-Zn 500, respectively. For chlorophene, the removal efficiency had the value of 78.79 and 77.87% on HNT-m 800 and HNT-Zn 800, respectively. Adsorption parameters of chloroxylenol and chlorophene were determined with inverse liquid chromatography methods: the adsorption equilibrium constants were determined with the peak division method and the adsorption capacity of the adsorbents was determined with the breakthrough curve method. Maximum adsorption capacity for the adsorption of chloroxylenol on HNT-m 800 was 5.48 mg·g−1 and on HNT-Zn 500 its value was 2.77 mg·g−1. For the adsorption of chlorophene on HNT-m 800 the value was 4.44 mg·g−1 and on HNT-Zn 800–2.5 mg·g−1. Halloysite-carbon composites can be successfully used as effective adsorbents for removing chloroxylenol and chlorophene from solutions in the flow system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The “emerging contaminants” include a very wide and diverse group of compounds such as: pesticides, pharmaceuticals, personal care products, fragrances, plasticizers, flame retardants, nanoparticles, perfluoroalkyl compounds, chlorinated paraffins, siloxanes, surfactants and their metabolites, gasoline additives, etc. [1, 2]. Their presence and concentration in the environment are not monitored or defined with legal terms. Among them, personal care products (PCPs) play a significant role [3]. PCPs receive little attention, despite the fact they are used in large amounts throughout the world. In contrast to many other known contaminants, most PCPs are disposed or discharged into the environment because of continual infusion into the aquatic environment. The most common sources of PCPs contamination are industrial/domestic sewage systems and wet-weather runoff. The main concerns connected with the detection of PCPs in the aquatic environment consist in their persistence, bioaccumulation, and toxicity to aquatic life and, indirectly, human life [4, 5]. PCPs contain mainly antiseptics, biocides, cosmetics, modified hormones, steroids, perfumes, shampoos, UV filters [3, 6]. There are several active substances used in cosmetics and disinfecting products. Triclosan, chloroxylenol and chlorophene can be found among the most frequently used ones [6, 7]. Triclosan (phenol derivative) (Fig. 1a) is a common antibacterial agent which has been incorporated into about 700 different personal care products (toothpastes, deodorants, hand sanitizers, soaps, and medical skin creams [8]) at a concentration level of 0.1–0.3% [9]. This compound can be highly toxic toward aquatic and land organisms. It can also lead to bacterial resistance [10, 11]. Triclosan can transform into other, more toxic organic compound, e.g.chlorodioxins, chlorophenol, 2,4-dichlorophenol or chloroform, when it is present in the oxidating environment or UV light [12, 13]. Since there have been many studies that confirm that triclosan can cause potential risks, it has been replaced with other antiseptics, for instance chloroxylenol [14]. Chloroxylenol (Fig. 1b) is another phenolic derivative with antiseptic and disinfectant properties [15, 16], which is widely used as a preservative for industrial and cosmetic purposes [17]. The main use of chloroxylenol includes wound-cleaning applications, household antiseptics, and antibacterial soaps. This compound is also used to control microorganisms, fungi, and algae in emulsions, adhesives, paints, and washing tanks [18]. In the experiment, chloroxylenol was found in surface river water at a concentration level of 0.03–1.20 µg/dm3 [19]. As a biocide, chloroxylenol can be potentially toxic to human health and environment, for instance it is skin and eye irritant. Its toxicity ranges from low to high, depending on test conditions and species [15, 18]. Chlorophene (Fig. 1c) is also a phenolic compound that belongs to a group of antiseptics and disinfectants [6, 11, 20, 21]. It is most commonly used at a concentration of 3–10% (wt.%) in households and hospitals for disinfecting and general cleaning. In industrial and farming environments, chlorophene has found its application as an active ingredient in disinfectants formulations [22]. Chlorophene was found in the influent at a concentration of 0.71 µg/dm3 and in the effluent (130 µg/dm3). As a representant of the group of emerging contaminants, chlorophene is believed to have negative impact on the environment, human life, and wildlife. Therefore, it is crucial to develop methods of water treatment technologies and purifying aquatic environment from triclosan, chloroxylenol, and chlorophene [23, 24].
“Emerging contaminants”, including triclosan, chloroxylenol, and chlorophene, were removed from the water using many different methods: biodegradation [10, 15, 25], oxidation [22], biocatalysis [9], filtration [23], advanced oxidation processes [24, 26,27,28], and adsorption [29,30,31,32,33,34,35]. Low cost, high efficiency and the ease of carrying out the process make adsorption a desirable method of removing triclosan, chloroxylenol, and chlorophene from water [16]. So far, studies on the adsorption of triclosan, chloroxylenol and chlorophene have been carried out, mainly with the use of carbon or polymer adsorbents (Table 1). Based on the knowledge available to us, there are no works describing the adsorption of these pollutants using mineral-carbon materials.
In our previous work, we presented the synthesis of halloysite-carbon adsorbents, in which sucrose was the carbon precursor, and the use of these composites to remove non-steroidal anti-inflammatory drugs from water [36, 37].
Based on our experience, we decided to develop study of mineral-carbon materials. Therefore, the aim of this study was to obtain halloysite-carbon composites through two different methods, using bleached halloysite and halloysite nanotubes as template and microcrystalline cellulose acting as carbon precursor. Another objective was to use these composites as adsorbents to remove chloroxylenol as well as chlorophene from the solution.

Molecular structure of triclosan (a), chloroxylenol (b), and chlorophene (c) [4]
2 Experimental
2.1 Materials and reagents
Bleached halloysite (H) was delivered from the “Dunino” strip mine located in Legnica, Poland. Halloysite nanotubes (HNT) were acquired from Merck KGaA, Darmstadt, Germany. Microcrystalline cellulose was purchased from Chem Point Sp. z o.o., Cracow, Poland. Chloroxylenol and chlorophene were, in turn, obtained from Merck KGaA, Darmstadt, Germany. Methanol (for HPLC) was acquired from Merck KGaA, Darmstadt, Germany. Physicochemical properties of chloroxylenol and chlorophene are presented in Table 2.
2.2 The synthesis of halloysite-carbon adsorbents
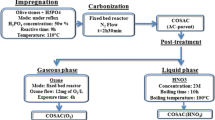
Bleached halloysite (H) was prepared as previously described in ref. [36]. Halloysite-carbon adsorbents were obtained using halloysite with a grain fraction of 0.2–0.32 mm. They were prepared through two different methods. The first method consisted in dissolving microcrystalline cellulose in a reagent known as the “Cross-Bewan reagent” (zinc chloride(II) dissolved in a 37% (wt. %) hydrochloric acid). Next, halloysite was added and mixed together in an Erlenmeyer flask. Then, it was centrifuged, dried, washed with deionized water to remove chlorides, dried again and carbonized in nitrogen atmosphere. The obtained composites were labelled as H-Zn 500, HNT-Zn 500, H-Zn 800, and HNT-Zn 800. The second method consists in milling halloysite and microcrystalline cellulose together in a ball mill and then carbonization composites in nitrogen atmosphere. Particular conditions of synthesis are described in Table 3. The obtained composites are labelled as H-m 500, HNT-m 500, H-m 800, and HNT-m 800.
2.3 The characterization of adsorbents
SEM/EDS analysis was performed at the Institute of High Pressure Physics of the Polish Academy of Sciences with the Zeiss Ultra Plus scanning electron microscope. The microscope is equipped with 2 detectors of secondary electrons: the standard one located in the SE2 chamber and the intra-column InLens. It is also equipped with the Bruker Quantax 400 EDS microanalysis system containing an ultra-fast (up to 300 kconts/s) detector with energy resolution of 127 eV and an active surface of 30 mm2.
The parameters of the porous structure were determined on the basis of low-temperature (− 196 °C) nitrogen adsorption-desorption isotherms using the ASAP 2020 volume adsorption analyzer developed by Micromeritics (Norcross, GA, USA) (Structural Research Laboratory of Jan Kochanowski University in Kielce). Before carrying out the adsorption tests, the samples of halloysite and halloysite-carbon composites were degassed under the reduced pressure for 2 h at 200 °C. Specific surface area was determined using the BET method (Brunauer-Emmett-Teller - SBET) on the basis of the obtained nitrogen adsorption isotherms in the range of relative pressures ranging from 0.05 to 0.2. The total pore volume (Vt) was determined from one point of the adsorption isotherm, which corresponded to the relative pressure p/p0 = 0.99. The volume of micropores (Vmi) with respect to halloysite and halloysite-carbon composites was determined with the DFT (Density Functional Theory). Pore size distribution functions were calculated using the non-local density functional theory (NLDFT) method and the calculations were made using the numerical program SAIEUS (Micromeritics) [43, 44]. External surfaces of the adsorbents (Sext) were determined in the range of αs from 2 to 8. The mesopore volume (Vme) was obtained by subtracting the micropore volume (Vmi) from the total pore volume (Vt). Mesoporosity was calculated from the ratio of the mesopore volume (Vme) to the total pore volume (Vt) [45, 46].
External surfaces of adsorbents were calculated using the following formula [47]:
where: S – external surface (m2/g), ɳ – slope of αs graph (cm3 STA/g), SBET, ref – specific surface area of the reference adsorbent (m2/g), v0.4, ref – adsorbed amount at a relative pressure of 0.4 for the reference adsorbent (cm3 STA/g). The reference adsorbent was in the form Cabot BP 280 unrepresented carbon black [47].
Infrared absorption spectra (4000 − 600 cm−1) were recorded with a Perkin-Elmer Spectrum 400 FT-IR/FT-NIR spectrometer. The device contains a monolithic diamond crystal necessary to register reflection spectra and ATR adapter. For each sample, 40 scans with a resolution of 4 cm−1 were performed. Before the measurement, each sample had been dried and milled in an agate mortar.
Boehm’s method was used to determine functional groups on the surface of halloysite-carbon adsorbents. For this purpose, a 0.2 g sample of each composite was placed in a 100 cm3 conical flask. Then, 25 cm3 of 0.1 M solution of sodium bicarbonate (NaHCO3), sodium hydroxide (NaOH) and sodium ethoxide (C2H5Ona) or 25 cm3 of 0.05 M solution of sodium carbonate (Na2CO3) and hydrochloric acid (HCl) were added to each adsorbent. The solutions of NaHCO3, Na2CO3, NaOH and C2H5OH were used to determine acidic groups (carboxyls, lactones, phenols and carbonyls, respectively), and the solution of hydrochloric acid was used to determine basic groups. The mixtures were shaken for 48 h and filtered. Next, 10 cm3 of every mixture was pipetted and the excess of acid and base was titrated by 0.05 M solution of NaOH or HCl, respectively. During titration, methyl orange was used as an indicator. Boehm’s titration method is based on the assumption that C2H5ONa and NaOH neutralizes different strength levels of acidic sites (weak, mild, and strong). Na2CO3 neutralizes mild and strong acidic sites and NaHCO3 neutralizes only strong acidic sites. Therefore, C2H5ONa neutralizes carboxyl, lactonic, phenolic and carbonyl groups, NaOH neutralizes carboxyl, lactonic and phenolic groups, Na2CO3 neutralizes carboxyl and lactonic groups, and NaHCO3 – only carboxyl groups. The amount of the hydrochloric acid, which was consumed by the adsorbents, was used to calculate the concentration of basic sites [48,49,50].
The pH value of point zero charge (pHpzc) of H–m 800 and H–Zn 800 composites was performed with the mass titration method. For this purpose, 150 cm3 of 0.01 M solution of NaCl was placed in 8 conical flasks. Then, pH was adjusted to initial values between 2 and 12 by adding HCl (0.1 M, 1 M HCl) or NaOH (0.1 M, 1 M, 2 M) solutions. 0.15 g of H–m 800 and H–Zn 800 was placed in a 100 cm3 conical flask. 50 cm3 of 0.01 M solution of NaCl of different pH values was added to adsorbents and stirred for 4 h at 25 °C and the final pH of each solution was recorded. The dependence of the initial pH on the final pH was plotted. The pHpzc value of each adsorbent was determined from the graph at the point of pH, at which the curve crosses the line pHfinal = pHinitial [50,51,52].
2.4 Adsorption measurements
2.4.1 Adsorption in batch system
Batch adsorption experiments of chlorophene and chloroxylenol on halloysite and halloysite-carbon composites were performed to select the best adsorbent for chlorophene and chloroxylenol removal. The experiments were conducted in a 100 cm3 conical flask. The concentration of adsorbate was measured with the spectrophotometric method (using the UV Shimadzu UV-1800 spectrophotometer). Contact time of the adsorbent and adsorbate was 24 h. The adsorbents dosage was 0.05 g for chlorophene and 0.1 g for chloroxylenol. Concentration level of the adsorbate was 20 mg∙dm−3. The temperature during experiment was 25 °C, whilst the mixing rate was equal 120 rpm. After the given adsorption time, the adsorbate solution was separated from the adsorbent with the use of cup-type centrifuge. Adsorbate concentration was determined at 220 nm for chlorophene and chloroxylenol.
Removal efficiency (R, %) was calculated using the following Eq. (2) [53]:
where: C0 and Ce (mg∙dm−3) – initial and equilibrium concentrations of chlorophene and chloroxylenol solutions.
2.4.2 Adsorption in the flow system
Maximum adsorption capacity was determined using the Breakthrough Curves (BC) method, while the adsorption isotherms were determined using the Peak Division (PD) method (Inverse Liqiud Chromatography). The chromatograph was equipped with a precision pump, which ensures the steady flow of the liquid phase (which was, in that case, adsorbate solution), the six-port valve systems for switching the chromatographic column (on/off) in order to regulate liquid phase flow, and the high-sensitivity detector necessary for determining solute concentration [54].
The Breakthrough Curve method consists in continuous registering of changes in the adsorbate concentration recorded at the column outlet during the flow of the adsorbate solution. In order to determine the response time (flow from the column inlet into the detector) of the liquid phase, which contains the adsorbate, the reference column with a nonadsorbing bed was used. The concentration of the adsorbate was measured at the outlet of the reference column from the beginning of the analysis until it reached the plateau. Both the mass of the adsorbed substances in the column with the adsorbent and in the reference column were calculated based on the Eq. (3) [54]:
where: a – quantity of adsorbed substance i on the adsorbent (mg·g−1), C – adsorbate concentration (mg·cm−3), F – liquid phase flow (cm3·min−1), \({t}_{e}^{a}\) and \({t}_{e}^{ref}\) – end time of the analysis of adsorbate concentration in the adsorbent column (min) and in the reference column (min), respectively, \({t}_{0}^{a}\) and \({t}_{0}^{ref}\) – start time of adsorbate concentration analysis in the adsorbent column (min) and in the reference column (min), respectively, m – mass adsorbent (g) [54].
The Peak Division method involves dividing the total absorption surface onto parallel parts (L) of the chromatogram baseline by measuring the surface area of each segment. The adsorption value, ai of the adsorbate, i, was calculated according to equation given below (4) [54]:
where: ai – quantity of the adsorbed substance i on the adsorbent (mg·g−1), n – mass of substance i applied on the adsorbent (mg), SLs – area of L that is part of the total adsorption surface (mV·min), m – adsorbent mass (g), SP – adsorbate peak area (mV·min) [54].
Equilibrium concentration of substance i in the liquid phase (Ci) corresponds to adsorption values ai of substance i and is calculated based on the Eq. (5) [54]:
where: Ci – equilibrium concentration of substance i in the liquid phase (mg·cm−3), n – mass of substance i applied on the adsorbent (mg), hI – height of the peak part I (mV), F – liquid phase flow (cm3·min−1), SP – adsorbate peak area (mV·min) [54].
Adsorption isotherm can be determined using the PD method based on a single chromatographic peak. It was possible by using the CDPS program (Computer Data Processing System – CDPS) [54]. CDPS was applied to calculate the adsorption area, the area of each segment, and the area of the adsorption peak together with the retention time. This system also allows to choose the number of segments, by which peak area can be divided. Calculations of equilibrium concentration Ci of substance i in the liquid phase and the amount of the adsorbed adsorbate ai were carried out based on the data obtained from the peak profile division. Therefore, relations ai= f(Ci) become a function describing an adsorption isotherm. The number of points in the isotherm is the number of adsorption areas divided into L segments [54].
In order to obtain adsorption parameters, the Dionex UltiMate 3000 Series chromatography system (Thermo Fisher Scientific, Inc.), equipped with a triple pump, and UV-Vis detector was used. Measurement details carried out using the BC and PD method are described in Refs [55, 56].
Measurements of the adsorption capacity and adsorption measurements for chloroxylenol and chlorophene on the given adsorbent were performed using a 10 cm long chromatographic column with an internal diameter of 0.8 mm (Knauer, Germany). The adsorbent weight was approx. 0.4 g for both experiments. Adsorption measurements were carried out at 25 °C.
Measurements of the adsorption capacity of adsorbent were conducted for chloroxylenol and chlorophene solution concentration of 10–50 mg∙dm−3. Before each measurement of the sorption capacity, the column was conditioned by forcing methanol solution (p.a. HPLC) with a flow rate of 0.5 ml∙min−1 for an hour. The reference column, with the same parameters as above, was filled with silanized granules. Solutions of individual adsorbates were dosed into the columns prepared in this manner.
Adsorption measurements were carried out as follows: 50 µl of adsorbate solution with a concentration of chloroxylenol: 1000–1400 mg∙dm−3 and chlorophene: 800–1200 mg∙dm−3. The column was conditioned for 1 h prior to measurements using methanol solution at a flow rate of 0.5 ml∙min−1. 50 µl of adsorbate were successively dosed into the column prepared in this manner, using an automatic injector.
In all measurements, chromatographic peaks were recorded using the UV detector with UV wavelength of 220 nm for chloroxylenol and chlorophene.
3 Results and discussion
3.1 Characterization of adsorbents
SEM images of H, HNT, and halloysite-carbon composites are presented in the Figs. 2 and 3. The morphology of the H sample was platy, blocky and tubular. HNT featured typically tubular morphology. In the SEM images of halloysite-carbon composites one can see carbon particles on the halloysite surface. The presence of carbon is confirmed by the EDS analysis (Table 4). The highest carbon content is observed in the HNT-Zn 500 and HNT-Zn 800 samples, equal to 30.00 and 32.26%, respectively. Carbon more readily covers the HNT surface than the H surface. Zinc content is higher for materials carbonized in 500 °C than for composites carbonized at 800 °C.

SEM images of H, H–Zn 500, H–Zn 800, H-m 500, and H–m 800

SEM images of HNT, HNT-Zn 500, HNT-Zn 800, HNT-m 500, and HNT-m 800
Figure4 and 5 present nitrogen adsorption/desorption isotherms (Fig 4a and Fig 5a) and pore size distribution (Figs. 4b and 5b) for all adsorbents. According to the IUPAC classification, experimental isotherms for the halloysite and halloysite-carbon adsorbents are type IV, which is typical for mesoporous materials. H1 hysteresis loops, observed on the isotherms, confirm that all tested materials were mesoporous. Pore volume distribution functions show two maxima at the presented curve, which correspond to the micropores and mesopores, respectively. The heights and dispersion, which indicate the micropores’ and mesopores’ share in the total porosity, are different for the tested materials. The largest proportion of micropores is observed for H-Zn 500, H-m 500, HNT-Zn 500, and HNT-Zn 800 (the highest peak), whilst the smallest for the non-modified halloysite (the lowest peak). The largest proportion of mesopores is present for H–Zn 800 and HNT-Zn 800. HNT-carbon composites have higher values of parameters of the porous structure than H-carbon composites.

Nitrogen adsorption/desorption isotherms and pore size distribution for H and its H-carbon composites

Nitrogen adsorption/desorption isotherms and pore size distribution for HNT and HNT-carbon composites
The following parameters of the porous structure are presented in Table 5: specific surface area, determined by the BET method (SBET), external surface area, total pore volume (Vt), the volume of ultrapores (VultraDFT) and micropores (VmicroDFT), the values of the average micropore (wmiDFT) and average mesopore (wmesoDFT) dimension determined with the DFT method, and also the volume of mesopores (Vmeso), used to obtain mesoporosity of materials. Micropore and mesopore dimensions are obtained from the maxima of the DFT distribution function. The largest specific surface area, external surface area, total pore volume, and the volume of ultrapores and micropores are observed for the HNT-Zn 500 and HNT-Zn 800 composites. Micropore dimensions vary between 0.76 and 0.82 nm, the value of mesopore dimensions is 10.85–13.53 nm for all materials. All halloysite-carbon composites developed porosity in comparison to the non-modified halloysite.
The ATR FTIR spectra of H, HNT, H-carbon, and HNT-carbon composites are shown in Figs. 6 and 7. The spectra of H and HNT samples show the following vibration bands: the bands at 3695 and 3622 (or 3625) cm−1 are assigned to two Al2OH-stretching bands (each OH-hydroxyl being linked to two Al atoms). The bands at 1028, 1006 and 689 or 686 cm−1 are assigned to perpendicular stretching vibrations of Si–O–Si. The bands observed at 911 or 909 cm−1 are attributed to the O–H deformation of inner-surface hydroxyl groups, and the O–H deformation of inner hydroxyl groups, respectively. Other bands at 791 (795) and (751) 750 cm−1 can be assigned to O–H translation vibrations of halloysite O–H units. Compared to the spectrum of HNT, the spectrum of H shows a significant increase in the intensity of all bands [57]. The spectra of H-carbon and HNT-carbon composites differ substantially from the H and HNT spectra. In addition, the bands are not observed in the region of 3700 − 3600 cm−1 for the H-carbon and HNT-carbon composites and the band at 1028 cm−1 is broadened and moved to the region of 1032–1065 cm−1. That suggests the change mostly on the inner (alumina octahedral sheet), but also on the outer (silica tetrahedron sheet) surfaces of halloysite, which may be caused by the presence of carbon on the halloysite surface.

FTIR spectra of H and H-carbon composites

FTIR spectra of HNT and HNT-carbon composites
Boehm’s method was used to determine the concentration of functional groups on the H-carbon and HNT-carbon composite surface (Table 6). The highest content of total basic functional groups was confirmed in HNT-m 800 and H-m 500 composites, while the greatest amount of total acidic groups was found for the HNT-Zn 800 and HNT-Zn 500 composites. H-carbon and HNT-carbon composites showed high concentration of carbonyl and phenolic groups, medium concentration of total basic and carboxyl groups as well as low concentrations of lactonic functional groups. High concentration of carbonyl groups is crucial, since it may promote the adsorption of phenol derivatives, such as chloroxylenol and chlorophene, by a donor-acceptor mechanism [58]. Composite preparation temperature is not a significant factor affecting the concentration of acidic and basic functional groups.
The dependence of the initial pH on the final pH for the H-Zn 800 and H-m 800 is illustrated in Figs. 8 and 9. The pHPZC value is equal 7.25, and 6.82, respectively. Therefore, the surface of adsorbents is positively charged in the case of both adsorbents in deionized water (pH about 6).

Point of zero charge (pHPZC) of the H-Zn 800, determined with the mass titration method

Point of zero charge (pHPZC) of the H-m 800, determined with the mass titration method
3.2 Adsorption experiments
3.2.1 Adsorption in batch system
Removal efficiencies of chloroxylenol and chlorophene for H, HNT, H-carbon and HNT-carbon composites are presented in Figs. 10 and 11. The results show that all H-carbon and HNT-carbon adsorb significantly better both adsorbates than H and HNT. HNT-carbon composites are more effective adsorbents in removing chloroxylenol and chlorophene from the solution than H-carbon composites. It may be caused by the different morphology of the H and HNT samples. HNT-composites have higher carbon content and higher values of the parameters of the porous structure than H-composites, which probably affects removal efficiencies of chloroxylenol and chlorophene from water. HNT-m 800 is the best adsorbent for chloroxylenol and chlorophene. For further research, two best adsorbents for every adsorbate were chosen: HNT-m 800 and HNT-Zn 500 for chloroxylenol and HNT-m 800 and HNT-Zn 800 for chlorophene.

Removal efficiency of chloroxylenol for H, HNT, H-carbon and HNT-carbon composites (mass of adsorbents – 0.1 g, concentration of adsorbate solution – 20 mg∙dm−3, contact time – 24 h, temperature – 25 °C)

Removal efficiency of chlorophene for H, HNT, H-carbon and HNT-carbon composites (mass of adsorbents – 0.05 g, concentration of adsorbate solution – 20 mg∙dm−3, contact time – 24 h, temperature – 25 °C)
3.2.2 Adsorption in the flow system
Maximum adsorption capacity was determined with the Breakthrough Curve (BC) method. Maximum adsorption capacity was calculated with the following values: for the adsorption of chloroxylenol on HNT-m 800 was 5.48 mg·g−1 and on HNT-Zn 500 was 2.77 mg·g−1, and for the adsorption of chlorophene on HNT-m 800 was 4.44 mg·g−1 and on HNT-Zn 800–2.5 mg·g−1.
Adsorption isotherms were determined using the Peak Division (PD) method based on a single chromatographic peak. Fitting experimental adsorption data to the most commonly adsorption models (Freundlich, Langmuir (one-center), Langmuir (multi-center), Temkin, and Dubin-Raduszkiewicz) was checked using non-linear regression (the Levenberg–Marquardt least squares method with the OriginLab software). The best correlation of experimental data is obtained for the Langmuir adsorption model (multi-center) (Eq. 6), for both chloroxylenol and chlorophene on all adsorbents. Equation parameters and correlation coefficients R2 for the adsorption of chloroxylenol on HNT-m 800 and HNT-Zn 500 and chlorophene on HNT-m 800 and HNT-Zn 800 are collected in Table 7.. Adsorption isotherms determined using the Peak Division (PD) method are presented in Fig. 12.
where: qe – the amount of a solute adsorbed per gram of the adsorbent at equilibrium (mg·g−1); KL – Langmuir (multi-center) isotherm constant (dm3·mg−1)1/n; qm – maximum monolayer coverage capacity (mg·g−1); n – adsorption model index; Ce – equilibrium concentration of a solute in the solution (mg·dm− 3) [59].

Adsorption isotherms determined using the Peak Division (PD) method
Adsorption constant value for chloroxylenol on HNT-m 800 is 0.79 (dm3·mg−1) and on HNT-Zn 500–0.24 (dm3·mg−1). Adsorption constant value for chlorophene on HNT-m 800 is 0.48 (dm3·mg−1) and this value for chloroxylenol on HNT-Zn 800 has the value of 0.42 (dm3·mg−1). The value of parameter n is similar for all adsorbents and it is fractional, which indicates the adsorption mechanism of chloroxylenol and chlorophene with a different number of adsorptive centers on the surfaces of the adsorbents. The parameter n is less than 1 (between 0.74 and 0.83) for all the studied materials. Therefore, more than one adsorbate molecule can occupy adsorption center.
4 Conclusions
The synthesis of halloysite-carbon composites through two different methods was performed with microcrystalline cellulose as carbon precursor. The first method consisted in dissolving the cellulose in the Cross-Bewan reagent and impregnating the halloysite with cellulose dissolved in this reagent, and the second in milling the halloysite and cellulose and suspending it in water. The second method did not require the use of additional chemical reagents, it was more environmentally friendly.
SEM/EDS analysis confirms the presence of carbon on the surface of halloysite, showing that the syntheses are successful. Specific surface area determined with the BET method, total pore volume and the volume of micropores are larger for all halloysite-carbon composites compared to non-modified halloysite. The largest values of these parameters can be observed for the HNT-Zn 500 and HNT-Zn 800 composites.
The conditions of adsorption experiments (the value of pHPZC of hallosite-carbon adsorbents equals pH ~ 6, and the value of pKa of chloroxylenol and chlorophene) confirm the presence of non-ionized form of adsorbate molecules during the whole adsorption process. The highest concentration levels of carbonyl groups can be observed on the surface of all halloysite-carbon adsorbents. They can promote the adsorption of such form of chloroxylenol and chlorophene using the donor-acceptor mechanism, as for phenol derivatives of carbon adsorbent surface.
Comparing the adsorption properties of the non-modified halloysite with the obtained composites, one can see that halloysite-carbon adsorbents are significantly more effective materials for removing chloroxylenol and chlorophene. HNT-carbon composites adsorbed chloroxylenol and chlorophene from the solution in the batch adsorption system better than for H-composites probably due to the higher carbon content and higher parameter values of the porous structure. Carbon content on the halloysite surface is the main factor facilitating the adsorption process of chloroxylenol and chlorophene.
The most effective adsorbents were used to determine the maximum adsorption capacity and isotherms parameters using inverse liquid chromatography. The best correlation experimental data was obtained for the multi-center Langmuir adsorption model. Maximum adsorption capacity was determined with the Breakthrough Curve (BC) method.
Maximum adsorption capacity (qmax) for the adsorption of chloroxylenol on HNT-m 800 was twice higher than on HNT-Zn 500, and for the adsorption of chlorophene on HNT-m 800 was also much higher than this value on HNT-Zn 800. The value of qmax was higher for chloroxylenol on HNT-m 800 in comparison to the one obtained for chlorophene on the same adsorbent. This value was slightly higher for chloroxylenol on HNT-Zn 500 compared to chlorophene on HNT-Zn 800.
Adsorption constant value (KL) for chloroxylenol adsorption on HNT-m 800 was more than three times higher than on HNT-Zn 500, and for chlorophene adsorption on HNT-m 800 this parameter was slightly higher than this value on HNT-Zn 800. The value of KL for chloroxylenol on HNT-m 800 was significantly higher in comparison to the one obtained for chlorophene on the same adsorbent. Adsorption constant value for chlorophene on HNT-Zn 800 was almost twice as high as this value for chloroxylenol on HNT-Zn 500.
Adsorbents obtained by milling cellulose with halloysite proved to be the best materials for removing chlorophene and chloroxylenol from aqueous solutions confirming that this method of obtaining them can be used instead of the method using zinc chloride.
Halloysite as natural, cheap and non-toxic mineral can be the template for carbon, which can lead to obtaining new composites with interesting properties. They can be successfully used as effective adsorbents for removing chloroxylenol and chlorophene from the solution in the flow system.
Data availability
Not applicable.
References
Stefanakis, A.I., Becker, J.A.: A review of emerging contaminants in water: Classification, sources, and potential risks. In: McKeown, A.E., Bugyi, G. (eds.) Impact of Water Pollution on Human Health and Environmental Sustainability, pp. 55–80. IGI Global, Hershey (PA) (2015)
Sauvé, S., Desrosiers, M.: A review of what is an emerging contaminant. Chem. Cent. J. (2014). https://doi.org/10.1186/1752-153X-8-15
Gogoi, A., Mazumder, P., Tyagi, V.K., Chaminda, G.G.T., An, A.K., Kumar, M.: Occurrence and fate of emerging contaminants in water environment: a review. Groundw. Sustain. Dev. 6, 169–180 (2018). https://doi.org/10.1016/j.gsd.2017.12.009
Daughton, C.G., Ternes, T.A.: Pharmaceuticals and personal care products in the environment: agents of subtle change? Environ. Health Perspect. 107, 907–938 (1999). https://doi.org/10.1289/ehp.99107s6907
Ebele, A.J., Abdallah, M.A.E., Harrad, S.: Pharmaceuticals and personal care products (PPCPs) in the freshwater aquatic environment. Emerg. Contam. 3, 1–16 (2017). https://doi.org/10.1016/j.emcon.2016.12.004
Nassar, H.N., Younis, S.A.: From priority contaminants to emerged threat: Risk and occurrence-based analysis for better water management strategies in present and future. In: Fosso-Kankeu, E. (ed.) Nano and bio-based Technologies for Wastewater Treatment: Prediction and Control Tools for the Dispersion of Pollutants in the Environment, 1st edn., pp. 41–103. Wiley, New York (2019)
El-Badawy, F.M., El-Desoky, H.S.: Quantification of chloroxylenol, a potent antimicrobial agent in various formulations and water samples: environmental friendly electrochemical sensor based on microwave synthesis of graphene. J. Electrochem. Soc. 165, 694–707 (2018). https://doi.org/10.1149/2.0391814jes
Quan, B., Li, X., Zhang, H., Zhang, C., Ming, Y., Huang, Y., Xi, Y., Weihua, X., Yunguo, L., Tang, Y.: Technology and principle of removing triclosan from aqueous media: a review. Chem. Eng. J. 378, 122185 (2019). https://doi.org/10.1016/j.cej.2019.122185
Bilal, M., Barceló, D., Iqbal, H.M.N.: Persistence, ecological risks, and oxidoreductases-assisted biocatalytic removal of triclosan from the aquatic environment. Sci. Total Environ. 735, 139194 (2020). https://doi.org/10.1016/j.scitotenv.2020.139194
Chen, X., Nielsen, J.L., Furgal, K., Liu, Y., Lolas, I.B., Bester, K.: Biodegradation of triclosan and formation of methyl-triclosan in activated sludge under aerobic conditions. Chemosphere. 84, 452–456 (2011). https://doi.org/10.1016/j.chemosphere.2011.03.042
Verma, K.S.: Occurrence and transformation of pharmaceutical and antibacterial compounds in the environment. Dissertation, Mississippi State University (2010)
Lee, D.G.: Removal of a synthetic broad-spectrum antimicrobial agent, triclosan, in wastewater treatment systems: a short review. Environ. Eng. Res. 20, 111–120 (2015). https://doi.org/10.4491/eer.2014.081
Dhillon, G.S., Kaur, S., Pulicharla, R., Brar, S.K., Cledón, M., Verma, M., Surampalli, R.Y.: Triclosan: current status, occurrence, environmental risks and bioaccumulation potential. Int. J. Environ. Res. Public. Health 12, 5657–5684 (2015). https://doi.org/10.3390/ijerph120505657
Sreevidya, V.S., Lenz, K.A., Svoboda, K.R., Ma, H.: Benzalkonium chloride, benzethonium chloride, and chloroxylenol—three replacement antimicrobials are more toxic than triclosan and triclocarban in two model organisms. Environ. Pollut. 235, 814–824 (2018). https://doi.org/10.1016/j.envpol.2017.12.108
Ghanem, K.M., Al.-Fassi, F.A., Al.-Hazmi, N.M.: Optimization of chloroxylenol degradation by Aspergillus Niger using Plackett-Burman design and response surface methodology. Afr. J. Biotechnol. 11, 15040–15048 (2012). https://doi.org/10.5897/AJB12.1953
Zhou, Y., Cheng, G., Chen, K., Lu, J., Lei, J., Pu, S.: Adsorptive removal of bisphenol A, chloroxylenol, and carbamazepine from water using a novel β-cyclodextrin polymer. Ecotoxicol. Environ. Saf. 170, 278–285 (2019). https://doi.org/10.1016/j.ecoenv.2018.11.117
Guess, W.L., Bruch, M.K.: A review of available toxicity data on the topical antimicrobial, chloroxylenol. J. ToxicoL-Cut & Ocular Toxicol. 5, 233–262 (1986). https://doi.org/10.3109/15569528609036300
Capkin, E., Ozcelep, T., Kayis, S., Altinok, I.: Antimicrobial agents, triclosan, chloroxylenol, methylisothiazolinone and borax, used in cleaning had genotoxic and histopathologic effects on rainbow trout. Chemosphere. 182, 720–729 (2017). https://doi.org/10.1016/j.chemosphere.2017.05.093
Choi, D., Oh, S.: Removal of chloroxylenol disinfectant by an activated sludge microbial community. Microbes Environ. 34, 129–135 (2019). https://doi.org/10.1264/jsme2.ME18124
Stawarz-Janeczek, M., Kryczyk-Poprawa, A., Muszyńska, B., Opoka, W., Pytko-Polończyk, J.: Disinfectants used in stomatology and SARS-CoV-2 infection. Eur. J. Dent. 15, 388–400 (2021). https://doi.org/10.1055/s-0041-1724154
Yang, G., Fan, M., Zhang, G.: Emerging contaminants in surface waters in China—a short review. Environ. Res. Lett. 9, 074018–074030 (2014). https://doi.org/10.1088/1748-9326/9/7/074018
Zhang, H.: Metal oxide-facilitated oxidation of antibacterial agents. Dissertation, Georgia Institute of Technology (2004)
Sasi, S., Rayaroth, M.P., Aravindakumar, C.T., Aravind, U.K.: Occurrence, distribution and removal of organic micro-pollutants in a low saline water body. Sci. Total Environ. 749, 141319–141329 (2020). https://doi.org/10.1016/j.scitotenv.2020.141319
Bolobajev, J., Goi, A.: Sonolytic degradation of chlorophene enhanced by Fenton-mediated oxidation and H•-scavenging effect. Chem. Eng. J. 328, 904–914 (2017). https://doi.org/10.1016/j.cej.2017.07.043
Dhir, B.: Removal of pharmaceuticals and personal care products by aquatic plants. In: Prasad, M.N.V., Vithanage, M., Kapley, A. (eds.) Pharmaceuticals and Personal care Products: Waste Management and Treatment Technology. Emerging Contaminants and micro Pollutants, pp. 321–340. Elsevier, Amsterdam (2019). https://doi.org/10.1016/B978-0-12-816189-0.00014-7
Mohammed, A.J., Kadhum, A.A.H., Ba-Abbad, M.M., Al.-Amiery, A.A.: Optimization of solar photocatalytic degradation of chloroxylenol using TiO2, Er3+/TiO2, and Ni2+/TiO2 via the Taguchi orthogonal array technique. Catalysts. 6, 163–178 (2016). https://doi.org/10.3390/catal6100163
Xu, L., Wang, J.: Degradation of 4-chloro-3,5-dimethylphenol by a heterogeneous Fenton-like reaction using nanoscale zero-valent iron catalysts. Environ. Eng. Sci. 30, 294–301 (2013). https://doi.org/10.1089/ees.2012.0025
Brillas, E.: A review on the degradation of organic pollutants in waters by UV photoelectro-Fenton and solar photoelectro-Fenton. J. Braz Chem. Soc. 25, 393–417 (2014). https://doi.org/10.5935/0103-5053.20130257
Bi, W., Wang, M., Yang, X., Row, K.H.: Facile synthesis of poly (ionic liquid)-bonded magnetic nanospheres as a high-performance sorbent for the pretreatment and determination of phenolic compounds in water samples. J. Sep. Sci. 37, 1632–1639 (2014). https://doi.org/10.1002/jssc.201400142
Khori, N.K.E.M., Hadibarata, T., Elshikh, M.S., Al.-Ghamdi, A.A., Salmiati, Yusop, Z.: Triclosan removal by adsorption using activated carbon derived from waste biomass: Isotherms and kinetic studies. J. Chin. Chem. Soc. 65, 951–959 (2018). https://doi.org/10.1002/jccs.201700427
Tong, Y., Mayer, B.K., McNamara, P.J.: Triclosan adsorption using wastewater biosolids-derived biochar. Environ. Sci. 2, 761–768 (2016). https://doi.org/10.1039/C6EW00127K
Yoo, D.K., An, H.J., Khan, N.A., Hwang, G.T., Jhung, S.H.: Record-high adsorption capacities of polyaniline-derived porous carbons for the removal of personal care products from water. Chem. Eng. J. 352, 71–78 (2018). https://doi.org/10.1016/j.cej.2018.06.144
Cova, T.F., Murtinho, D., Aguado, R., Pais, A.A.C.C., Valente, A.J.M.: Cyclodextrin polymers and cyclodextrin-containing polysaccharides for water remediation. Polysaccharides. 2, 16–38 (2021). https://doi.org/10.3390/polysaccharides2010002
Mo, C., Faheem, M., Aziz, S., Jian, S., Xue, W., Yuyang, T., Shuang, D., Guangshan, Z.: Hydroxyl porous aromatic frameworks for efficient adsorption of organic micropollutants in water. RSC Adv. 10, 26335–26341 (2020). https://doi.org/10.1039/D0RA04222F
Bhadra, B.N., Yoo, D.K., Jhung, S.H.: Carbon-derived from metal-organic framework MOF-74: A remarkable adsorbent to remove a wide range of contaminants of emerging concern from water. Appl. Surf. Sci. 504, 144348–144356 (2020). https://doi.org/10.1016/j.apsusc.2019.144348
Szczepanik, B., Rędzia, N., Frydel, L., Słomkiewicz, P., Kołbus, A., Styszko, K., Dziok, T., Samojeden, B.: Synthesis and characterization of halloysite/carbon nanocomposites for enhanced NSAIDs adsorption from water. Materials. 12, 3754–3776 (2019). https://doi.org/10.3390/ma12223754
Szczepanik, B., Banaś, D., Kubala-Kukuś, A., Szary, K., Słomkiewicz, P., Rędzia, N., Frydel, L.: Surface properties of halloysite-carbon nanocomposites and their application for adsorption of paracetamol. Materials. 13, 5647–5662 (2020). https://doi.org/10.3390/ma13245647
Dou, R., Zhang, J., Chen, Y., Feng, S.: High efficiency removal of triclosan by structure-directing agent modified mesoporous MIL-53(Al). Environ. Sci. Pollut Res. Int. 24, 8778–8789 (2017). https://doi.org/10.1007/s11356-017-8583-7
Kasprzyk-Hordern, B., Dinsdale, R.M., Guwy, A.J.: The removal of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs during wastewater treatment and its impact on the quality of receiving waters. Water Res. 43, 363–380 (2009). https://doi.org/10.1016/j.watres.2008.10.047
Wick, A., Marincas, O., Moldovan, Z., Ternes, T.A.: Sorption of biocides, triazine and phenylurea herbicides, and UV-filters onto secondary sludge. Water Res. 45, 3638–3652 (2011). https://doi.org/10.1016/j.watres.2011.04.014
https://: pubchem.ncbi.nlm.nih.gov/compound/Chloroxylenol. Accessed 13 November 2022
https://: pubchem.ncbi.nlm.nih.gov/compound/2-Benzyl-4-chlorophenol. Accessed 13 November 2022
Jedynak, K., Repelewicz, M., Kurdziel, K., Wideł, D.: Removal of orange II from aqueous solutions using micro-mesoporous carbon materials: Kinetic and equilibrium studies. Desal. Water Treat. 190, 294–311 (2020). https://doi.org/10.5004/dwt.2020.25588
Jedynak, K., Charmas, B.: Preparation and characterization of physicochemical properties of spruce cone biochars activated by CO2. Materials. 14, 3859–3875 (2021)
Choma, J., Jedynak, K., Górka, J., Jaroniec, M.: Morfologia i właściwości adsorpcyjne mezoporowatych węgli otrzymywanych metodą miękkiego odwzorowania z użyciem azotanu cynku oraz nanocząstek cynku i tlenku cynku. Ochrona Środowiska. 61, 265–280 (2012)
Choma, J., Jedynak, K., Jamioła, D., Jaroniec, M.: Wpływ temperatury karbonizacji na adsorpcyjne i strukturalne właściwości mezoporowatych węgli otrzymanych metodą miękkiego odwzorowania. Ochrona Środowiska. 34, 3–8 (2012)
Kruk, M., Jaroniec, M., Gadkaree, K.P.: Nitrogen adsorption studies of novel synthetic active carbons. J. Colloid Interfac. Sci. 192, 250–256 (1997). https://doi.org/10.1006/jcis.1997.5009
Wu, H., Lu, W., Chen, Y., Zhang, P., Cheng, X.: Application of Boehm titration for the quantitative measurement of soot oxygen functional groups. Energy Fuels. 34, 7363–7372 (2020). https://doi.org/10.1021/acs.energyfuels.0c00904
Pawlicka, A., Doczekalska, B.: Determination of surface oxygen functional groups of active carbons according to the Boehm’s titration method. For. Wood Technol. 84, 11–14 (2013)
Chaouki, Z., Hadri, M., Nawdali, M., Benzina, M., Zaitan, H.: Treatment of a landfill leachate from Casablanca city by a coagulation-flocculation and adsorption process using a palm bark powder (PBP). Sci. Afr. 12, e00721–e00736 (2021). https://doi.org/10.1016/J.SCIAF.2021.E00721
Kumar, T.K.M.P., Mandlimath, T.R., Sangeetha, P., Sakthivel, P., Revathi, S.K., Kumar, S.K.A., Sahoo, S.K.: Highly efficient performance of activated carbon impregnated with ag, ZnO and Ag/ZnO nanoparticles as antimicrobial materials. RSC Adv. 5, 108034–108043 (2015). https://doi.org/10.1039/C5RA19945J
Jiao, Y., Han, D., Lu, Y., Rong, Y., Fang, L., Liu, Y., Han, R.: Characterization of pine-sawdust pyrolyic char activated by phosphoric acid through microwave irradiation and adsorption property toward CDNB in batch mode. Desal. Water Treat. 77, 247–255 (2017). https://doi.org/10.5004/dwt.2017.20780
Szczepanik, B., Słomkiewicz, P., Garnuszek, M., Czech, K.: Adsorption of chloroanilines from aqueous solutions on the modified halloysite. Appl. Clay Sci. 101, 260–264 (2014). https://doi.org/10.1016/j.clay.2014.08.013
Słomkiewicz, P.M., Szczepanik, B., Garnuszek, M., Rogala, P., Witkiewicz, Z.: Determination of adsorption equations for chloro derivatives of aniline on halloysite adsorbents using inverse liquid chromatography. J. AOAC Int. 100, 1715–1726 (2017). https://doi.org/10.5740/jaoacint.17-0237
Słomkiewicz, P.M.: Wyznaczanie stałych równowagi adsorpcji metodą inwersyjnej chromatografii gazowej. Aparatura Badawcza i Dydaktyczna. 9, 109–117 (2004)
Słomkiewicz, P.M.: Zastosowanie Chromatografii Gazowej w Badaniach Kinetyki Katalitycznej Syntezy Eterów z Alkenów i Alkoholi. Wydawnictwo Akademii Świętokrzyskiej, Kielce, Poland (2007)
Szczepanik, B., Słomkiewicz, P., Garnuszek, M., Czech, K., Banaś, D., Kubala-Kukuś, A., Stabrawa, I.: The effect of chemical modification on the physico-chemical characteristics of halloysite: FTIR, XRF, and XRD studies. J. Mol. Struct. 1084, 16–22 (2015). https://doi.org/10.1016/j.molstruc.2014.12.008
Húmpola, P., Odetti, H., Moreno-Piraján, J.C., Giraldo, L.: Activated carbons obtained from agro-industrial waste: Textural analysis and adsorption environmental pollutants. Adsorption. 22, 23–31 (2016). https://doi.org/10.1007/s10450-015-9728-y
Haghtalab, A., Nabipoor, M., Farzad, S.: Kinetic modeling of the Fischer-Tropsch synthesis in a slurry phase bubble column reactor using Langmuir-Freundlich isotherm. Fuel Process. Technol. 104, 73–79 (2012). https://doi.org/10.1016/j.fuproc.2011.07.005
Funding
This work was supported by Ministry of Science and Higher Education, Poland (research project SUPB.RN.21.188). Laura Frydel acknowledges financial support through the project „AKCELERATOR ROZWOJU Uniwersytetu Jana Kochanowskiego w Kielcach” (Development Accelerator of the Jan Kochanowski University of Kielce), co-financed by the European Union under the European Social Fund, with no. POWR.03.05.00-00-Z212/18.”
Author information
Authors and Affiliations
Contributions
LF performed the experiments, designed the conception, wrote the manuscript and discussed the results; PS made ILC calculation and participated in the discussion of results; BS discussed the results and participated in the preparation and final revision of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
Nothing to declare.
Ethical approval.
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Frydel, L., Słomkiewicz, P.M. & Szczepanik, B. The adsorption studies of phenol derivatives on halloysite-carbon adsorbents by inverse liquid chromatography. Adsorption 30, 185–199 (2024). https://doi.org/10.1007/s10450-023-00396-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-023-00396-w