
Abstract
On-surface synthesis of C–C covalent low-dimensional nanomaterials is a promising method of obtaining structures with tailored and novel physicochemical and electric properties. In this contribution, the Monte Carlo simulation approach was proposed to predict the topology of metal–organic (MO) intermediates formed in the Ullmann homocoupling of halogenated isomers of tetracene. The coarse-grained model of polyaromatic hydrocarbons (PAH) haloderivatives and divalent copper adatoms on a metallic crystal surface (111) was used, where locations of substituents in the molecules were encoded as active centres with directional C–Cu interactions. The computations were performed for various structural isomers of tetracene, from disubstituted to tetrasubstituted units. As a result, diverse superstructures were obtained, such as dimers, trimers, and other oligomers, chains and ladders, and metal–organic (MO) networks, both chiral and achiral. Additionally, for the prochiral linkers, simulations of the racemic mixtures were performed. Our study provided useful insight into the influence of substituents’ position and the carbon backbone’s size on the topology of the modelled precursor architectures.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The bottom-up approach to nanomaterials synthesis provides a tailorable and practical pathway for the production of customized novel polymers [1,2,3,4,5]. One of the most promising methods of block-by-block synthesis is the on-surface transformation of organic molecules, which not only provides polymerization routes unattainable in other techniques but also allows for direct imaging of the reaction products [6,7,8,9,10]. Adsorption of monomers on a catalytic surface decreases their mobility, increasing the chance for linkage of molecules which under normal, bulk conditions would not connect. The reduced number of molecular degrees of freedom facilitates bond formation between adsorbed species and helps the monomers achieve specific relative configurations. Because of these beneficial features, the on-surface synthesis can be effectively used in areas such as the synthesis of low-dimensional polymers, gas storage, electronics, nano-optics, catalysis, chemical sensing, and material engineering [9,10,11].
Among many on-surface covalent synthesis methods, the on-surface Ullmann coupling exhibits especially utile attributes [10, 12,13,14,15]. This process is a modification of the conventional in-solution Ullmann aryl-aryl reaction. In surface-assisted coupling, halogenated aromatic compounds (aryl halides) are adsorbed on a catalytic metallic surface—most commonly Au(111), Ag(111), Cu(111)—and usually heated or exposed to light [16]. Under these conditions, C–X bonds dissociate, creating highly reactive aryl radicals. When the radicals start to combine, covalently bonded carbon superstructures emerge and grow. In some instances, the covalent bonding is preceded by the formation of metal–organic (MO) intermediates via self-assembly [17,18,19]. C–Cu bonds in such MO precursors are weak and labile enough to be broken and rearranged during the reaction, which allows for optimization of morphology, self-healing, and error correction of defects. The arrangement of linkers in the intermediate MO constructs is usually preserved in the final product, as at elevated temperatures, metal–organic bonds are directly converted into covalent carbon–carbon ones. Understanding and predicting the formation of metal–organic intermediates is, therefore, an important objective in the designing and fabrication of organic polymers with unique properties.
In the development of novel low-dimensional organic polymers, extensive theoretical and experimental studies of surface-confined polymerization have been conducted for various monomers [6,7,8,9, 11]. Those investigations focused mainly on the coupling of terphenyl derivatives [20,21,22], PAHs (polyaromatic hydrocarbons) [6, 7, 12, 13, 23], triazines [24,25,26], and other compounds [27,28,29] on catalytic Ag, Au and Cu surfaces under UHV (ultra-high vacuum) conditions. The results of the cited examples proved that it is possible to control and optimize the self-assembly of the MO intermediates and to tune the properties of the final polymer via smart modification of the intrinsic properties of the monomers at play. For example, changes in the halogenation pattern of the organic linkers resulted in a wide array of ordered structures, from chains, ladders, and ribbons [11,12,13, 30, 31] through oligomers [32, 33] to porous networks [14, 25, 26, 34].
Although the advancement in nanoscale imaging techniques such as scanning tunnelling microscopy (STM) has enabled the determination of the pathways of polymerization reactions running on surfaces [11,12,13, 25, 26, 30,31,32,33,34], selection of the optimal monomeric building block has still been problematic in certain cases. This refers especially to the number and intramolecular distribution of halogen substituents in PAHs monomers, aiming at the synthesis of the polymer with predefined architecture. To facilitate the optimization of the monomeric units, computer-aided methods like quantum mechanical (QM) modelling, molecular dynamics (MD), and Monte Carlo (MC) simulations have often been applied [35,36,37,38,39,40,41,42,43,44,45]. While QM calculations have been used mainly to determine energetics and mechanisms of intermolecular bonding [35,36,37,38], the other methods have been much less widespread, and they have drawn considerable attention quite recently, in spite of better scaling properties and being relatively less complicated [39,40,41,42,43,44,45,46].
Among diverse computational chemistry tools, the coarse-grained MC simulations turned out to be an effective way of predicting the architecture of metal–organic intermediates, as confirmed by the corresponding experimental data—obtained, for example, with STM imaging of adsorbed overlayers [11,12,13,14]. Those results proved that the simplified coarse-grained modelling of aromatic adparticles (naphthalene, anthracene and phenanthrene) [47,48,49,50,51] and metal adatoms on solid substrates is a suitable and sufficiently accurate representation of the real systems enabling correct reproduction of complex 2D molecular patterns. Accordingly, in this contribution, we continue our research on the Ullmann coupling of halogenated PAHs and focus on the isomers of the longer linear aromatic hydrocarbon comprising four fused benzene rings, tetracene (t) [47,48,49,50,51]. As was observed in the previous studies, the main structural features of the obtained metal–organic assemblies were highly dependent on the number and position of halogen substituents in the PAH building blocks. To examine how these factors affect structure formation in the tetracene-based adsorbed systems, herein, we model the on-surface self-assembly of differently halogenated isomers of t and bivalent metal atoms on a (111) crystalline surface. The key objective of the present study is to identify groups of isomorphic self-assembled structures resulting from a given type of halogenation.
2 The model and simulations
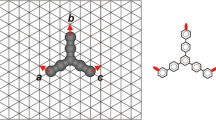
The MC model adopted in the present study was based on our earlier research on the substituted PAHs, in which the coarse-graining method was used for the smaller PAH units [47,48,49,50,51]. The tetracene molecules were treated as rigid linear rods consisting of four connected segments representing aromatic rings. The metallic (111) surface was designed as a collection of equivalent adsorption sites arranged in a triangular lattice with spacing equal to 1. The geometry of the lattice corresponded to the structure of a (111) crystalline surface of typical catalytically active metals used in the Ullmann coupling reaction (copper, silver or gold). Each phenyl ring of t was able to occupy one adsorption site. The intersegment distance was set to \(\sqrt{3}\), so that the segments of an adsorbed molecule occupied the next nearest vertices of the lattice. To prevent the molecules from overlapping, steric hindrance conditions were additionally imposed; that is, the next nearest-neighbour sites surrounding the segments of a given t unit were not accessible to foreign molecular segments. Metal adatoms were represented by single segments. Figure 1. portrays the assumptions described above and the set of interactions used in the model.

A visual representation of the model used in simulations (see text for details); as an example, 1,3,6,11-tetra substituted tetracene was shown. Purple arrows determine the directions of the metal-mediated intermolecular interactions corresponding to the selected substitution locations, numbered from 1 to 12. Red, blue and green arrows denote possible colinear (same colour) linker-metal-linker interactions. Both surface enantiomers of the tetracene derivative (R and S) are depicted
In the proposed model, only the metal–ligand interactions and simple volume-exclusion conditions were assumed for simplicity. The directional interactions originating from active centres of the t-linker corresponded to the distribution of halogen atoms in a given molecule. The naming convention, consistent with the IUPAC nomenclature, was adopted to describe the substitutional isomers used in the simulations. [52] The molecules of tetracene were denoted as tabcd, where t is an abbreviation of “tetracene” and a…d stand for the halogen locants. For two-digit locants, the underscore was used for clarity. The range of linker-metal interaction was limited to the nearest-neighbour sites of the virtual carbon atoms forming benzene rings of t (numbered 1- 12 in Fig. 1). In other words, the metal-linker interaction was possible only for the specific distance equal to two lattice spacings, measuring form the centre of a given molecular segment (i.e. from the underlying lattice vertex). Metal atoms were allowed to create only single (with energy \(\varepsilon\)) and double connections with the linker. In the latter case, only straight metal-linker-metal (i.e. → ● ←) configurations were considered, contributing with \(2\varepsilon\) to the energy of the adsorbed phase. The energy of non-linear twofold 120º nodes was assumed to be \(\varepsilon\), reflecting steric repulsion between the contributing molecules. Moreover, for convenience, the energy of interaction between a molecule of t and the surface was set equal to zero since the surface was mapped as energetically homogenous.
The simulations were conducted in the canonical ensemble for the constant: lattice area (rhomboid fragment with side L = 200 sites), number of particles N (including \({N}_{l}\) linkers and \({N}_{m}\) atoms), and temperature, T [53, 54]. The conventional Metropolis sampling algorithm was implemented in the simulation algorithms [53]. Periodic boundary conditions were imposed in both planar directions to eliminate finite-size effects. The starting point of our simulations was the lattice with randomly distributed \(N = {N}_{l}+ {N}_{m}\) molecules and metal atoms. The values of \({N}_{l}\) and \({N}_{m}\) were chosen in a way such that the assumed surface coverage, \(\theta =\frac{{4{N}_{l} + N}_{m}}{{L}^{2}}\) segments/site, was reached. To equilibrate the adsorbed phase state, a series of trial MC moves were made, in which the position and orientation of the components were randomly changed. First, a linker molecule or metal atom was picked at random. Then for this component, the potential energy \({U}_{o}\), to which all of the reckoned directional metal-linker interactions contributed according to the adopted restrictions (see Fig. 1), was calculated for the actual (old) state. Specifically, the immediate surrounding of two neighbouring adsorption sites was considered for local energy calculation, where for a chosen particle, the program performed a search in the main directions of the lattice for compatible directional interactions. Next, the selected linker or metal atom was translated to a new random position. In the case of a linker, the molecule was additionally rotated randomly by a multiple of 60 degrees. If the newly chosen lattice sites were already occupied, the adsorbed configuration was set back to the original state. In the opposite case, the potential energy, \({U}_{n}\), was computed in the new position, analogously to \({U}_{o}\). These two energies were used to determine the acceptance probability \(\mathrm{p }=\mathrm{ min}[1,\mathrm{ exp}(-\frac{{U}_{n}-{U}_{o}}{\mathrm{kT}})]\), where \(k\) stands for the Boltzmann constant. The calculated value of p was compared with a pseudorandom number, r lying between 0 and 1. For \(p > r,\) the new configuration was accepted; otherwise, the trial move was abandoned, and the system was reset to the old configuration. Remind that in our model, the energy factor \({U}_{n}-{U}_{o}\) Thus, the acceptance probability was solely dependent on the adsorbate–adsorbate interactions, as the surface was energetically homogeneous. The above procedure constitutes one MC step, which was repeated multiple times (typically, \(N \times {10}^{6}\)) at a given temperature. To ensure thermodynamic control over the modelled systems, the adsorbed monolayers were subjected to a temperature gradient, where the temperature was lowered from \({T}_{start} = 0.51\) to \({T}_{fin} = 0.01\) in 500 equal steps. The energies and temperatures used in the simulations were expressed in units of \(\varepsilon\) and \(\left|\varepsilon \right|/k\), respectively. Specifically, the total energy of the adsorbed phase was calculated by reckoning metal atoms connected with zero, one, and two linker molecules in both 120º and linear nodes, with contributions 0, ε, ε and 2ε, respectively. The last ten per cent of the total number of MC steps was used to calculate average values at a given temperature, including the degree of metal coordination, average potential energy, heat capacity and so forth. These characteristics were presented as averages over ten independent systems with the same macroscopic parameters (θ, L and T). A consistent colouring scheme was used in the graphical representations of the modelled systems—orange for copper atoms, grey for the enantiomer R and blue for the enantiomer S of a given halogenated derivative of t. In the simulations, we always used 400 linker molecules (\({N}_{l}\)) mixed with the stoichiometric number of metal atoms, \({N}_{l}\), which was dependent on the number of halogens in a given isomer. These calculations were performed for the interaction energy ε equal to -1. Ordered phases observed in the simulations were characterized by the density, d, which was defined as the average number of adsorbed segments per unit cell area with a linear size of the cell expressed in lattice constant units.
3 Results and discussion
Because of the increased number of carbon atoms and thus locants as compared to shorter acenes, halogenated tetracenes make a large group of structural isomers. Moreover, some of the considered tectons exhibit prochirality—when confined on a plane, they become chiral and can occur in the mirror-image configurations R and S (see Fig. 1). For these reasons, we selected only a few most interesting cases and when applicable (prochiral linkers), modelled both enantiopure (only R) and racemic systems (R + S). The choice of racemic composition in the calculations was motivated by the naturally occurring unbiased adsorption of prochiral molecules on achiral surfaces, resulting in equal chances for adsorption in configurations S and R. On the other hand, the enantiopure overlayers were studied to examine whether the single enantiomers are able to create structures which are significantly different from their racemic counterparts. Since experimental studies of enantiopure systems comprising prochiral molecules would require additional functionalization to force these units to adopt one adsorption configuration (R or S), the results reported herein can be helpful in the preliminary selection of molecular platforms with the largest potential of creating new interesting homochiral architectures and, thus, covalent polymers.
3.1 Disubstituted tetracene derivatives
As a rule, to be able to form an extended polymeric structure, a monomer must contain at least two reactive centres. The same is true for the considered tetracene derivatives. In most cases, for the dihalogenated isomers, elongation of the backbone did not change the topology of the obtained structures, as compared to the shorter acenes, including naphthalene (n) and anthracene (a) [47, 49]. Analogous halogenation patterns in the molecules of n, a and t resulted in the precursor aggregates, which were very similar in terms of spatial molecular organization and connectivity. Selected examples of these connections, calculated for the achiral and prochiral tetracene tectons, are shown in Fig. 1.
In this Fig. 2, the final snapshots of the t17, t5_11, t1_10, and t29 systems were presented, both enantiopure and racemic in the case of the first two prochiral isomers. Starting with the achiral tectons first, let us focus on t1_10. This isomer, having two active centres providing parallel interaction directions on the same side of the backbone, was not surprisingly found to create only “sandwich” dimers, where each molecule and metal atom were fully coordinated. The other possible way of binding, that is in chains, was only scarcely observed, being rather a defect than a trace of competing phase. The primary source of this result was the energetically optimal, strong linkage of a pair of complementary molecular units, such that the quasi-cyclic dimers were formed with no undercoordinated elements. The self-compatibility effect of these linkers can also be noticed in the dependencies presented in Fig. 3 and Fig. S1 of the Supporting Info—showing the coordination of metal atoms and the net system energy as functions of temperature, respectively. When comparing the creation of bonds by t1_10 to the other disubstituted isomers, it can be seen in Fig. 3 that the peak corresponding to monocoordinated metal atoms (blue curve) is lower (max. 0.35), and its maximum occurs at the higher temperature (0.25), as compared to the remaining dihalogenated isomers of t. Moreover, for t1_10, the curve plotted for dicoordinated metal atoms is steeper and shifted towards higher temperatures. These observations indicate that the sandwich dimers are created yet at higher temperatures and are highly stabilized when the temperature drops. Specifically, for the remaining tectons, the dominant form are the labile chains which can easily dissociate terminal metal atoms or molecules at moderate temperatures. When the temperature decreases, this process becomes gradually limited, so the chains and closed ring-type structures (t17, t5_11, t29) solidify. The mechanism of facilitated formation of the sandwich t1_10 dimers is also supported by Fig. S1, where the corresponding energy curve reaches a plateau at a temperature higher than for the other isomers.

Adsorbed structures created by the molecules of t17, t5_11 (enantiopure: 400 R and racemic: 200 R + 200 S), t1_10, and t29 (400 R) mixed with 400 metal atoms. The insets show characteristic motifs of the simulated assemblies. L = 200, θ = 0.05, T = 0.01

Effect of temperature on the fraction of 0-, 1- and 2-coordinated metal atoms, calculated for the systems comprising 400 molecules of t17, t5_11, t1_10, and t29 mixed with 400 metal atoms. Results calculated for the racemic mixtures (200 R + 200 S) are denoted with dashed lines. L = 200, θ = 0.05
The second considered achiral isomer is t29, with two halogens placed, similarly to t1_10, on the opposite ends of the backbone but forming an angle of 120°. In the course of the simulation, this tecton started to form large cyclic hexamers and less abundant long chains. Although the looped forms (hexameric and larger) are energetically more stable, since there are no uncoordinated (terminal) linkers and metal atoms, they occupy a considerable part of the surface. When sufficiently abundant, these forms prevent the closing of the remaining chains so that they stay open, meandering between the looped forms. The shape of those chain oligomers varies depending on the (random) sequence of nodal motifs, each of which can be either straight or 120° bent. As depicted in Fig. 3, the effective coordination of metal atoms in the overlayers comprising t29 requires a stronger lowering of the temperature, as compared to t1_10. This can be seen in the fraction of metal atoms connected to one linker (blue curve) peaking at a temperature of about 0.17 (vs 0.22 for t1_10). At this point, the chains start to close and create cyclic forms, which result in a rapid increase in the number of fully coordinated metal atoms (red curve, coordination 2). As can be seen in Fig. S1, at low temperatures, the corresponding energy curve reaches the plateau, which is placed slightly below the maximal value of 2. This effect comes directly from the quite numerous presence of monocoordinated terminal molecules in the winded metal–organic chains.
Moving from the achiral tetracene derivatives to prochiral ones, t17 and t5_11, the corresponding results are presented in the top part of Fig. 2. These snapshots demonstrate the influence of the distance between halogen substituents attached on the opposite sides of the backbone of t, on the topology of the self-assembled constructs. Both isomers used in the calculations were centrosymmetric, with the substituted positions located in t17 at the first and fourth rings, and in t5_11—at the second and third rings. In both cases, the assigned interaction directions were antiparallel to each other. Because of these similarities, the self-assembly in the corresponding enantiopure (R) systems produced comparable results, creating long, linear chains where the contributing enantiomers were oriented in parallel. The resulting chains were aligned in one direction due to the blocked growth in the remaining directions (with a potential crossing of the chains). To quantify molecular alignment in the obtained structures, we calculated the associated order parameter, δ (see the Supporting Info), which is equal to 1 for entirely uniformly oriented molecules and takes values close to zero when the possible orientations (here, three) are equally populated. As presented in Fig. 4, the order parameter for those systems at low temperatures is close to 1, which confirms the previous observations. For the corresponding racemic mixtures, the obtained structures were also chains but winded and bent. Since there were no steric or energetic constraints imposed on the bienantiomeric motifs, the units R and S were free to mix and create any sequence. As shown in Fig. 2, the predicted structures comprise randomly distributed R–S connections as well as longer enantiopure fragments. The less stepped shape of the chain formed by rac-t5_11 originates from the position of the active centres in R (and in S), which are less distant in comparison to t17. Despite this difference, the linkers in both chains are still oriented in one primary way, as confirmed in Fig. 4. The order parameter plotted therein for enantiopure and racemic overlayers of t5_11 and t17 reaches values close to 1 at low temperatures.

Effect of temperature on the order parameter calculated for the enantiopure (black lines) and racemic (blue lines) systems comprising t17 and t5_11. In the simulations, 400 molecules of R and 200 R + 200 S (racemates) mixed with 400 metal atoms were used for each isomer. θ = 0.05
3.2 Trisubstituted tetracene derivatives
Addition of a third active site to the backbone of t vastly altered topology of the modelled systems and routes of their self-assembly. To illustrate this, we selected five tetracene derivatives which created qualitatively different structures with the most interesting properties. Let us remind that among the trihalogenated isomers of t, there are no achiral linkers, so in the following, both enantiopure and racemic overlayers are discussed in each case.
Considering the enantiopure snapshots presented in Fig. 5, the molecules of t129(R) and t29_11(R) create similar ribbon-like superstructures consisting of six-membered rounded units connected by two tetracene molecules. The observed similarities can be explained by taking into account the analogous structure of these enantiomers, in which each pair out of the three interaction directions creates an angle of 60°. Moreover, both terminal rings of t129 and t29_11 are functionalized. The only difference between these units is the location of the active middle centre, being moved from the first benzene ring (t129) to the second one (t29_11). The comparison of the energy curves (Fig. S3), Voronoi diagrams (Fig. S4), and coordination curves (Fig. 6, top) calculated for these systems confirms that the corresponding structural features and self-assembly scenarios exhibit numerous similarities. However, the assemblies formed in the racemic overlayers of the two considered isomers are diametrally different. The mixed enantiomers of t129 produced closed hexagonal rings with two additional linkers inclused in each ring. Compared to t29, the rings formed by t129 are more sterically stable since more molecules are packed in one ring, so chances for chain growth are reduced (most of the molecules are consumed by the rings). This increased stability comes from the energetic effects associated with the triple coordination of each linker in the ring of t129. Specifically, such coordination results in the average energy per linker reaching its maximum value of 3 at low temperatures, as it can be seen in Fig. S3. For the achiral tecton t29, the maximum energy (2) was not achieved due to the already mentioned more numerous presence of chains in the overlayer (see Fig. S1). The local mixing in the rings of t129 was racemic, with every aggregate consisting of 4R and 4S enantiomers. In a single ring oligomer of this tecton, one linker molecule interacts via a metal atom with one linker of the same chirality and with two mirror-image linkers. Accordingly, the fraction of R-S nodes in the rac-t129 overlayer should be around 0.67. To illustrate this, in the bottom part of Fig. 6, the fraction of metal atoms bonded homochirally (R-R or S–S) and heterochirally (R-S) was presented. The curve plotted for a fraction of R-S bonds in the t129 system reaches a plateau of about 0.63, which is in close agreement with the theoretical value. The slightly lower value obtained from the simulations can be associated with structural defects, such as the distorted, not fully closed, rings from Fig. 5. In the case of the isomer t29_11, the racemic overlayer comprises highly defected, fragmented ribbons with the basic rounded structural units, analogous to those occurring in the corresponding enantiopure system. As illustrated in Fig. 5, the linkage of R and S in the ordered rounded units is rather limited, resulting in the recreation of the homochiral fragments observed already for t29_11(R). The fraction of R-S nodal connections calculated for this racemate confirms this effect, as at low temperatures it reaches about 0.3, indicating partial demixing of the enantiomers.

Adsorbed structures created by the molecules of t129 and t29_11 (enantiopure: 400 R, racemic: 200 R + 200 S) mixed with 600 metal atoms. The insets show characteristic motifs of the simulated assemblies. L = 200, θ = 0.05, T = 0.01

Top: Influence of temperature on the fraction of 0-, 1- and 2-coordinated metal atoms calculated for the systems comprising 400 molecules of t129 and t29_11 (enantiopure: 400 R, racemic: 200 R + 200 S) and 600 metal atoms. θ = 0.055. Data obtained for the racemic mixtures are plotted with dashed lines. Bottom: Effect of temperature on the fraction of metal atoms dicoordinated homochirally (R–R or S–S) and heterochirally (R–S) metal atoms obtained for rac-t129 and rac-t29_11
Figure 7 presents the results of the self-assembly of the three remaining isomers: t127, t138, and t1_10_11 in both enantiopure and racemic modes. In the case of the first building block, the enantiomer R created a partially ordered network with periodic fragments. This partial ordering is easier to notice using the Voronoi diagram shown in Fig. S6. The regular porous network with a rhombic unit cell (cell side of length 26) comprises void spaces delimited by 12 linker molecules. Due to the large area of these pores and, consequently, very low density of the corresponding porous phase—equal to 0.056, the network cannot propagate freely over the entire surface under the parameters assumed in the calculations. Because of that, smaller irregular pores made of 9 and 10 linkers were formed in the superstructure, disturbing the periodicity of the network. When the other enantiomer of t127, S, was introduced to the adsorbed overlayer, the network architecture broke up completely, transforming into an irregular collection of six-membered tripod-shaped motifs with alternating R/S sequences connected in diverse ways (see the inset). Even though the presence of t127(S) led to the disordering of the racemic system, the temperature dependencies of the associated energetic and structural parameters were nearly identical as in the case of the enantiopure t127 overlayers (see Fig. S5 and Fig. 8).

Adsorbed structures created by the molecules of t127, t138 and t1_10_11 (enantiopure: 400 R, racemic: 200 R + 200 S) mixed with 600 metal atoms. The insets show characteristic motifs of the simulated assemblies. L = 200, θ = 0.055, T = 0.01

Effect of temperature on the fraction of 0-, 1- and 2-coordinated metal atoms calculated for the systems comprising 400 molecules of t127, t138, and t1_10_11 (enantiopure: 400 R, racemic: 200 R + 200 S) mixed with 600 metal atoms. The results obtained for the racemic mixtures are plotted with dashed lines. θ = 0.055
In the case of the next tecton, t138, the simulations predicted the aperiodic porous networks shown in Fig. 7. These irregular networks were formed in the enantiopure as well as in the racemic overlayers and consisted of regular hexagonal pores mixed with numerous deformed pores. The obtained results are comparable to the networks created by the smaller PAH derivatives with an analogous arrangement of the active centres (n136 and a136). As seen in Fig. 7, the presence of the S enantiomer induces additional structural heterogeneity in the network, introducing pores with new shapes. Although the racemic superstructure comprises a series of pores that were not present in the enantiopure system, the energy plots for t138 and rac-t138 are nearly identical (see Fig. S5). The same is true for the corresponding coordination plots (Fig. 8), highlighting similar mechanisms of the enantiopure and racemic self-assembly of this building block.
The simulations run for the last of discussed tectons, t1_10_11, resulted in the creation of ladders with zipper-like antiparallel linker configuration. Basic structural units in these ladders, with two parallel linkers and two metal atoms, are stabilized by two linker-metal-linker interactions and these motifs are further connected via a single metal to form the ladders. The low-energy connection within each unit (\(2\times 2\varepsilon\)) causes the shortening of the emerging structures and an abundance of small, often defective oligomers. Specifically, since the interaction in the basic units is twice as strong as between them, erroneous structures are hard to repair, especially at low temperatures. This effect is confirmed by the coordination curves calculated for t1_10_11(R), as shown in Fig. 8. In this figure, we can observe that the fraction of monocoordinated metal atoms reaches a maximum at a temperature that is higher in comparison to other trisubstituted acenes and also that at elevated temperatures the complete (twofold) coordination of the metal atoms is more effective for this isomer. Despite these initial effects, further formation of ladders, when T decreases, is inhibited: the corresponding curves are flattened, and a noticeable bump at T = 0.14 can be observed on the plot of the fraction of monocoordinated metal atoms. This result comes directly from the formation of dimeric R-R units in which pairs of metal atoms can be entrapped, each metal creating one bond with one contributing molecule, as schematically shown in the inset to Fig. 8. The above effect is also illustrated with the corresponding snapshots of the adsorbed overlayer of taken t1_10_11(R), included in the Supporting Information (Fig. S7).
The presence of the enantiomer t1_10_11(S) completely changed the morphology of the adsorbed overlayer. When in the racemate, the linkers were found to be no longer able to create extended structures, and they preferred binding into R-S dimers instead. Like for the achiral t1_10, the enantiomers of t1_10_11 are highly compatible with each other thanks to the three pairs of collinear interaction directions. Because of this compatibility, the energy of the racemic systems reaches a plateau at high temperatures (Fig. S5), and all of the metal atoms become fully coordinated quite quickly (Fig. 8). In consequence, at the stoichiometric metal-linker proportion used here, the racemic overlayer of t1_10_11 contains only the R-S dimers with uniform internal structure.
3.3 Tetrasubstituted tetracene derivatives
The most complex molecules studied in this work are the tetrahalogenated tetracenes. The abundance of possible isomers and the ability to create periodic networks make these building blocks especially promising materials for 2D covalent polymers. To explore these beneficial features, we selected eleven representative examples of tetracene-based monomers. In Fig. 9, we presented low-temperature snapshots of the structures created by the four enantiomers R of t1238, t1239, t1247 and t1249, together with the results obtained for the corresponding racemic mixtures.

Adsorbed structures created by 400 molecules of t1238, t1239, t1247, and t1249 (enantiopure: 400 R, racemic: 200 R + 200 S) mixed with 800 metal atoms. The insets show characteristic motifs of the simulated assemblies. L = 200, θ = 0.06, T = 0.01
Starting with t1238(R), our simulations predicted the homochiral porous network comprising hexagonal star-shaped pores, each surrounded by six smaller, parallelogram void spaces. The arrangement of the linkers on the surface created chamfered hexagonal tiling. The simulated heteroporous phase was characterized by a rhombic unit cell with a side equal to √337 and a density of 0.103. The presence of the other enantiomer of t1238 vastly disturbed the structure formation, resulting in irregular superstructures with randomly distributed enantiomers. The outcome of the self-assembly of the following linker, t1239, was a highly regular pattern with only one kind of triangular void spaces. In this case, the obtained superstructure resembled 36 triangular tessellation. The network has a density of 0.118 and a rhomboid unit cell with a side smaller than for t1238, i.e., 7√3. Despite different topologies of the enantiopure ordered porous structures formed by t1238 and t1239, the result of the racemic self-assembly of the latter molecule (distorted aggregates with numerous defects) was similar to that of the first one, highlighting the disorganizing role of the corresponding mirror-image conformers.
In the case of t1247(R), the calculations resulted in the homochiral porous network with six-armed star-shaped pores. This periodic heteroporous pattern, resembled the (4.6*π/6)3 star tiling, with each star-shaped void space surrounded by six smaller, rectangular ones. The unit cell of the resulting architecture was rhombic, and its side measured 19. The density of the aforementioned phase was equal to 0.096. Regarding the racemic mixture t1247, the presence of the enantiomer S largely disrupted the morphology of the adsorbed overlayer, like for the previous racemates. However, a repeating motif of sandwich dimers, often connected into triangular structures with internal hexagonal void space can be observed in this case, as noticeable in the corresponding inset to Fig. 9. In the case of the pure isomer t1249(R), two polymorphic structures were observed in the simulations. These distinct phases, denoted here as A and B, differed significantly in topology and density. Specifically, network A is a homoporous, brickwall and dense (d = 0.144) structure characterized by an elongated rectangular unit cell with sides lengths equal to 10√3 and 4. The other possible phase, B, contains void spaces of three types—the largest, hexagonal, surrounded by six linkers; medium, trigonal, generated by three linkers; and the smallest, rectangular, created only by two linkers. These pores are arranged in a wavy double-layered kagome-like structure which has a density of 0.115, and a rhombic unit cell with side 2√151. Although any of the aforementioned networks were not observed in racemic systems of t1249, the enantiomers R and S combined into alternative plait architectures richer in one of these components. In spite of the regular structure, these aggregates contain numerous undercoordinated linker molecules and metal atoms, as can be seen in Fig. 10 and Fig. S8. In fact, for every tetrasubstituted isomer mentioned in this paragraph, the corresponding coordination and energy curves indicate that ordering in the R/S systems is hindered, and it results in many defects. In particular, any of the twofold metal coordination curves plotted in Fig. 10 (red dashed lines) do not reach the maximum value of 1 at low temperatures.

Effect of temperature on the fraction of 0-, 1- and 2-coordinated metal atoms calculated for the systems comprising 400 molecules of t1238, t1239, t1247, and t1249 mixed with 800 metal atoms. The results obtained for the racemic mixtures are plotted with dashed lines. L = 200, θ = 0.06
In Fig. 11, we presented another four examples of molecular units able to self-assemble into ordered structures with interesting topologies, that is, t139_12, t1357, t189_11, and t2358. The first molecule is the only tetrasubstituted tetracene isomer of this work which did not create a network when adsorbed as a single enantiomer. Instead, it formed ordered ladders with relatively big rhombic pores. As presented in Figs. 12 and S10, the formation of chains is slower than in the case of networks. The results obtained for the racemate of t139_12 are exceptional among the studied systems, as for this tecton, the complete chiral resolution was observed, as seen in Fig. 11. The main reason for this effect is the shape incompatibility of the enantiomers R and S which cannot connect to form any extended structure. The chiral separation predicted for rac-t139_12 can also be seen in the results presented in Fig. 13, where the fraction of metal dicoordinated homo- and heterochirally was plotted against temperature. Specifically, when T drops below 0.2, the fraction of R-S tends to zero, which is markedly different from all of the corresponding results of this study. On the other hand, this behaviour was observed in our previous works [47,48,49,50,51] on the smaller PAH derivatives.

Adsorbed structures created by 400 molecules of t139_12, t1357, t189_11 and t2358 mixed with 800 metal atoms (enantiopure: 400 R, racemic: 200 R + 200 S) mixed with 800 metal atoms. The insets show characteristic motifs of the simulated assemblies. L = 200, θ = 0.06, T = 0.01

Effect of temperature on the fraction of 0-, 1- and 2-coordinated metal atoms calculated for the systems comprising 400 molecules of t139_12, t1357, t189_11, and t2358 (enantiopure: 400 R, racemic: 200 R + 200 S) mixed with 800 metal atoms. The results obtained for the racemic mixtures are plotted with dashed lines. θ = 0.06

Effect of temperature on the fraction of metal atoms dicoordinated homochirally (R-R or S–S) and heterochirally (R–S) obtained for the racemic systems of t139_12, t1357, t189_11 and t2358. θ = 0.06
In the case of the next isomer, t1357, the simulations of the enantiopure overlayer resulted in the hexagonal hierarchical network. Two main types of pores—hexagonal and C3-symmetric fan-shaped—can be identified in this ordered superstructure which is characterized by the density d = 0.135 and rhombic unit cell with a side equal to 16. Easily noticeable structural elements of the network are the pairs of molecules (dimers) longitudinally shifted relative to each other. Regarding the racemic self-assembly of t1357, the network architecture was preserved, but the periodicity was partially lost. In this case, the homochiral dimers R-R and S–S these units were randomly mixed, but interestingly, they were able to support the periodic pattern of hexagonal pores. On the other hand, the formation of the R–S dimers was highly unfavourable, as shown in Fig. 13. As it can be seen therein, the fraction of R-metal-S nodes calculated for this racemate stabilizes at about 0.23 when the temperature goes to zero. This result is in line with simple statistical predictions assuming that a single enantiomer (either R or S) always creates two metal-mediated bonds with like enantiomer (homochiral dimers), and the two remaining bonds have a random nature, involving zero, one or two, unlike enantiomers. This calculation gives a 25% chance for the occurrence of the R-metal-S link between a single enantiomer and its neighbourhood.
The outcome of the calculations run for the enantiopure overlayer of t189_11 (R) was the hexagonal network with large pores and a low density of 0.077. This superstructure was characterized by a rhombic unit cell with a side measuring 8√7 lattice spacings. For the corresponding racemate, we observed drastic changes in the morphology of the mixed overlayer. Precisely, the adsorbed enantiomers created mainly tetrameric structures with a 1:1 R-S ratio and sparse larger forms usually comprising two such interconnected tetramers. As a single racemic tetramer of t189_11 contains seven metal atoms, out of which six are coordinated heterochirally, the fraction of these metal atoms should be high and equal to about 0.87. According to Fig. 13, in this case, the corresponding curve reaches a plateau of 0.73 only. The lower-than-expected fraction of homochirally bonded metal atoms can be attributed to the presence of numerous monocoordinated at the edges of the tetramers as well as to the less abundant metal atoms which mediate bonding of the tetramers via homochiral interactions (see the inset to the corresponding panel of Fig. 11.
The last molecule depicted in Fig. 11, t2358(R), self-assembled into the chiral network made of triangular void spaces of two sizes. The density of this phase was equal to 0.101, and the side of the rhombic unit cell measured √229. The simulations of the corresponding R/S mixture predicted the formation of random aggregates being defective islands with brickwall-type structure. As shown in Fig. 12, in this system, the fraction of mono-coordinated metal atoms at low temperatures is relatively high and reaches about 0.2, which correlates well with the presence of dispersed domains having such metal atoms at peripheries. The positioning of enantiomers in the aggregates occurring in the racemic overlayer of t2358 was random, as visible in Fig. 13. At low temperatures, the fraction of heterochirally bonded metal atoms plotted therein oscillates around 0.5, which means equal chances for a metal atom to be coordinated homo- or heterochirally.
The last set of snapshots, collected in Fig. 14., was obtained for the molecules of t158_11, t15_10_11, and t169_12. In the case of enantiopure self-assembly of the first tecton, t158_11(R), dichotomous structure formation was observed in the simulations. One possible homochiral phase, A, was of the brick-wall type with a density equal to 0.120. The other one, B, comprised pores of two types (hexagonal and triangular), and it was similar to the Kagome lattice with rounded hexagons. The latter polymorph, B, was less dense than A, with a density of 0.078. In both cases, the unit cells were determined, that is, a parallelogram with sides of lengths 12 and 4√7 and an angle of 41°, and a rhombus with a side equal to 20. For the corresponding racemic system, the regularity of the enantiopure polymorphs was no longer present. In the mixed overlayer, random arrangements of small fragments of the enantiopure networks were observed, including brick-wall oligomers and small triangular pores. According to the energy (Fig. S12) and coordination (Fig. 15) curves, the resulting racemic assembly has many defects, and, as a result, its components are not fully coordinated.

Adsorbed structures created by 400 molecules of t158_11, t15_10_11 and t169_12 (enantiopure: 400 R, racemic: 200 R + 200 S) mixed with 800 metal atoms. The insets show characteristic motifs of the simulated assemblies. L = 200, θ = 0.060, T = 0.01

Effect of temperature on the fraction of 0-, 1- and 2-coordinated metal atoms calculated for the systems comprising 400 molecules of t158_11, t15_10_11 and t169_12 (enantiopure: 400 R, racemic: 200 R + 200 S) mixed with 800 metal atoms. The results obtained for the racemic mixtures are plotted with dashed lines. θ = 0.06
The enantiomer t15_10_11(R) was revealed to be one more tecton able to create the brick-wall type network. Even denser than before (0.144), this network has the unit cell in the shape of a parallelogram with sides equal to 5√3 and √91 and an angle equal to 57°. Changing the composition of the adsorbed phase to racemic resulted in the formation of completely different, much less ordered structures. For this racemate, isolated bimolecular R/S aggregates and larger but dispersed connections thereof were dominant. These forms were structurally similar to the dimers observed for the rac-1_10_11. This effect is not surprising as the tectons t15_10_11 and 1_10_11 have three halogen atoms in the same positions 1, 10 and 11. The only difference is the locant number 5 in the tetrasubstituted isomer, providing the additional active centre, which allows the R-S dimers for further linkage and creation of mixed chains. The effect of temperature on the energetic and structural parameters characterizing the enantiopure and racemic self-assembly of t15_10_11 was illustrated in Fig. 15 and S12.
The last molecule studied here, t169_12, was found to create the heteroporous network when enantiopure adsorption was assumed. The resulting network consisted of clover-leaf and hexagonal pores arranged in a spatially repetitive pattern visible in Fig. 14. With a density of 0.111, this network was characterized by a rhombic unit cell with a side length of √313. When the opposite enantiomer, S, was added to the adsorbed phase, a significant topology change took place. In this case, complex phase coexistence was observed with three types of structures occurring in this system. Those were the narrow enantiopure domains R and S with new morphology and the (locally) ordered domains of mixed network structures. The inset to the corresponding panel of Fig. 14 presents a magnified fragment of this ordered non-racemic network with 2:1 R/S proportions, density equal to 0.151 and the parallelogram unit cell with side lengths 16 and 19. For comparison, the energy and coordination curves calculated for the isomer t169_12 were presented in Fig. 15.
As for most of the isomers probed in this study, the predicted structures, to the best of our knowledge, have not been realized experimentally. It was thus not always possible to make a direct comparison. However, the Ullmann coupling of the isomer t5_11 on the Au(111) surface has been reported by Basagni et al. [16] and those studies (performed at much higher surface coverage of tetracene) demonstrated the formation of densely packed chains also observed in our simulations. Regarding the remaining PAH isomers, structurally similar assemblies have been obtained with chemically different molecules but equipped with analogous sets of interaction directions (interaction directions). This includes, for example, chains formed by dibenzopyrene, dichlorobianthryl and coronene [55,56,57] and networks comprising hexa-peri-hexabenzocoronene, dibromodimethylnaphthalene and hexaaminobenzene/pyrene tetraone. [58,59,60] Such structures have also been observed in the corresponding MC [40, 41, 45,46,47,48,49,50,51, 61] and MD [42,43,44] simulations.
4 Summary and conclusions
The results presented in this work demonstrated how the coarse-grained Monte Carlo simulations could be utilized to predict the structure of metal–organic intermediates formed in the surface-assisted Ullmann coupling of tetracene derivatives. The calculations performed for the set of differently di-, tri-, and tetrasubstituted isomers revealed that the architecture of the investigated precursors could be controlled by a suitable choice of the number and intramolecular distribution of halogen atoms. The predicted metal–organic intermediate structures varied in topology, from oligomers and chains for molecules with two active centres to ladders and aperiodic networks for the majority of trisubstituted tetracenes to diverse periodic networks for most linkers with four reactive sites. Although in most cases, the presence of additional benzene ring(s) in the tetracene linker did not significantly alter the morphology of assemblies obtained for shorter molecules (namely, naphthalene and anthracene), it enabled additional substitution patterns which open new self-assembly pathways and possibilities of designing 2D covalent polymers. Our calculations took into account also the prochiral character of the halogenated tectons, which is inherent to the vast majority of the isomers tetracene. For that reason, the simulations were performed for both enantiopure and racemic mixtures to identify the most important effects associated with the presence of the other enantiomer in the adsorbed phase. These calculations showed, for example, the significant dissimilarity between the enantiopure and racemic systems, including the formation of phases with entirely different morphologies. In most cases, the introduction of the opposite optical isomer into the adsorbed overlayer led to the substantial disordering and creation of numerous local defects. Among the obtained results, the exceptional case was the racemic assembly of the isomer t139_12, for which the chiral resolution was observed, preserving the architecture of the homochiral connections.
The theoretical predictions reported herein for differently substituted tetracenes confirm that many motifs observed in the simulations are typical for the analogously halogenated shorter molecules with linear aromatic backbone. In this light, it can be concluded that for the acenes, the structure formation is more dependent on the substitution pattern than on molecular length. As the number of possible halogen distributions grows strongly with a molecular size of a given PAH, practical testing of the performance of the various distributions is hardly feasible. Because of that, computer simulations seem to be a helpful tool for understanding, designing and predicting new nanopolymers with targeted properties. As we showed, the simplified model of metal–organic self-assembly preceding the covalent Ullmann coupling was able to identify basic types of molecular superstructures which can be expected to occur in this on-surface process. We believe that these findings will facilitate the preliminary screening of molecular libraries to select PAH platforms capable of the creation of adsorbed precursors and, in consequence, covalent polymers with predefined architectures and functions.
Availability of data and materials
Data used in this paper is available to view by contacting authors.
References
Yang, Y., Schäfer, C., Börjesson, K.: Detachable all-carbon-linked 3D covalent organic framework films for semiconductor/COF heterojunctions by continuous flow synthesis. Chemistry (2022). https://doi.org/10.1016/j.chempr.2022.05.003
Vaseghi, Z., Nematollahzadeh, A.: Nanomaterials. In: Green Synthesis of Nanomaterials for Bioenergy Applications. Wiley, New York, pp. 23–82 (2020). https://doi.org/10.1002/9781119576785.ch2.
Moores, A.: Bottom up, solid-phase syntheses of inorganic nanomaterials by mechanochemistry and aging. Curr. Opin. Green Sustain. Chem. (2018). https://doi.org/10.1016/j.cogsc.2018.05.004
Sealy, C.: Bottom-up approach yields 2D nanomaterials by the kilo. Mater. Today 54, 5–6 (2022). https://doi.org/10.1016/j.mattod.2022.03.007
Abid, N., Khan, A.M., Shujait, S., et al.: Synthesis of nanomaterials using various top-down and bottom-up approaches, influencing factors, advantages, and disadvantages: a review. Adv. Colloid Interface Sci. 300, 102597 (2022). https://doi.org/10.1016/j.cis.2021.102597
Mallada, B., Chen, Q., Chutora, T., et al.: Resolving atomic-scale defects in conjugated polymers on-surfaces. Chemistry (2022). https://doi.org/10.1002/chem.202200944
Biswas, K., Urgel, J.I., Xu, K., et al.: On-surface synthesis of a dicationic diazahexabenzocoronene derivative on the Au(111) surface. Angew. Chem. Int. Ed. 133, 25755–25760 (2021). https://doi.org/10.1002/ange.202111863
Clair, S., De Oteyza, D.G.: Controlling a chemical coupling reaction on a surface: tools and strategies for on-surface synthesis. Chem. Rev. 119, 4717–4776 (2019). https://doi.org/10.1021/acs.chemrev.8b00601
Evans, A.M., Strauss, M.J., Corcos, A.R., et al.: Two-dimensional polymers and polymerizations. Chem. Rev. (2022). https://doi.org/10.1021/acs.chemrev.0c01184
Shen, Q., Gao, H.Y., Fuchs, H.: Frontiers of on-surface synthesis: from principles to applications. Nano Today (2017). https://doi.org/10.1016/j.nantod.2017.02.007
Chen, J., Zhang, J., Zou, Y., et al.: PPN (poly-: peri -naphthalene) film as a narrow-bandgap organic thermoelectric material. J. Mater. Chem. A 5, 9891–9896 (2017). https://doi.org/10.1039/c7ta02431b
Moreno, C., Panighel, M., Vilas-Varela, M., et al.: Critical role of phenyl substitution and catalytic substrate in the surface-assisted polymerization of dibromobianthracene derivatives. Chem. Mater. 31, 331–341 (2019). https://doi.org/10.1021/acs.chemmater.8b03094
Moreno, C., Vilas-Varela, M., Kretz, B., et al.: Bottom-up synthesis of multifunctional nanoporous graphene. Science (80) 360, 199–203 (2018). https://doi.org/10.1126/science.aar2009
Eichhorn, J., Nieckarz, D., Ochs, O., et al.: On-surface Ullmann coupling: the influence of kinetic reaction parameters on the morphology and quality of covalent networks. ACS Nano 8, 7880–7889 (2014). https://doi.org/10.1021/nn501567p
Lackinger, M.: Surface-assisted Ullmann coupling. Chem. Commun. (2017). https://doi.org/10.1039/c7cc03402d
Basagni, A., Ferrighi, L., Cattelan, M., et al.: On-surface photo-dissociation of C-Br bonds: towards room temperature Ullmann coupling. Chem. Commun. 51, 12593–12596 (2015). https://doi.org/10.1039/c5cc04317d
Judd, C.J., Haddow, S.L., Champness, N.R., et al.: Ullmann coupling reactions on Ag(111) and Ag(110); substrate influence on the formation of covalently coupled products and intermediate metal-organic structures. Sci. Rep. 7, 14541 (2017). https://doi.org/10.1038/s41598-017-13315-1
Barton, D., Gao, H.Y., Held, P.A., et al.: Formation of organometallic intermediate states in on-surface Ullmann couplings. Chem. Eur. J. 23, 6190–6197 (2017). https://doi.org/10.1002/chem.201605802
Eichhorn, J., Strunskus, T., Rastgoo-Lahrood, A., et al.: On-surface Ullmann polymerization via intermediate organometallic networks on Ag(111). Chem. Commun. 50, 7680–7682 (2014). https://doi.org/10.1039/c4cc02757d
Ebeling, D., Zhong, Q., Schlöder, T., et al.: Adsorption structure of mono- and diradicals on a Cu(111) surface: chemoselective dehalogenation of 4-bromo-3″-iodo-p-terphenyl. ACS Nano 13, 324–336 (2019). https://doi.org/10.1021/acsnano.8b06283
Fan, Q., Wang, C., Han, Y., et al.: Surface-assisted formation, assembly, and dynamics of planar organometallic macrocycles and zigzag shaped polymer chains with C-Cu–C bonds. ACS Nano 8, 709–718 (2014). https://doi.org/10.1021/nn405370s
Lu, H., Wenlong, E., Cai, L., et al.: Dissymmetric on-surface dehalogenation reaction steered by preformed self-assembled structure. J. Phys. Chem. Lett. 11, 1867–1872 (2020). https://doi.org/10.1021/acs.jpclett.9b03688
Palma, C.-A., Diller, K., Berger, R., et al.: Photoinduced C-C reactions on insulators toward photolithography of graphene nanoarchitectures. J. Am. Chem. Soc. 136, 4651–4658 (2014). https://doi.org/10.1021/ja412868w
Cai, L., Huang, Y., Wang, D., et al.: Supramolecular tiling of a conformationally flexible precursor. J. Phys. Chem. Lett. 13, 2180–2186 (2022). https://doi.org/10.1021/acs.jpclett.2c00147
Hao, L., Ning, J., Luo, B., et al.: Structural evolution of 2D microporous covalent triazine-based framework toward the study of high-performance supercapacitors. J. Am. Chem. Soc. 137, 219–225 (2015). https://doi.org/10.1021/ja508693y
Wei, S., Zhang, F., Zhang, W., et al.: Semiconducting 2D triazine-cored covalent organic frameworks with unsubstituted olefin linkages. J. Am. Chem. Soc. 141, 14272–14279 (2019). https://doi.org/10.1021/jacs.9b06219
Castro-Esteban, J., Albrecht, F., Fatayer, S., et al.: An on-surface Diels-Alder reaction. Angew. Chem. Int. Ed. 60, 26346–26350 (2021). https://doi.org/10.1002/anie.202110311
Shi, H., Liu, Y., Song, J., et al.: On-surface synthesis of self-assembled monolayers of benzothiazole derivatives studied by STM and XPS. Langmuir 33, 4216–4223 (2017). https://doi.org/10.1021/acs.langmuir.7b00674
Sun, K., Fang, Y., Chi, L.: On-surface synthesis on nonmetallic substrates. ACS Mater. Lett. 3, 56–63 (2021). https://doi.org/10.1021/acsmaterialslett.0c00452
Saywell, A., Browning, A., Rahe, P., et al.: Organisation and ordering of 1D porphyrin polymers synthesised by on-surface glaser coupling. Chem. Commun. (2016). https://doi.org/10.1039/C6CC03758E
Adisoejoso, J., Li, Y., Liu, J., et al.: Two-dimensional metallo-supramolecular polymerization: toward size-controlled multi-strand polymers. J. Am. Chem. Soc. 134, 18526–18529 (2012). https://doi.org/10.1021/ja308480x
Zhang, X., Shen, Q., Ding, H., et al.: On-surface synthesis of thiophene-containing large-sized organometallic macrocycles on the Ag(111) surface. J. Phys. Chem. C 125, 11454–11461 (2021). https://doi.org/10.1021/acs.jpcc.1c01540
Su, J., Fan, W., Mutombo, P., et al.: On-surface synthesis and characterization of [7]triangulene quantum ring. Nano Lett. 21, 861–867 (2021). https://doi.org/10.1021/acs.nanolett.0c04627
Sánchez-Sánchez, C., Brüller, S., Sachdev, H., et al.: On-surface synthesis of BN-substituted heteroaromatic networks. ACS Nano 9, 9228–9235 (2015). https://doi.org/10.1021/acsnano.5b03895
Di Giovannantonio, M., Tomellini, M., Lipton-Duffin, J., et al.: Mechanistic picture and kinetic analysis of surface-confined Ullmann polymerization. J. Am. Chem. Soc. 138, 16696–16702 (2016). https://doi.org/10.1021/jacs.6b09728
Björk, J., Hanke, F., Stafström, S.: Mechanisms of halogen-based covalent self-assembly on metal surfaces. J. Am. Chem. Soc. 135, 5768–5775 (2013). https://doi.org/10.1021/ja400304b
Voznyy, O., Dubowski, J.J., Yates, J.T., et al.: The role of gold adatoms and stereochemistry in self-assembly of methylthiolate on Au(111). J. Am. Chem. Soc. 131, 12989–12993 (2009). https://doi.org/10.1021/ja902629y
Gouron, A., Le Mapihan, K., Camperos, S., et al.: New insights in self-assembled monolayer of imidazolines on iron oxide investigated by DFT. Appl. Surf. Sci. 456, 437–444 (2018). https://doi.org/10.1016/j.apsusc.2018.06.119
Gdula, K., Nieckarz, D.: On-surface self-assembly of metal-organic architectures: insights from computer simulations. J. Phys. Chem. C 124, 20066–20078 (2020). https://doi.org/10.1021/acs.jpcc.0c04597
Nieckarz, K., Nieckarz, D.: Monte Carlo simulations of the metal-directed self-assembly of Y-shaped positional isomers. Crystals 12, 492 (2022). https://doi.org/10.3390/cryst12040492
Nieckarz, D., Nieckarz, K.: Computer-aided design of hierarchically organized 2D metal-organic networks (2022).
Baran, Ł: Coarse-grained modeling of on-surface self-assembly of mixtures comprising Di-substituted polyphenyl-like compounds and metal atoms of different sizes. ACS Omega 6, 25193–25200 (2021). https://doi.org/10.1021/acsomega.1c02857
Baran, Ł, Dąbrowska, K., Rżysko, W., et al.: Molecular dynamic study of model two-dimensional systems involving Janus Dumbbells and spherical particles. Condens. Matter Phys. 24, 33401 (2021). https://doi.org/10.5488/CMP.24.33401
Baran, Ł, Dyk, K., Kamiński, D., et al.: Influence of the substitution position in the tetratopic building blocks on the self-assembly process. J. Mol. Liq. 346, 117074 (2021). https://doi.org/10.1016/j.molliq.2021.117074
Fadeeva, A.I., Gorbunov, V.A., Stishenko, P.V., et al.: Melting of Fe-terephthalate layers on Cu(1 0 0) surface with randomly distributed point defects. Appl. Surf. Sci. 545, 148989 (2021). https://doi.org/10.1016/j.apsusc.2021.148989
Fadeeva, A.I., Gorbunov, V.A., Solovyeva, O.S., et al.: Homologous series of flower phases in metal-organic networks on Au(111) surface. J. Phys. Chem. C (2020). https://doi.org/10.1021/acs.jpcc.0c02527
Lisiecki, J., Szabelski, P.: Surface-confined metal-organic precursors comprising naphthalene-like derivatives with differently distributed halogen substituents: a Monte Carlo model. J. Phys. Chem. C 124, 20280–20293 (2020). https://doi.org/10.1021/acs.jpcc.0c06726
Lisiecki, J., Szabelski, P.: Designing 2D covalent networks with lattice monte carlo simulations: precursor self-assembly. Phys. Chem. Chem. Phys. 23, 5780–5796 (2021). https://doi.org/10.1039/d0cp06608g
Lisiecki, J., Szabelski, P.: Halogenated anthracenes as building blocks for the on-surface synthesis of covalent polymers: structure prediction with the lattice Monte Carlo method. J. Phys. Chem. C 125, 15934–15949 (2021). https://doi.org/10.1021/acs.jpcc.1c03973
Lisiecki, J., Szabelski, P.: Theoretical modeling of the metal-organic precursors of anthracene-based covalent networks on surfaces. ChemPhysChem (2022). https://doi.org/10.1002/cphc.202100877
Lisiecki, J., Szabelski, P.: Monte Carlo simulation of the surface-assisted self-assembly of metal-organic precursors comprising phenanthrene building blocks. Colloids Surf. A (2022). https://doi.org/10.1016/j.colsurfa.2022.129177
Favre, H.A., Powell, W.H.: Nomenclature of Organic Chemistry. Royal Society of Chemistry, Cambridge (2013)
Frenkel, D., Smit, B.: Understanding Molecular Simulation, Elsevier, San Diego, pp. 23–61, 111–137 (2002). https://doi.org/10.1016/B978-012267351-1.
Allen, M.P., Tildesley, D.J.: Computer Simulation of Liquids. Oxford University Press, Oxford (2017). https://doi.org/10.1093/oso/9780198803195.001.0001.
Fogel, Y., Zhi, L., Rouhanipour, A., et al.: Graphitic nanoribbons with dibenzo[e, l]pyrene repeat units: synthesis and self-assembly. Macromolecules 42, 6878–6884 (2009). https://doi.org/10.1021/ma901142g
Jacobse, P.H., Simonov, K.A., Mangnus, M.J.J., et al.: One precursor but two types of graphene nanoribbons: on-surface transformations of 10,10′-dichloro-9,9′-bianthryl on Ag(111). J. Phys. Chem. C 123, 8892–8901 (2019). https://doi.org/10.1021/acs.jpcc.8b12209
Wang, Z., Liu, M., Chen, S., et al.: On-surface synthesis of gold–coronene molecular wires. Chem. Commun. 56, 11239–11242 (2020). https://doi.org/10.1039/D0CC04540C
Zhang, L., Li, J., Qiu, S., et al.: Synthesis and self-assembly of a D3h symmetric polycyclic aromatic hydrocarbon into a rigid 2D honeycomb network. New J Chem 41, 3260–3264 (2017). https://doi.org/10.1039/C6NJ03962F
Liu, J., Xia, B., Lin, N.: Controlling the reaction steps of bifunctional molecules 1,5-dibromo-2,6-dimethylnaphthalene on different substrates. J. Phys. Chem. C 122, 13001–13008 (2018). https://doi.org/10.1021/acs.jpcc.8b04651
Riaño, A., Strutyński, K., Liu, M., et al.: An expanded 2D fused aromatic network with 90-ring hexagons. Angew. Chem. Int. Ed. 61, e202113657 (2022). https://doi.org/10.1002/anie.202113657
Ibenskas, A., Tornau, E.E.: Modeling of high-temperature ordered structures with weak intermolecular C-H···F and C–H···N bonds. J. Phys. Chem. C 125, 19560–19569 (2021)
Acknowledgements
This work was supported by the National Science Centre, Poland research grant 2018/31/B/ST4/01759
Funding
This work was supported by the National Science Centre, Poland research grant 2018/31/B/ST4/01759.
Author information
Authors and Affiliations
Contributions
JL and PS wrote the main manuscript text. JL ran simulations and prepared figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have no competing interests to declare that are relevant to the content of this article.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lisiecki, J., Szabelski, P. On-surface Ullmann coupling of halo-derivatives of arenes: Monte Carlo simulations for tetracene. Adsorption 30, 201–219 (2024). https://doi.org/10.1007/s10450-023-00395-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-023-00395-x