
Abstract
Reforming of fossil fuels coupled with carbon capture and storage has the potential to produce low-carbon H2 at large scale and low cost. Adsorption is a potentially promising technology for two key separation tasks in this process: H2 purification and CO2 capture. In this work, we present equilibrium adsorption data of H2 and CH4 on zeolite 13X, in addition to the already established CO2 isotherms. Further, we carry out binary (CO2–CH4) and ternary (H2–CO2–CH4) breakthrough experiments at various pressures and temperatures to estimate transport parameters, assess the predictive capacity of our 1D column model, and compare different multi-component adsorption models. CO2 adsorbs strongly on zeolite 13X, CH4 adsorbs less, and H2 adsorbs very little. Thus, H2 breaks through first, CH4 second (first in the binary breakthrough experiments) and CO2 last. Linear driving force (LDF) mass transfer coefficients are estimated based on a single breakthrough experiment and mass transfer is found to be fast for H2, slower for CH4, and slowest for CO2. The LDF parameters can be used in a predictive manner for breakthrough experiments at varying pressures, temperatures, flows, and, though with lower accuracy, even compositions. Heat transfer inside the column is described well with a literature correlation, thus yielding an excellent agreement between simulated and measured column temperatures. Ideal and real adsorbed solution theories (IAST and RAST, respectively) both model the observed breakthrough composition profiles well, whereas extended isotherms are inferior for predicting the competitive behavior between CH4 and CO2 adsorption. This study provides the groundwork necessary for full cyclic experiments and their simulation.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
As climate change is becoming an increasingly pressing issue, hydrogen as clean, versatile, and carbon free energy carrier is receiving growing attention in politics, industry and the scientific community [10, 15, 16]. It can be used as feedstock, carbon-free fuel or energy carrier across a variety of sectors including industry, power generation, heating, and transportation [16].
Water electrolysis using renewable energy can produce H2 with little associated CO2 emissions. However, challenges like a high capital cost and limited availability of cheap, renewable energy so far hindered large-scale deployment [16, 36, 40]. Currently, only about 1% of H2 is produced from water electrolysis, whereas the vast majority is produced from fossil fuels. Reforming of natural gas (mainly steam methane reforming, SMR), accounts for about three-quarters of dedicated H2 production, coal gasification for 23% [16]. Fossil fuel based production, however, results in H2 with high associated CO2 emissions.
Coupling fossil fuel based H2 production with carbon capture and storage (CCS) is a feasible and logical option to deliver carbon neutral H2 at a large scale in the near future [18, 40, 47]. This approach is interesting not only as a bridging technology until H2 from water electrolysis becomes cheaper and renewable energy can be generated at a large enough scale and low cost, but will likely remain the more cost-competitive option in many places like Europe or Japan [16]. In addition, replacing the fossil fuel source with renewable fuels like biomass or biogas can enable negative emissions [1, 45].
A variety of options like cryogenic separation, absorption, membranes or adsorption, are discussed and even tested at pilot scale for coupling fossil- or biofuel based hydrogen production with CCS, but they typically require the addition of a dedicated CO2 separation stage [11, 17, 18, 47]. CO2 can be captured from the reformer flue gas; after reformer and water-gas shift reactor, but before entering the H2 purification through pressure swing adsorption (PSA) [32]; or from the H2-PSA tail-gas. As an alternative, we target the integration of H2 purification and carbon capture into a single, adsorption based unit thus reducing the number of required separation stages from two (H2-PSA and CO2 capture) to one, thereby likely also reducing capital cost and process complexity.
Such combined processes were introduced in the literature with the Gemini process developed by Air Products and Chemicals Inc. [21, 33, 38]. The Gemini process is an adsorption process with two trains of in total 8 or 9 columns, that are interconnected during part of the cycle, but undergo a completely different sequence of steps for regeneration. The process requires both a vacuum pump and a compressor to compress a CO2 rich stream back to feed pressure. Recently, [31] developed an adsorption process for co-purification of CO2 and H2 in an integrated gasification combined cycle plant (IGCC). Only part of the hydrogen in the inlet, however, can be separated as high purity product, whereas a big part is used at lower purity in the gas turbine.
We have recently developed different vacuum pressure swing adsorption (VPSA) cycles, that overcome the shortcomings of both Gemini 8/9 and [31]’s one-train process [42, 43]. The VPSA process requires a single train only, needs no re-compression, but only a vacuum pump, and allows for the co-purification of both products at high recovery: over 90% of the H2 can be separated at high purity sufficient for industrial application (99.9% purity, [47]) or even fuel cells (99.97% purity, [19, 47]) whilst CO2 is co-produced at over 90% recovery and 96% purity, thus reaching the target typically set for CCS applications [26, 46].
So far, our studies were based on modeling and optimization only. In this two-part series, we present the complementary experimental studies to
-
validate the column model for the operating conditions of interest,
-
assess its predictive capacity,
-
estimate important model parameters like transport coefficients based on experimental data,
-
establish a suitable model for multi-component adsorption,
-
demonstrate the technology at small-scale to pave the way towards a rapid advancement in technology readiness level.
In this first part of the series, we present isotherm measurements and binary (CO2–CH4) and ternary (CO2–CH4–H2) breakthrough studies at various pressures, temperatures and flows. Commercial zeolite 13X was chosen as adsorbent due to its good performance in modeling studies [42, 43], low cost and easy availability. We use one reference experiment to estimate mass transfer parameters and assess the validity of the correlation used to model heat transfer. The predictive capacity of the model is assessed by comparing experimental and simulation results at different pressures, temperatures, flows, and compositions using the transport parameters estimated based on the reference breakthrough experiment in combination with the predictive correlation for heat transfer. Different approaches for describing multi-component adsorption are compared including extended isotherms, ideal adsorbed solution theory, and real adsorbed solution theory. The isotherms and transport parameters established in this part will then be used to model the cyclic VPSA process studied in the second part of the series [43].
2 Experimental
2.1 Materials
For all experiments, pelleted zeolite 13X (Zeochem, Switzerland) was used with a particle size between 1.6 to 2 mm. The material properties are summarized in Table 1.
Pure gases (H2, 5.0; He, 4.6; CH4, 5.5) and gas mixtures (binary: CO2:CH4 = 50:50 mol%; ternary: CO2:CH4:H2 = 20:5:75 mol%) were obtained from Pangas (Switzerland). The binary mix has a relative error of 1% on both components, the other mix has a relative error of 2% on CH4 and 1% on CO2.
2.2 Experimental methods
2.2.1 Isotherm measurements
Adsorption isotherms were measured for CH4 and H2 at 25 °C, 45 °C,and 65 °C and pressures up to 30 bar using a Rubotherm magnetic suspension balance (MSB). A dual sinker configuration allows for the simultaneous measurement of the mass change of the sample during adsorption and of the bulk density. The procedure was as follows:
-
1.
Sample regeneration for at least 4 h at 400 °C under vacuum;
-
2.
Volume measurement using He (non-adsorbing) at high pressure (50–200 bar) and temperature (150–200 °C);
-
3.
Evacuation and heating to target isotherm temperature;
-
4.
Measurement of zero value;
-
5.
Isotherm measurement in desorption mode (some points were repeated in adsorption mode, to ensure that there is no hysteresis).
Measuring H2 adsorption on zeolite 13X was difficult due to its low density and adsorption. In order to obtain reproducible results, the zero value was measured after every single point (step 4) by evacuating the balance, similar to the procedure described in the literature [34]. The excess amount adsorbed, which is the measured quantity, was converted to the absolute amount adsorbed assuming a constant adsorbed phase density equal to the liquid density at the boiling point, as often done in the literature [9, 13, 14, 27, 34]. Both the setup and the evaluation procedure are explained in detail in the literature [28, 30].
2.2.2 Breakthrough experiments
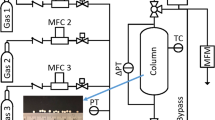
Breakthrough experiments were carried out in the laboratory setup shown in Fig. 1. The setup is in principle the same as described in the literature [23]. It consists of two double-jacketed columns that can be actively heated/cooled with two thermostats (Huber Kältemaschinenbau AG, Switzerland). Only one is shown in Fig. 1, as only one at a time is operated for the breakthrough studies.

Experimental setup for breakthrough studies
The column pressure can be controlled with two back pressure regulators (Bronkhorst High-Tech B.V., the Netherlands). The inflow is controlled with different mass flow controllers (MFC) and the outflow is measured with three mass flow meters (MFM, all from Bronkhorst High-Tech B.V.). A mass spectrometer (MS, Pfeiffer Vacuum AG, Schweiz) downstream of the MFMs is used to analyze the composition of the gas phase. Column inlet and outlet pressure are measured with pressure sensors (PI, Keller, Germany) and the temperature inside the column is monitored at 5 axial positions (10, 35, 60, 85 and 110 cm from the column bottom) at the center of the column (\(r = 0\)). The setup dimensions are summarized in Table 1.
Breakthrough experiments were carried out for a binary and a ternary mixture at various pressures, temperatures, and flows as summarized in Table 2. A generic equimolar CO2–CH4-mix has been chosen for the binary mixture to assess competition between CO2 and CH4. The composition of the ternary mixture is based on typical steam methane reformer gas after water–gas shift with CH4 as representative impurity.
Before all experiments, both columns were packed with zeolite 13X. Before each experiment, the relevant column was regenerated by heating to 240 °C and applying a vacuum for a minimum of 2 h. After a maximum of three subsequent experiments per column and before the first experiment, a more thorough regeneration was performed by heating to 240 °C for at least 12 h. The lower regeneration temperature compared to the isotherm measurements is due to equipment limitations. No difference was found between these different regeneration procedures. Directly before starting a set of two experiments (one in each column), the MS was calibrated using the feed mixture for binary breakthrough experiments and both feed mixture and binary mixture for ternary breakthrough experiments. Next, the regenerated column was pressurized with helium to the adsorption pressure. Then, the inlet was switched to the feed mixture. The temperatures and pressures within the column, the outflow velocity and the composition after the column were monitored continuously. Each experiment was repeated at least once, and in most cases in the other column, so as to ensure reproducibility also using different columns.
3 Modeling
3.1 Adsorption isotherms and heat of adsorption
3.1.1 Single component adsorption isotherms
Like in the case of the isotherm model for the CO2 isotherms measured previously in our laboratory on the same zeolite [14], we fitted a temperature dependent Sips isotherm equation to the isotherm measurements to describe single component adsorption:
with the following temperature dependence of the maximum capacity \(q^{{\infty }}_i\)
of the affinity coefficient \({b_i}\),
and of the exponent \({s_i}\)
\(T_{(\text {ref})}\) is the (reference) temperature, \(P_i\) is the pressure of component i, \(q_i\) is the absolute adsorbed amount, R is the universal gas constant and \(A_{1,i}\) to \(C_{2,i}\) are constant parameters. For parameter estimation, we performed a least-squares minimization coupling Matlab’s fmincon local optimizer with the global search algorithm. Whenever possible without significantly reducing the quality of the fit, we eliminated the temperature dependence of the maximum capacity or of the Sips exponent or of both.
Isosteric heat of adsorption The isosteric heat of adsorption \(\Delta H_i^{\text {iso}}\) was determined from the isotherm measurements at different temperatures using the Clausius Clapeyron equation:
To compute the isosteric heat, data points at different temperatures, but the same loading are required. Therefore, we first fitted the experimental results at each temperature to a suitable isotherm, then calculated isosteric data points at the three different temperatures and plotted \(\ln P\) over 1/T. The temperature-averaged isosteric heat was then evaluated from the slope of straight lines fitted to these points. To avoid extrapolation, only loadings with data points for all three temperatures were considered.
3.1.2 Multi-component adsorption
For modeling multi-component adsorption, we compared three different approaches, namely the use of extended isotherms, the real adsorbed solution theory (RAST), and the ideal adsorbed solution theory (IAST).
Extended isotherms are convenient to use in process modeling and optimization, as they provide an explicit equation for multi-component adsorption based on single component isotherm data only. For the Sips isotherm, the extended isotherm for N components is given as [32]
Another well known approach for describing multi-component- adsorption is the adsorbed solution theory introduced by Mofarahi and Bakhtyari [25], which treats adsorption equilibria as phase equilibria between adsorbed phase as solution in equilibrium with the gas phase:
\(x_i\) is the mole fraction in the adsorbed phase (hence \(\sum _i x_i = 1\)), \(y_i\) is the mole fraction in the gas phase, \(\gamma _i\) is the activity coefficient in the adsorbed phase, \(P_i^0(\pi )\) is the pressure of pure component i evaluated at the spreading pressure \(\pi\) and temperature T of the mixture. The spreading pressure has to be equal for all components at the same temperature and can be evaluated through integration from
With \(q_i^0(P_i^0)\) being the single component equilibrium adsorbed amount at the same T and \(\pi\) of the mixture. For the Sips isotherm equation used in this work, this results in
with the reduced surface potential \(\Pi\). Finally, the total amount adsorbed, \(q_\text {t}\), can be calculated as
and the amount adsorbed for each component, \(q_i\), as
In the special case where the adsorbed phase can be approximated as ideal solution, the activity coefficients are unity and the theory simplifies to the ideal adsorbed solution theory (IAST). The main advantage of IAST is that it requires single component isotherm data only, which makes it a theory that is widely used for adsorbent characterization and process modeling [48]. In contrast to extended isotherms, however, in general no closed form of the solution exists, thus requiring numerical solution for given gas phase composition hence increasing computational time. In addition, it is known to be less accurate for adsorbents with heterogeneous surfaces, such as zeolite 13X [2, 48].
For non-ideal adsorbed phase solutions, the real adsorbed solution theory (RAST) is used. In contrast to IAST, RAST requires an estimate of the activity coefficients \(\gamma _i\) based on multi-component experimental data. Here, we use the equation suggested by [49] as model for the activity coefficients (Eq. 12), with the temperature dependence originally suggested in his work for the binary interaction parameters \(\varLambda _{ij}\) (Eq. 13).
\(\lambda _{ij}\) is the i, j-interaction energy and \(\nu _i\) the molar volume of species i. \(\overline{\lambda _{i}}\) has to be estimated by fitting experimental data. It is noted that the dependence of \(\varLambda _{ij}\) on the spreading pressure is neglected here, as discussed in the literature [14], thus eliminating one fitting parameter. Nevertheless, for a ternary system, six parameters would need to be estimated. We do not directly measure multi-component adsorption, but estimate \(\overline{\lambda _{i}}\) from cyclic experiments as described in part two of this series [41] and discussed in the results section (Sect. 4.2.4). To reduce the number of parameters that need to be estimated by fitting with this indirect approach, we simplified the theory as much as possible whilst still ensuring a reasonable agreement between experimental data and simulation results. Therefore, we set all \(\Lambda _{ij}\) equal to unity for all binary mixtures that contain H2. Thus, we assume that all binary mixtures with H2 behave ideal, i.e. according to IAST (Eq. 12 in this case yields an activity coefficient of unity). This assumption may not result in an accurate prediction of H2 co-adsorption, but as H2 adsorption is low on zeolite 13X, we do not expect it to hinder the uptake of strongly adsorbing components like CO2 and CH4 notably independent of the model used for describing multi-component adsorption. Furthermore, we found in a previous study that an accurate description of hydrogen adsorption is of little importance for correctly predicting the separation performance of VPSA, as long as the other components feature a significantly higher affinity towards the adsorbent, and that even the assumption that no hydrogen is adsorbed at all results in almost the same separation performance [44]. In addition, we assume that the interaction energy between CO2 and CH4 is the mean of the interaction between the individual species, as suggested in the literature [32]:
Which results in \(\overline{\lambda _{i}}=(\lambda _{ji}-\lambda _{ii}) = -(\lambda _{ij}-\lambda _{jj})=-\overline{\lambda _{j}}\). With these simplifications, the single parameter left to estimate is \(\overline{\lambda _{i}}\). This was estimated not based on breakthrough results, but on cyclic experiments [41], and a value of \(\overline{\lambda _{1}}=-4\) kJ/mol was used for all simulations.
3.2 Process modeling and estimation of transport coefficients
A one dimensional adsorption bed model based on mass and energy balance equations was used for the estimation of transport parameters, and for the comparison of experimental and simulation results. The following simplifying assumptions were made:
-
the gas phase is described with the ideal gas law,
-
the pressure drop is described with the Ergun equation (Eq. 20) ,
-
diffusion is neglected [5],
-
thermal equilibrium is reached between adsorbent particles and gas phase,
-
axial conductivity of the column wall is neglected,
-
mass transfer is described using a linear driving force (LDF) approximation,
-
constant, average values are used for the heat of adsorption \(\Delta H_i^{\text {iso}}\), the heat capacity of wall \(C_\text {w}\), adsorbent \(C_\text {s}\), adsorbed phase \(C_\text {ads}\), and gas phase \(C_\text {g}\), the LDF coefficients \(k_i\) and the viscosity \(\mu\) of the gas phase.
These result in the overall mass balance
the component mass balance
the component mass balance in the adsorbed phase with the adsorbed phase concentration, \(q_i\), and the adsorbed phase concentration in equilibrium with the bulk gas, \(q_i^*\)
the energy balance for the column
the energy balance for the wall
and the momentum balance, i.e. the Ergun equation
\(c_{(i)}\) is the fluid phase concentration (of component i), \(\varepsilon _\text {t}\) and \(\varepsilon _\text {b}\) are the total and bed void fraction, u is the superficial velocity, T and \(T_\text {w}\) are the column and wall temperature, \(h_\text{L}\) is the heat transfer coefficient from the column inside to the wall and \(h_\text {w}\) that from the wall to the thermofluid. The important modeling parameters are summarized in Table 1. A more detailed explanation of the model can be found in previous works [4, 5, 35].
When comparing experimental results with simulation results, the piping system of the experimental setup has to be considered, as it results in a time difference between the flow through the MFC and the entry of the column, as well as after the column exit and the MFMs and the MS. Whereas the first could be accounted for with a simple time shift between simulation results and experimental data, the second has to be modeled, as the time difference is not constant, but depends on the outflow, which changes over time. Thus, we model the upstream and downstream piping as an isothermal plugflow reactor as in [4], i.e. neglecting diffusion. The downstream piping is assumed to be at high (adsorption) pressure until the BPR, and at ambient pressure thereafter, thus the pipe consists of two distinct sections with constant pressure and velocity.
3.2.1 Transport parameters and parameter estimation
The external heat transfer coefficients depends on the thermostat and thermofluid used, as well as on the column geometry. We use the same value as estimated in a previous work by [23], i.e. \(h_{\text {w}}\) = 220 W/(m K). Heat transfer from the column to the wall was modeled following the approach suggested in [39] based on the column inlet conditions. The effective radial thermal conductivity, \(\lambda _{\text {e}}\), and the heat transfer to the column wall, \(\alpha _{\text {w}}\), are lumped together in a single coefficient \(h_\text{L}\):
with the Biot number \(\text {Bi}=\alpha _\text {w}d_\text {i}/(2\lambda _{\text {e}})\). Both \(\lambda _{\text {e}}\) and \(\alpha _{\text {w}}\) have a static and a convective component that was calculated according to [39].
Mass transfer coefficients for CO2 and CH4 were estimated directly using one binary experiment, and then used to predict all other breakthrough experiments. For the ternary breakthrough experiments, the mass transfer coefficient of H2 is needed in addition to those of CO2 and CH4. As mass transfer of H2 is typically fast in zeolite 13X (and many other adsorbents), i.e. much faster than for any other component, it often cannot be estimated based on experimental data [7, 29]. Also in this work, fitting results were inconclusive, so that we chose a high value of 1 s\(^{-1}\) for all experiments. In addition, the separation performance of cyclic adsorption processes exhibits little sensitivity with respect to the mass transfer coefficient of H2 [44], and therefore a very accurate estimate is not critical process modeling and optimization.
In addition to estimating the mass transfer coefficients, the inlet velocity is allowed to vary within a few percentage points to account for the range of uncertainty of the MFCs, i.e. ± 2% of the full scale [4, 23]. Mass transfer coefficients and inlet velocity are estimated by minimizing the objective function
with the measured mole fraction \(y_{j,k}\) and temperature \(T_{j,k}\), and the corresponding simulated values \(\hat{T}_{j,k}\) and \(\hat{y}_{j,k}\). Note that weighting factors were added as suggested in the literature [23], such that all concentration measurements together have the same weight as all temperature measurements, and unity is added to the denominator of the molar fraction term to avoid dividing by zero. As for the isotherm fitting, we used Matlab’s fmincon local optimizer coupled with its global search algorithm.
4 Results
4.1 Adsorption isotherms
The measured CH4 and H2 adsorbed amounts are shown together with the estimated adsorption isotherms in Figs. 2 and 3. All corresponding isotherm parameters are provided in Table 3. We previously measured CO2 adsorption isotherms on the same material [14]; the corresponding parameters are provided in the same table.

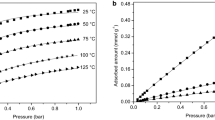
CH4 adsorption isotherms on zeolite 13X. Lines indicate the fitted Sips isotherms. The low pressure region up to 1 bar is shown separately. The dotted lines correspond to literature isotherms on zeolite 13X at 45 °C from [8] (blue), [24] (green), [29] (yellow), and [7] (purple) (Color figure online)

As shown in the figures, the agreement between the fitted Sips isotherm model and the measurements is very good. As mentioned in Sect. 3.1, we eliminated the temperature dependence of the maximum capacity and of the Sips exponent whenever possible. The H2 isotherms can be described very well with a temperature independent maximum capacity and Sips exponent, whereas for CH4, the temperature dependence of the maximum capacity is necessary to improve the model accuracy. The CH4 isotherms are linear in the low pressure region, but approach the maximum capacity at 30 bar (Fig. 2). On the contrary, H2 exhibits a much lower affinity towards zeolite 13X and its isotherms are almost linear over the whole pressure range (Fig. 3). The adsorption isotherms reported here are within the range reported in the literature for CH4 [7, 8, 24, 29] and at the upper bound of the range reported for H2 [7, 29].
CO2 has an even higher affinity toward zeolite 13X than CH4, and also a higher maximum capacity than both CH4 and H2 ([14], Table 3). This order is reflected in the isosteric heats of adsorption shown in Fig. 4 with a high isosteric heat for CO2, a low isosteric heat for H2 and a medium one for CH4. The values for all three components are in line with the range reported in the literature [6, 24, 29]. Note that to estimate parameters based on the breakthrough experiments, a constant heat of adsorption was used. Therefore, we computed the average over the entire loading range for each component, thus resulting in an isosteric heat of − 36.3 kJ/mol for CO2, of − 18.8 kJ/mol for CH4 and − 10.7 kJ/mol for H2.

Isosteric heat of adsorption on zeolite 13X, averaged over temperature (25 °C, 45 °C and 65 °). For computing the isosteric heat of adsorption for CO2, the isotherms as reported in [14] up to 65 °C were used
4.1.1 Multi-component adsorption
Figure 5 shows the adsorbed amount of CO2 and CH4 calculated with the different models over the total pressure for the exemplary case of the binary mixture used in this work.

Co-adsorption of an equimolar CO2-CH4 mixture on zeolite 13X modeled without competition (dash-dotted lines), with extended Sips isotherms (dashed lines), with IAST (dotted lines) and with RAST (solid lines, \(\overline{\lambda _{1}}=-4\) kJ/mol, [41]). For CO2, lines calculated with IAST and without competition overlap
As shown in the figure, the adsorbed amount is the highest for both components when considering no interaction at all (dash-dotted lines).
The use of extended Sips isotherms (dashed lines) leads to a significant reduction in predicted adsorbed amount for both components. Note that the effect on CH4 is more pronounced than that on CO2 due to the high affinity of CO2 toward zeolite 13X. In contrast to that, CH4 adsorption starts affecting CO2 adsorption only at higher pressures. Above 7 bar, even an overall decrease in adsorbed amount of CO2 is predicted with increasing pressure.
Both IAST (dotted lines) and RAST (solid lines) predict only an insignificant reduction in CO2 adsorption capacity, meaning that CH4 does not affect CO2 adsorption at all (IAST) or only very little (RAST). On the other hand, CH4 adsorption reduces drastically in the presence of CO2. Essentially no CH4 co-adsorption is predicted by IAST, whereas RAST predicts a still low, but significant co-adsorption of CH4 in the presence of CO2.
4.2 Breakthrough experiments
4.2.1 Reference experiment and parameter estimation
The reference binary breakthrough experiment A3 (see Table 2) is illustrated in Fig. 6 with the measured temperature profiles at the top and the measured composition after the column outlet at the bottom. Simulations with extended isotherms are indicated with dashed lines, those using IAST with dotted lines, and those using RAST with solid lines. This experiment was used to estimate the mass transfer parameters; heat transfer was modeled using the correlation suggested in [39].

Reference binary breakthrough experiment A3 at \(T=\) 25 °C, \(P=\) 10 bar and \(\dot{V}=\) 20 cm\(^{3}\)/s (at P and T) with CO2:CH4 = 50:50 mol% feed mix. Simulations with extended Sips isotherms (dashed lines), IAST (dotted lines), and RAST (solid lines). Simulations with IAST and RAST essentially overlap
As CO2 and CH4 adsorb, the released heat of adsorption gives rise to two distinct temperature fronts that propagate through the column. CH4 is the weaker adsorbing component and therefore propagates faster. Its moderate heat of adsorption gives rise to a temperature increase of about 30 K that propagates through the column. After 140 s and few seconds after it reaches TI5 10 cm below the column outlet, CH4 breaks through and pure CH4 is produced. Note that for TI1 (10 cm from the column bottom), the temperature increase due to CH4 adsorption precedes that of CO2 adsorption only by a few seconds, such that two separate temperature peaks cannot be distinguished. The difference increases the further through the column the CH4 front propagates.
CO2 adsorbs more and has a higher affinity and heat of adsorption than CH4, and its adsorption therefore results in a larger temperature increase of about 100 K. The temperature increase for TI1 is the highest, because the column does not have time to cool down after CH4 adsorption. It decreases the further up the column CO2 adsorption occurs, due the increased time difference between CH4 and CO2 adsorption fronts and thus longer time for cooling down. After 400 s, i.e. roughly 260 s of production of pure CH4, CO2 breakthrough occurs. The shape of the breakthrough curve is initially governed by mass transfer and it is steep, but flattens quickly and is governed by heat rather than mass transfer in the later stage, as the temperature inside the column decreases slowly and additional CO2 is adsorbed. It takes more than 800 s longer until the outlet composition reaches the equimolar inlet composition once the column temperature reaches 25 °C everywhere.
The model predicts both composition and temperature profiles very well. Both the height and timing of the temperature fronts is reproduced almost exactly by the model, and so are the composition profiles. The simulated temperatures show a slightly faster cooling (especially visible for TI1), which can be due to the positioning of the thermocouples: they are mounted in the center of the column at \(r=0\), where radial heat transfer is worse than closer to the wall. The 1D model, however, does not account for any difference in radial heat transfer. It is evident, however, that both IAST and RAST (dotted and solid lines, respectively) predict the time when CH4 breakthrough occurs and the shape of the CO2 breakthrough front better than extended isotherms.
For all further predictions, we use the mass transfer coefficients estimated based on this reference experiment using IAST as summarized in Table 4. Mass transfer is slower for CO2 (\(k_\text {CO2}\) = 0.06 s\(^{-1}\)) than for CH4 (\(k_\text {CH4}\) = 0.2 s\(^{-1}\)). The value for CO2 is similar to that found for the same zeolite 13X and CO2-\(\text {N}_{2}\) mixtures [23] and for pure CO2 [3, 29], but about an order of magnitude higher than the values reported in [7, 20, 37]. Literature values for CH4 are higher than the value estimated here, i.e. around 1 s\(^{-1}\) [3, 29]. Despite those differences, the parameters estimated here, i.e. for the system and conditions of interest, were used in this work to ensure consistency. Where the studies provide the reciprocal diffusion time constant, we converted it to a LDF coefficient based on the equation Global CCS Institute [12]. For all multi-component models, the feed velocity was allowed to vary slightly to account for the uncertainty of the MFCs, and thus better predict the experiments. The deviation between the setpoint of the MFC and the final fitted velocity thus can differ for each experiment and repetition, and also for IAST, RAST and extended isotherms. For extended isotherms, CH4 breakthrough is predicted later than when it occurs experimentally (Fig. 6, dashed lines). This could as well be interpreted as a correct prediction of CH4 breakthrough in combination with the prediction of an early CO2 breakthrough. Due to the large temperature increase upon CO2 adsorption, however, the fitting of the velocity favors the correct prediction of CO2 adsorption over that of CH4 adsorption. On the one hand, when comparing extended isotherms and IAST/RAST, it is clear that extended isotherms result in a higher CH4 adsorption and thus possibly a later breakthrough thereof (Fig. 5). However, for the major part of the breakthrough, CH4 sees a clean column as it propagates faster than CO2, thus the competition is not expected to influence the time until CH4 breaks through significantly. On the other hand, extended isotherms also predict a lower CO2 co-adsorption in the presence of CH4 (Fig. 5), which reduces the time until CO2 breaks through. Therefore, the interpretation suggested above, that the late CH4 breakthrough is an artefact of fitting the velocity, and actually the model can predict the CH4 breakthrough well, but underestimates CO2 adsorption when using extended isotherms, seems reasonable.
4.2.2 Prediction of binary breakthrough experiments

Binary breakthrough experiments A1, A2, A4, A5, A6 and A7 using mass transfer parameters as estimated based on the reference breakthrough experiment A3 and heat transfer coefficients estimated according to [39]. Middle: error in estimated flow \(\Delta u = u_\text {fit}-u_\text {set}\) relative to setpoint for extended Sips isotherms, IAST and RAST. IAST (dotted lines), RAST (solid lines) and extended Sips isotherms (dashed lines). For Exp. A1, all simulations essentially overlap
To assess the predictive capability of the column model, the same mass transfer coefficients as estimated based on the reference experiment (Table 4) are used to predict the other binary breakthrough experiments at changing velocities, pressures and temperatures (Table 2). The results are illustrated in Fig. 7. Only one repetition is shown for each combination of temperature, pressure and flow, as all experiments featured an excellent reproducibility.
As for the reference breakthrough experiment, the overall agreement between simulation results and experimental data is very good. Timing, shape and height of the temperature fronts are predicted well using the literature heat transfer correlation [39]. The correlation predicts a higher heat transfer in the case of higher Reynolds numbers, i.e. higher pressures and flows (Fig. 8). As before, it is clear that IAST (dotted lines) and RAST (solid lines) result in better predictions than extended isotherms (dashed lines): depending on the temperature and flow, extended isotherms overestimate the time until CH4 breaks through. The difference is barely noticeable for low pressures and high temperatures, so conditions where there is little competition, but significant for high pressures. However, when using IAST or RAST, the agreement is excellent.

Heat transfer coefficient for different binary and ternary breakthrough experiments depending on Reynolds number calculated according to [39] based on the inlet composition, pressure, temperature and velocity
Note that the simulation results shown in Fig. 7 are fully predictive, except for the uncertainty of the mass flow controller and thus of the feed velocity. The relative deviations of the velocity from the setpoint are shown in Fig. 7 (middle) and differ for extended Sips isotherms, IAST and RAST. In all cases, the estimated velocity for extended Sips isotherms (Fig. 7 middle, dashed lines) is the lowest, thus further supporting the assumption that indeed CH4 breakthrough can be modeled well, but that fitting favors a correct prediction of CO2 breakthrough over that of CH4 breakthrough. In fact, if the velocity for extended Sips isotherms were to be increased to that of RAST or even IAST, the breakthrough curves simulated with extended isotherms would shift to the left, thereby resulting in a better prediction of CH4 breakthrough, but a simulated CO2 breakthrough earlier than that measured.
4.2.3 Prediction of ternary breakthrough experiments
Figure 9 shows a typical ternary breakthrough experiment (Exp. A6) for a feed mixture representative of shifted syngas produced from steam reforming of natural gas (CO2:CH4:H2 = 20:5:75 mol%). For all ternary experiments, the same mass transfer parameters as estimated based on the binary case were used (Table 4) to test the predictive capability of the model, and a high value of 1 s\(^{-1}\) was chosen for H2. An enlarged view is provided in Fig. 10. The heat transfer coefficients were calculated for each experiment according to [39]. Due to the lower kinematic viscosity of the gas phase, the Reynolds numbers are lower for experiments at the same pressure, temperature and flow as for corresponding binary experiments, but the specific heat capacity (per mass of gas) is much higher, so as the heat transfer coefficients are higher than for the binary case (Fig. 8).

Exemplary ternary breakthrough experiment B6 at \(T=\) 45 °C, \(P=\) 10 bar and \(\dot{V}=\) 20 cm\(^{3}\)/s (at P and T) inflow with a 20:5:75 CO2:CH4:H2 mix (mol%). Simulations with extended Sips isotherms (dashed lines), IAST (dotted lines), and RAST (solid lines) essentially overlap
Out of the three components in the feed, hydrogen adsorbs the least and therefore breaks through first. The weak adsorption (low affinity, low heat of adsorption) gives rise to the very fast propagation of a temperature front with an increase in temperature of approximately 2 °C) (see Fig. 10). H2 breaks through after only about 50 s. CH4 is the second component break through. The temperature increase related to CH4 adsorption is much lower than for the case of the binary breakthrough experiments due to its concentration of only 5% in the feed, but nevertheless more pronounced than for H2 as it can clearly be seen in the figure. Due to the lower concentration and a convex CH4 isotherm shape, CH4 breaks through later than for the binary case, in this case after around 220 s. Thus, pure hydrogen is produced for a period of about 170 s. CO2 adsorbs strongly and has a high heat of adsorption, thus giving rise to the propagation of a high temperature front with a temperature increase of approximately 90 K, so slightly lower than for the binary case due to the lower partial pressure. It breaks through after over 1150 s, so much later than for the binary case, due to the lower concentration but high affinity of CO2 already at low partial pressures. In contrast to the binary case, the temperature increase is similar for all thermocouples, because there is less heat released as CH4 adsorbs and sufficient time for cooling down before the CO2 front reaches the same position.
Comparing the experimental measurements with the simulation results demonstrates that also in this case the predictive correlation for heat transfer works rather well. The model predicts a temperature increase slightly below that measured for CO2 at the center of the column, as expected for a 1D model that does not account for radial temperature profiles. The temperature increase due to CH4 adsorption is slightly overestimated (Fig. 9, insert), which could be related to the use of an average heat of adsorption, that is higher than the value for low loadings (Fig. 4).

Exemplary ternary breakthrough experiment B6. Simulations with IAST (dotted lines) and transfer parameters estimated based on reference binary breakthrough experiment (dotted lines) or transfer parameters estimated based on this experiment (dash-dotted lines, Table 4)
The shape of the composition profiles indicates that the mass transfer coefficients from the binary case slightly underestimate mass transfer in the ternary case, which can be appreciated even better in the enlarged view shown in 10. Indeed, the simulated CO2 and CH4 composition fronts are flatter than the ones measured experimentally, and so is the shape of the temperature front related to CH4 adsorption. To test this hypothesis, we also estimated mass transfer coefficients based directly on this ternary experiment, including also that of H2. They are larger than for the binary case for all components, as can be expected from the gas composition: diffusion in H2-rich mixtures is typically faster than in CO2 or CH4-rich mixtures [22] (Table 4). The simulation results with these higher transport parameters are shown in Fig. 10 with dash-dotted lines in comparison to the simulation results with the binary transport parameters (IAST only for both cases). Indeed, both the shape of the CH4 breakthrough as well as that of the corresponding temperature front can be predicted better with the ternary transport parameters. However, as we aim at using a constant set of transport parameters also for predicting cyclic operation, we are interested in finding parameters that work reasonably well to predict mass transfer over a wide range of compositions (and including streams that consist mainly of CO2), pressures, and temperatures and thus decided to continue using the parameters estimated based on the reference binary experiment.
The additional breakthrough experiments and simulations using the mass transfer coefficients as estimated based on the binary reference experiment and the literature heat transfer correlation [39] are illustrated in figure 11.

Ternary breakthrough experiments B1, B2, B3, B4, B5 and B7 using mass transfer parameters as estimated based on the reference binary breakthrough experiment A3 and heat transfer coefficients estimated according to [39]. Simulations with extended Sips isotherms (dashed lines), IAST (dotted lines) and RAST (solid lines). A big difference between the different multi-component models is visible only for high pressure (Exp. B5) whereas for the other simulations, the curves essentially overlap
Overall, the agreement is rather good. Especially the heat transfer correlation works very well and almost exactly predicts the temperature fronts. However, the duration until CH4 breakthrough occurs is slightly overestimated in some cases. This can be related to the use of slightly lower mass transfer coefficients than estimated based on the reference ternary breakthrough experiment. By estimating both heat and mass transfer parameters based on each individual experiment or by adjusting the heat of adsorption to the loading range of each experiment, the quality of the fits could be improved further, but we consider the agreement satisfactory and deem a confirmation of the predictive capability of the model more important than an exact agreement between experiment and simulation with individually estimate parameters, that are difficult to generalize for with the aim of simulating cyclic experiments.
Note that for ternary breakthrough experiments, there is barely any difference between the simulation results with the three different multi-component adsorption models, in strong contrast to the binary case. This is related to the lower CO2 and CH4 concentrations and thus partial pressures in the feed, at which competition plays a smaller role (see Fig. 5). Only at higher total pressures, i.e. for Exp. B5 at 25 bar, the difference is considerable. In this case, IAST and RAST predict the experiment better than extended isotherms, with the latter predicting an even later CH4 breakthrough.
4.2.4 Real adsorbed solution theory
Note that we here successfully predict the breakthrough experiments using IAST and even reach a good agreement for many experiments with the use of extended isotherms; therefore it does not seem necessary to include a more complex multi-component model such as RAST in the analysis. However, breakthrough experiments only contain a limited amount of information about competitive behavior. In contrast, in cyclic experiments, the choice of competitive isotherms is challenged over a much wider range of conditions and compositions, hence cyclic experiments provide much more information on competitive behavior than simple breakthrough studies. They cover compositions from 0 to almost 100% for all three components, pressures between 0.1 bar and 25 bar, and temperatures as low as 5 °C. Thus, such experiments can easily reach the limit of either extended isotherms or IAST, even though those performed well in predicting breakthrough experiments. In our case, neither approach can predict the cyclic experiments sufficiently well. Contrary to IAST prediction, which indicates a very low CH4 co-adsorption in the presence of any adsorbed CO2 and thus results in an almost immediate CH4 breakthrough during adsorption for a column with residual adsorbed CO2 (as is the case in any cyclic experiment where the column is not fully regenerated), in the cyclic experiments it was possible to contain CH4 within the column and produce a pure stream of H2 during high pressure adsorption [41]. When using extended isotherms, the speed at which the CO2 front propagates through the column is overestimated. Hence, based on the cyclic experiments described in part two of this work, we found that the agreement between experimental and simulation results can be improved a lot when using RAST. However, it is clear that the multi-component model, that predicts adsorption well for cyclic experiments still should be valid for breakthrough studies, as those included in this analysis. As shown before, RAST performs much better than extended Sips isotherms and comparable to IAST in all cases, thus demonstrating that it is a good model not only for the cyclic experiments shown in part two of this series, but also for breakthrough experiments. In contrast to that, the limit of extended isotherms is already reached for some of the binary breakthrough experiments, whereas IAST performs well for breakthrough studies but reaches its limit when simulating cyclic experiments, for which an accurate description of co-adsorption of CH4 in the presence of pre-adsorbed CO2 is essential.
4.2.5 Uncertainty in feed velocity
The feed velocity was treated as a fitting parameter for all examined cases. Figure 12 shows a comparison between the estimated velocity and the setpoint (a) as well as between the resulting relative error and the error expected for the MFCs (2% of the full scale) (b). All experiments and repetitions as well as fits with all three multi-component models are included. Almost 90% of all estimated velocities are within the expected error (Fig. 12b) with maximum deviations of just over 10% from the setpoint velocity. The outlier shown in red with a deviation of four times the specified uncertainty of the MFC corresponds to experiment A3 fitted using extended isotherms. As discussed above, the model predicted a later CH4 breakthrough than measured experimentally for this experiment when using extended isotherms. When increasing the velocities in the simulation up the range of the MFC specifications, both CH4 and CO2 breakthrough would be predicted to occur earlier, thus eventually resulting in a correct prediction of CH4 breakthrough, but an early prediction of CO2 breakthrough. The large deviation from the expected uncertainty of the MFC for this experiment thus further supports this as the correct interpretation. Note that this effect can be seen particularly well for Exp. A3, as the inflow is close to the maximum range of the MFC, and thus has a low expected relative error of just above 2%.

a error in estimated flow relative to setpoint; b flow error relative to error specified for MFC; all points outside the specified error of the MFC are shown in light grey, the corresponding velocity errors are shown in a in light grey. All experiments and repetitions, and the fits with IAST, RAST and extended isotherms are included. The outlier in red in both figures corresponds to Exp. A3 fitted using extended isotherms (Color figure online)
5 Conclusions
In this work, CH4 and H2 equilibrium isotherms were measured on pelleted zeolite 13X at various temperatures and up to 30 bar, thus complementing prior CO2 isotherm measurements. CO2 adsorbs strongly on zeolite 13X, CH4 less and H2 very little, which results in a high selectivity of CO2 over both CH4 and H2 on the one hand, but also a high selectivity of CH4 over H2 on the other hand. Thus, zeolite 13X is a suitable material for H2 purification (H2 as light product), or CO2 purification (CO2 as heavy product), or even for combined adsorption processes that target a co-purification of CO2 and H2 from a stream with an impurity like CH4, i.e. with an adsorption strength in between CO2 and H2.
Zeolite 13X was characterized further through binary (CO2–CH4) and ternary (H2–CO2-CH4) breakthrough experiments at a variety of temperatures, pressures and flowrates. Comparing experimental measurements with simulation results allows for an estimation of transport parameters and for an assessment of the prediction accuracy of the model. A high predictive capability of the model at different conditions is essential for accurately simulating and optimizing cyclic adsorption processes, which naturally cover a wide range of compositions at various temperatures, pressures and flowrates throughout the cycle.
In this work, we have demonstrated the high predictive capability of the 1D column model beyond previous studies. Most importantly, we have shown that after calibrating the model by estimating the mass transfer coefficients using a single breakthrough experiment only, the model can be used in a fully predictive manner for different temperatures, pressures, flowrates and with a slightly lower accuracy also for different compositions. In addition, a literature correlation for the heat transfer coefficient was identified that results in a good prediction of heat transfer over the whole experimental range.
For a good agreement between simulation results and experimental measurements, in addition to good estimates of heat and mass transfer parameters, also accurately modeling multi-component adsorption is key. A comparison of three different models for multi-component adsorption showed that extended Sips isotherms underpredict CO2 adsorption at high partial pressures and low temperatures. In contrast to that, both the ideal adsorbed solution theory and the real adsorbed solution theory result in a good agreement between experimental and simulation results over the whole range of experimental conditions. However, breakthrough studies alone are not sufficient to establish the best multi-component model, as the less adsorbing components (H2 and CH4) propagate faster through the column than CO2, and thus adsorb mainly on a CO2-free column. Hence, their uptake is not influenced through competition with CO2. This shows that breakthrough experiments alone, even though essential for estimating transport parameters, may not be sufficient to evaluate co-adsorption of the lighter component.
Overall, this work paves the way for a full experimental demonstration of and further model validation for cyclic adsorption processes. This is presented in the second part of this series, which focuses on vacuum pressure swing adsorption processes for H2 purification with integrated CO2 capture in the context of low-carbon hydrogen production from fossil fuels.
References
Antonini, C., Treyer, K., Streb, A., van der Spek, M., Bauer, C., Mazzotti, M.: Hydrogen production from natural gas and biomethane with carbon capture and storage-a techno-environmental analysis. Sustain. Energy Fuels 4, 2967–2986 (2020). https://doi.org/10.1039/D0SE00222D
Bartholdy, S., Bjørner, M.G., Solbraa, E., Shapiro, A., Kontogeorgis, G.M.: Capabilities and limitations of predictive engineering theories for multicomponent adsorption. Ind. Eng. Chem. Res. 52(33), 11552–11563 (2013). https://doi.org/10.1021/ie400593b
Brea, P., Delgado, J., Águeda, V.I., Gutiérrez, P., Uguina, M.A.: Multicomponent adsorption of \(\text {H}_{2}\), \(\text {CH}_{4}\), CO and CO2 in zeolites NaX, CaX and MgX. evaluation of performance in PSA cycles for hydrogen purification. Microporous Mesoporous Mater. 286, 187–198 (2019). https://doi.org/10.1016/j.micromeso.2019.05.021
Casas, N., Schell, J., Pini, R., Mazzotti, M.: Fixed bed adsorption of CO2 /\(\text {H}_{2}\) mixtures on activated carbon: experiments and modeling. Adsorption 18(2), 143–161 (2012). https://doi.org/10.1007/s10450-012-9389-z
Casas, N., Schell, J., Joss, L., Mazzotti, M.: A parametric study of a PSA process for pre-combustion CO2 capture. Sep. Purif. Technol. 104, 183–192 (2013). https://doi.org/10.1016/j.seppur.2012.11.018
Cavenati, S., Grande, C.A., Rodrigues, A.E.: Adsorption equilibrium of methane, carbon dioxide, and nitrogen on zeolite 13X at high pressures. J. Chem. Eng. Data 49(4), 1095–1101 (2004). https://doi.org/10.1021/je0498917
de Visser, E., Hendriks, C., Barrio, M., Mølnvik, M.J., de Koeijer, G., Liljemark, S., Gallo, Y.L.: Dynamis CO2 quality recommendations. Int. J. Greenhouse Gas Control 2(4), 478–484 (2008). https://doi.org/10.1016/j.ijggc.2008.04.006
Delgado, J.A., Águeda, V., Uguina, M., Sotelo, J., Brea, P., Grande, C.A.: Adsorption and diffusion of \(\text {H}_{2}\), CO, \(\text {CH}_{4}\), and CO2 in BPL activated carbon and 13X zeolite: evaluation of performance in pressure swing adsorption hydrogen purification by simulation. Ind. Eng. Chem. Res. 53(40), 15414–15426 (2014). https://doi.org/10.1021/ie403744u
Divekar, S., Arya, A., Hanif, A., Chauhan, R., Gupta, P., Nanoti, A., Dasgupta, S.: Recovery of hydrogen and carbon dioxide from hydrogen psa tail gas by vacuum swing adsorption. Sep. Purif. Technol. 254, 117113 (2021). https://doi.org/10.1016/j.seppur.2020.117113
Dreisbach, F., Staudt, R., Keller, J.: High pressure adsorption data of methane, nitrogen, carbon dioxide and their binary and ternary mixtures on activated carbon. Adsorption 5(3), 215–227 (1999). https://doi.org/10.1023/A:1008914703884
European Commission (2020) A hydrogen strategy for a climate-neutral Europe. https://ec.europa.eu/energy/sites/ener/files/hydrogen_strategy.pdf
Global CCS Institute (2020) Facilities database. https://co2re.co/FacilityData, accessed: 08/2020
Glueckauf, E.: Theory of chromatography part 10 formulæ for diffusion into spheres and their application to chromatography. J. Chem. Soc. Faraday Trans. I and II 51, 1 (1955). https://doi.org/10.1039/TF9555101540
Grande, C.A., Lopes, F.V., Ribeiro, A.M., Loureiro, J.M., Rodrigues, A.E.: Adsorption of off-gases from steam methane reforming (\(\text {H}_{2}\), CO2, \(\text {CH}_{4}\), CO and \(\text {N}_{2}\)) on activated carbon. Sep. Sci. Technol. 43(6), 1338–1364 (2008). https://doi.org/10.1080/01496390801940952
Hefti, M., Marx, D., Joss, L., Mazzotti, M.: Adsorption equilibrium of binary mixtures of carbon dioxide and nitrogen on zeolites ZSM-5 and 13X. Microporous Mesoporous Mater. 215, 215–228 (2015). https://doi.org/10.1016/j.micromeso.2015.05.044
Hydrogen Council: Global interest for hydrogen soars as hydrogen council grows to 90+ members. https://hydrogencouncil.com/en/newmemberannouncement2020-2/ (2020)
IEA: The future of hydrogen. https://www.iea.org/reports/the-future-of-hydrogen, Paris (2019)
IEAGHG: Reference data and supporting literature reviews for SMR based hydrogen production with CCS. http://documents.ieaghg.org/index.php/s/7Ii9WGEAufoMPvP/download, 2017–TR3 (2017a)
IEAGHG: Techno-economic evaluation of SMR based standalone (merchant) hydrogen plant with CCS. https://ieaghg.org/exco_docs/2017-02.pdf, 2017–TR3 (2017b)
ISO 14687-2:2012(E) (2012) Hydrogen fuel – Product specification – Part 2: Proton exchange membrane (PEM) fuel cell applications for road vehicles
Kamiuto, K., Goubaru, A., Ermalina, : Diffusion coefficients of carbon dioxide within type 13X zeolite particles. Chem. Eng. Commun. 193(5), 628–638 (2006). https://doi.org/10.1080/00986440500193970
Kumar, R., Kratz, W.C., Guro, D.E., Rarig, D.L., Schmidt, W.P.: Gas mixture fractionation to produce two high purity products by pressure swing adsorption. Sep. Sci. Technol. 27(4), 509–522 (1992). https://doi.org/10.1080/01496399208018897
Liu, J., Keskin, S., Sholl, D.S., Johnson, J.K.: Molecular simulations and theoretical predictions for adsorption and diffusion of CH\(_4\)/H\(_2\) and CO2 /CH\(_4\) mixtures in ZIFs. J. Phys. Chem. C 115(25), 12560–12566 (2011). https://doi.org/10.1021/jp203053h
Marx, D., Joss, L., Hefti, M., Mazzotti, M.: Temperature swing adsorption for postcombustion CO2 capture: Single- and multicolumn experiments and simulations. Ind. Eng. Chem. Res. 55(5), 1401–1412 (2016). https://doi.org/10.1021/acs.iecr.5b03727
Mofarahi, M., Bakhtyari, A.: Experimental investigation and thermodynamic modeling of CH\(_4\)/N\(_2\) adsorption on zeolite 13X. J. Chem. Eng. Data 60(3), 683–696 (2015). https://doi.org/10.1021/je5008235
Myers, A.L., Prausnitz, J.M.: Thermodynamics of mixed-gas adsorption. AIChE J. 11(1), 121–127 (1965). https://doi.org/10.1002/aic.690110125
NETL: Quality guidelines for energy system studies: CO2 impurity design arameters. https://doi.org/10.2172/1566771 (2019)
NIST Chemistry WebBook: NIST standard reference database number 69. https://doi.org/10.18434/T4D303 (2018)
Ottiger, S., Pini, R., Storti, G., Mazzotti, M.: Competitive adsorption equilibria of CO2 and \(\text {CH}_{4}\) on a dry coal. Adsorption 14(4–5), 539–556 (2008). https://doi.org/10.1007/s10450-008-9114-0
Park, Y., Ju, Y., Park, D., Lee, C.H.: Adsorption equilibria and kinetics of six pure gases on pelletized zeolite 13X up to 1.0 MPa: CO2, CO, \(\text {N}_{2}\), \(\text {CH}_{4}\), Ar and \(\text {H}_{2}\). Chem. Eng. J. 292, 348–365 (2016). https://doi.org/10.1016/j.cej.2016.02.046
Pini, R., Ottiger, S., Rajendran, A., Storti, G., Mazzotti, M.: Reliable measurement of near-critical adsorption by gravimetric method. Adsorption 12(5–6), 393–403 (2006). https://doi.org/10.1007/s10450-006-0567-8
Riboldi, L., Bolland, O.: Pressure swing adsorption for coproduction of power and ultrapure \(\text {H}_{2}\) in an IGCC plant with CO2 capture. Int. J. Hydrog. Energy 41(25), 10646–10660 (2016). https://doi.org/10.1016/j.ijhydene.2016.04.089
Ruthven, D.M.: Principles of Adsorption and Adsorption Processes. Wiley, New York (1984)
Ruthven, D.M., Farooq, S., Knaebel, K.S.: Pressure Swing Adsorption. VCH Publishers, New York (1994)
Schell, J., Casas, N., Pini, R., Mazzotti, M.: Pure and binary adsorption of CO2, \(\text {H}_{2}\), and \(\text {N}_{2}\) on activated carbon. Adsorption 18(1), 49–65 (2012). https://doi.org/10.1007/s10450-011-9382-y
Schell, J., Casas, N., Marx, D., Mazzotti, M.: Precombustion CO2 capture by pressure swing adsorption (PSA): Comparison of laboratory PSA experiments and simulations. Ind. Eng. Chem. Res. 52(24), 8311–8322 (2013). https://doi.org/10.1021/ie3026532
Schmidt, O., Gambhir, A., Staffell, I., Hawkes, A., Nelson, J., Few, S.: Future cost and performance of water electrolysis: an expert elicitation study. Int. J. Hydrog. Energy 42(52), 30470–30492 (2017). https://doi.org/10.1016/j.ijhydene.2017.10.045
Silva, J.A., Schumann, K., Rodrigues, A.E.: Sorption and kinetics of CO2 and \(\text {CH}_{4}\) in binderless beads of 13X zeolite. Microporous Mesoporous Mater. 158, 219–228 (2012). https://doi.org/10.1016/j.micromeso.2012.03.042
Sircar, S., Kratz, W.C.: Simultaneous production of hydrogen and carbon dioxide from steam reformer off-gas by pressure swing adsorption. Sep. Sci. Technol. 23, 2397–2415 (1988). https://doi.org/10.1080/01496398808058461
Specchia, V., Baldi, G., Sicardi, S.: Heat transfer in packed bed reactors with one phase flow. Chem. Eng. Commun. 4(1–3), 361–380 (1980). https://doi.org/10.1080/00986448008935916
Staffell, I., Scamman, D., Abad, A.V., Balcombe, P., Dodds, P.E., Ekins, P., Shah, N., Ward, K.R.: The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 12(2), 463–491 (2019)
Streb, A., Mazzotti, M.: Adsorption for efficient low carbon hydrogen production – part 2: cyclic experiments and model predictions. Adsorption (2021)
Streb, A., van der Spek, M., Joersboe, J., Asgari, M., Queen, W., Mazzotti, M.: Report on characterization of equilibria and transport phenomena in promising new adsorbents for CO2 /\(\text {H}_{2}\) separation. https://www.sintef.no/projectweb/elegancy/publications/, deliverable D1.1.2. of the ELEGANCY project (2019b)
Streb, A., Mazzotti, M.: Novel adsorption process for co-production of hydrogen and CO2 from a multicomponent stream - part 2: Application to SMR and ATR gases. Ind. Eng. Chem. Res. 59(21), 10093–10109 (2020)
Streb, A., Hefti, M., Gazzani, M., Mazzotti, M.: Novel adsorption process for co-production of hydrogen and CO2 from a multicomponent stream. Ind. Eng. Chem. Res. 58(37), 17489–17506 (2019a). https://doi.org/10.1021/acs.iecr.9b02817
Susmozas, A., Iribarren, D., Zapp, P., Linßen, J., Dufour, J.: Life-cycle performance of hydrogen production via indirect biomass gasification with CO2 capture. Int. J. Hydrog. Energy 41(42), 19484–19491 (2016). https://doi.org/10.1016/j.ijhydene.2016.02.053
Voldsund, M., Jordal, K., Anantharaman, R.: Hydrogen production with CO2 capture. Int. J. Hydrog. Energy 41(9), 4969–4992 (2016). https://doi.org/10.1016/j.ijhydene.2016.01.009
Walton, K.S., Sholl, D.S.: Predicting multicomponent adsorption: 50 years of the ideal adsorbed solution theory. AIChE J. 61(9), 2757–2762 (2015)
Wilson, G.M.: Vapor-liquid equilibrium. XI. A new expression for the excess free energy of mixing. J. Am. Chem. Soc. 86, 127–130 (1964). https://doi.org/10.1021/ja01056a002
Acknowledgements
ACT ELEGANCY, Project No 271498, has received funding from DETEC (CH), BMWi (DE), RVO (NL), Gassnova (NO), BEIS (UK), Gassco, Equinor and Total, and is cofunded by the European Commission under the Horizon 2020 programme, ACT Grant Agreement No 691712.This project is supported by the pilot and demonstration programme of the Swiss Federal Office of Energy (SFOE).
Funding
Open Access funding provided by ETH Zurich.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
This is the first part of a two-part contribution to the Special Issue of Adsorption dedicated to the memory of Dr. Shivaji Sircar, and honoring his multi-faceted pioneering contributions to Adsorption Science and Technology. For the senior author of this work, Dr. Sircar has been, during the last three decades, a role model and a mentor, with his scientific insight and inspiring ideas, as well as with his kindness and friendship. Dr. Sircar’s ideas have influenced everything from the foundations of adsorption to industrial practice, and his impact on the field of adsorption remains unsurpassed. He will be dearly missed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Streb, A., Mazzotti, M. Adsorption for efficient low carbon hydrogen production: part 1—adsorption equilibrium and breakthrough studies for H2/CO2/CH4 on zeolite 13X. Adsorption 27, 541–558 (2021). https://doi.org/10.1007/s10450-021-00306-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-021-00306-y