
Abstract
Quasielastic neutron scattering (QENS) allows measurement of the molecular displacements in time and space, from pico- to tens of nanoseconds and from Ångstroms to nanometers, respectively. The method probes dynamics from fast vibrational modes down to slow diffusive motion. Every scattering experiment leads to a dynamic structure factor \(S\left( {\vec Q,\omega } \right)\) or its spatial and temporal Fourier transform (van Hove correlation function \(G\left( {\vec r,t} \right)\)). This shows exactly where the atoms are and how they move. In this manuscript the basics of the QENS method are presented and a few examples highlighting the potentials of QENS are given: (i) diffusion of liquids and gases in nano- and mesoporous materials; (ii) hydrogen dynamics in a high temperature polymer electrolyte fuel cell (HT-PEFC) and (iii) influence of the surface interactions on polymer dynamics in nanopores.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Knowledge of dynamics on the molecular scale is equally important in material research and in biology. For solid state crystals, the dynamics is typically represented by vibrations and oscillations of atoms or defects in a crystal lattice. For soft matter systems, the dynamics of molecules or macromolecules (polymers) over large time and lengths scales are important to clarify their functional properties. Exploring both self-correlated and collective dynamics, the main functional principles can be understood.
Neutron scattering has great potential for studying the displacements of atoms and molecules over wide time and length scales. This important advantage of neutron scattering is exploited to allow investigation of the dynamics of atoms in a crystal lattice and chain dynamics of polymer macromolecules. Neutron scattering experiments measure the intensity scattered into a solid angle between Ω and Ω + ΔΩ as a function of the energy difference between the incident \(E\) and scattered \(E +\Delta E\) neutrons. In a wider sense, for a QENS experiment it is only necessary that the energy transfer \(\Delta E\) be small compared to the incident energy \(E\) of the scattered particles.
One of the pioneering works on QENS were performed on solid proton conductors [1, 2] combining incoherent inelastic neutron scattering spectroscopy (IINS) and quasi-elastic neutron scattering (QENS). Later advances in neutron instrumentation and in data analysis allowed to open new possibilities of the method for polymers and other soft matter system including biomacromolecules and proteins [3, 4]. In the last decade there are a number of works where QENS was used to study solid proton conductors based on Metal–Organic Frameworks (MOF) [5, 6].
This review contains a few examples demonstrating the power of the QENS method. In particular, we will focus on (i) measurements of the diffusion of liquid, gases and polymers in mesoporous materials; (ii) hydrogen dynamics in a high temperature polymer electrolyte fuel cell (HT-PEFC) and energy relevant materials. In the first chapter the main principles of QENS measurements and limitations of the method will be considered. In the following part the applications of QENS will be given. The potentials of QENS shall be discussed in connection with Pulsed Field Gradient (PFG) NMR and solid state NMR as complementary techniques, which are also presented in this thematic issue.
2 Principles of QENS measurement
Scattering of thermal neutrons yields information on the sample by measurement and analysis of the double differential cross section
i.e. the intensity of scattered neutrons with energy Ef into a given direction θ, where Ω is the solid angle. The energy transfer, i.e. the difference of kinetic energy before and after the scattering, ΔE = Ef–Ei is related to the angular frequency ω = \(\hbar\)ΔE.
The momentum transfer is Q, in which the wave vector is given by \(\vec Q = {\vec k_i} - {\vec k_f}\) where \({\vec k_i}\) and \({\vec k_f}\) are the wave vectors of the incoming and outgoing (scattered) neutrons and are related to the neutron wavelength \({k_{i,f}} = \frac{2\pi }{{{\lambda_{i,f}}}}\). Therefore \(\omega = \frac{{{\Delta }E}}{\hbar } = \left( {\frac{{{\hbar^2}k_f^2}}{2m} - \frac{{{\hbar^2}k_i^2}}{2m}} \right)\;\;\) and the neutron velocity is \({v_{i,f}} = \frac{{\hbar {k_{i,f}}}}{m}\). The energy transfer ∆E and ω can be determined either by measuring the neutron wavelength by diffraction or by measurements of the neutron velocities \({v_i}\) and \({v_f}\). Note that for all problems discussed in this article |\({\vec k_i}\)| ≈ |\({\vec k_f}\)| and therefore \(Q = \frac{4\pi }{{\lambda_i}}{\text{sin}}\left( {\frac{\theta }{2}} \right)\) = 2\({k_i}{\text{sin}}\left( {\frac{\theta }{2}} \right)\) can be assumed. Finally, bi denotes the scattering length of atom nucleus i and 〈…〉 is the ensemble average.
The unique features of neutrons are: (i) the isotope and spin dependence of bi, (ii) typical wavelengths of cold and thermal neutrons that match molecular and atomic distances and (iii) even slow motions of molecules cause neutron velocity changes that are large enough to be detectable. In particular neutron spin echo (NSE) is able to resolve changes of the neutron velocity Δv of the order of 10−5 νi. The intermediate scattering function S(Q,t) is defined as the Fourier transform of S(Q,\(\;\omega )\;\) and directly depends on the time dependent atomic positions. For nuclei of the same type we have:
where \({r_n}\left( t \right)\) is the position vector of an atom “n” (of altogether N atoms) at time t and \({r_m}\left( 0 \right)\) is that of an atom “m” at time 0. \({G_{pair}}\left( {r,t} \right)\) is the van Hove pair correlation function describing the probability to find an atom around a position \(r + dr\) if an atom has been at time 0 at the origin.
Chemically equivalent atoms may have a number of different scattering lengths that are randomly distributed in the sample. In particular the proton scattering length depends on the relative orientation of proton and neutron spin. The average value 〈bi〉 leads to coherent scattering, while the fluctuating part bi—〈bi〉 relates to incoherent scattering that determines the atom–atom self-correlation, i.e.:
where Gself is the self-part of the van Hove correlation function giving the probability to find a given nucleus i around the position \(r + dr\) if it was located at \(\;r\left( 0 \right)\) at t = 0. Thus, incoherent scattering provides information about the correlations between the positions of the same nucleus (usually hydrogen) at different times and is an ideal tool to directly measure diffusive processes. The van Hove correlation function corresponds, in PFG NMR, to the “mean propagator”, where the time and space scales are in the range of, respectively, milliseconds and micrometers [7, 8].
Applying the Gaussian approximation, i.e. assuming that the atomic displacement distribution functions are Gaussian, Eq. (4) transforms into
where \(\left\langle {r_i^2\left( t \right)} \right\rangle\) is the mean-square displacement of atom “i”. For most cases of diffusion in homogeneous media, the isotropic motion of a given atom (or single molecule) is well represented by the diffusion equation (Fick’s second law):
where \({D_{self}}\) is the self-diffusion coefficient. Function \(p\left( {\vec r,\;t} \right)\) represents the probability to find a given nucleus i around the position \(r + dr\) if it was located at \(\;r\left( 0 \right)\) at t = 0. If the atom is at the origin at time zero \({G_{self}}\left( {r,0} \right) = \;\delta \left( r \right)\), a Gaussian expression for the self-correlation function can be applied:
Fourier transform of the self-correlation function leads to the intermediate scattering function:
Self-diffusivity \({D_{self}}\) of the process under study may be introduced by the Einstein relation \(\left\langle {r_i^2\left( t \right)} \right\rangle = 6{D_{self}}t\). The Einstein relation is equivalent to Fick's first law (Eq. 7), where the self-diffusivity is introduced as a factor of proportionality between the concentration gradient of labelled molecules and their flux density.
The scattering function for the coherent scattering (Eq. 3) depends on the pair correlation function:
where the self-part was discussed above (see Eqs. 3–6) and the second term \({G_d}\left( {r,t} \right)\) connects the relative motion of different atoms as a function of time. The QENS experiment is following the change of local concentration gradient around the equilibrium. By this way the transport diffusivity Dt can be obtained [9] from the collective diffusion coefficient at the limit of small values of the momentum transfer Q.
The coherent scattering gives a valuable information about the transport diffusivity of a variety of molecules, including N2, O2, CO2, etc. in zeolites. In particular, the coherent QENS studies indicate that the transport diffusivity is rarely constant and that increasing or decreasing trends can be found as a function of concentration [10].
Most of the scientific interest relates to structure and dynamics in the mesoscopic regime [11,12,13]. Then, instead of exact atomic positions, a coarse grained description in terms of scattering length density Δρ(\(\vec r\),t) is used which considers the average scattering density of a specimen (e.g. polymer segment and a solvent molecule or guest molecule and host porous matrix). The scattering in the lower Q regime only depends on the scattering length density difference, the contrast Δρ(\(\vec r\),t) = ρ2—ρ1 where ρ1 and ρ2 are scattering length densities of different specimens. The related coherent scattering function is then:
The corresponding small angle neutron scattering (SANS) intensity is proportional to S(Q,t = 0). In most of the following we use \(Q = \left| {\vec Q} \right|\), since the scattering functions in question depend only on \(\left| {\vec Q} \right|\). At this level contrast variation and matching by hydrogen (h)/deuterium (d) labelling can be employed to enhance or suppress the contribution of a signal from selected subunits in a system. Only the motion of those structures that contribute to the SANS intensity are seen in the corresponding NSE experiment. Prominent examples of successful application of contrast variation are the investigations of the single chain dynamics of polymers in melts [14] or the investigation of d-polymers that contain only a h-labeled section, i.e. at the ends, at branching points [15] or at its center in a fully deuterated matrix [16].
As a prerequisite for studies into polymer dynamics, the relevant structural information needs to be available. On the mesoscopic space–time scale of interest SANS is the method of choice. There a well-collimated, monochromatic (within 5 to 20%) neutron beam hits the sample and the scattering profile is monitored by a 2D detector. Most relevant neutron sources feature at least one of such instruments [17]. Since this review focuses on investigations of dynamic features mainly by quasielastic neutron scattering (QENS), we briefly discuss the important instrument categories:
Time of Flight Spectroscopy (TOF) TOF-instruments measure the neutron velocities before and after scattering, in order to determine energy changes during scattering. This is performed by exposing the sample to monochromatic neutron pulses (typical band width δλ/λ ≅ 10−2). The beam pulses are then produced by a chopper system that opens the beam path periodically for a short moment. For cold neutrons typical frequencies are between 20 and 60 Hz with an opening ratio in the order of 1 out of 100. Typical pulse widths are in the order of tens of microseconds. Thereafter the neutron pulses hit the sample and are scattered. In this process some neutrons exchange kinetic energy with the moving atoms in the sample, thereby they change their velocities. After the scattering process the neutrons traverse a well-defined flight distance between the sample and the detectors. For an efficient TOF instrument it is mandatory to cover a solid angle as large as possible with detector tubes, which requires thousands of detectors. Neutrons that have gained energy at the sample will arrive earlier. Those that have lost energy will arrive later. The detected neutrons are stored in a histogrammic memory according to their arrival time. For each detector such a histogram vs. time is obtained. A monitor (M0) in the direct beam serves to normalize the histograms to the incoming neutron flux. The counting histogram obtained at the different scattering angles contains information on the double differential cross section to be determined. The method allows to reach a relative resolution 10−3. Typically such instruments are used to measure incoherent scattering from hydrogen containing polymers that reveals the self-correlation function of the moving hydrogen [18].
Backscattering (BS) Instruments are crystal spectrometers where crystal reflections determine the wavelength of the incoming and scattered neutrons. The highest energy resolution achievable by Bragg reflection from a crystal is obtained in backscattering. Then angular divergences only enter to second order and the wavelength spread of the Bragg reflected beam depends essentially on the uncertainty of the reciprocal lattice vector G. With perfect Si crystals typically \(\frac{\delta G}{G} \cong {10^{ - 4}}\) is achieved. Backscattering instruments operate with both the monochromator and the analyzer under backscattering conditions and reach relative energy resolutions in the order of 10−4. Energy variation is performed in exploiting the Doppler effect moving the monochromator in a periodic fashion [19]. With such a resolution it is possible to study e.g. proton motion on the nanoscale in polymeric electrolyte membranes [20] or segmental relaxation in polymer melts [21].
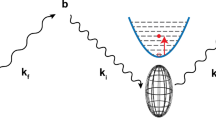
Neutron Spin Echo (NSE) An attempt to increase the energy resolution of TOF instruments far beyond the 1% level leads to an unacceptable loss of intensity. The neutron spin echo method which directly determines the run time difference between neutron tracks before and after the sample, improves the situation decisively [22]. NSE uses the neutron spin directions of each individual neutron as an individual stopwatch. The precession of the neutron spins in an external magnetic field effect the clockwork of this watch. The basic experimental setup of a NSE spectrometer is shown in Fig. 1 [23]. A beam of longitudinally polarized neutrons, i.e. neutrons with spins pointing into the beam direction enter the instrument and traverse a π/2-flipper at instrument entrance that rotates the neutron spins by 90° = π/2 such that they point perpendicular to the beam. Immediately after leaving the flipper the spins start to rotate around the longitudinal field generated by the primary precession coil. At the sample position the neutron spin may have performed a total of up to several 10,000 or 100,000 precessions. However, different neutrons with different velocities from the incoming 10% to 20% distribution have total precession angles that differ proportionally. For that reason the ensemble of neutron spins at the sample contains any spin direction on a disc perpendicular to the longitudinal field with virtually equal probability.

Spin rotations and setup of a generic NSE-spectrometer. Upper part: Spin rotation, middle part: magnitude of the magnetic field indicated by differently sized arrows, lower part: schematic setup of the JCNS-NSE-spectrometer. Copyright 2005 Springer
Nevertheless, each neutron is tagged by its velocity with an individual precession angle of its spin that may be viewed as the neutron’s own stopwatch. At the sample position there is π-flipper turning the spins during the neutron passage by 180° = π around an upward pointing axis. Thereby the flipper transforms a spin with precession angle αmod(2π) to − αmod(2π) = − αmod(− 2π) effectively reversing the sense of the spin clock. Then the neutrons enter the secondary part of the spectrometer, which is symmetric to the primary part. During the passage of the second main solenoid each spin, provided the sample did not change the neutron velocity, undergoes exactly the same number of precessions as in the primary coil. Due to the reversing action of the π-flipper this leads to the result that all neutron spins arrive at the same precession angle pointing upwards at the second π/2-flipper irrespective of the individual velocity. This effect is called spin echo in analogy to similar phenomena in NMR spin echo experiments [24]. Then finally a π/2-flipper turns these spins by 90° into the longitudinal direction, thereby freezing the acquired transverse spin polarization.
The analyzer transmits only neutrons with spin components parallel to the axis. Thus, the spin projection to the axis determines the probability that neutrons reach the detector. If the neutrons undergo a velocity change \({\Delta }{v_s}\;\) in the course of the scattering by the sample the final spin direction is no longer upwards and only the projection on the vertical axis is transmitted. Thus, the cosine of the final precession angle determines the analyzer transmission leading in fact to a cosine-modulating filter. The filter period is controlled by the magnetic field inside the main precession solenoids. The frequency ω in the dynamic structure factor S(Q,ω) is proportional to the energy transfer between neutron and sample \(\frac{\omega }{2\pi } = \frac{{m}}{2h}\left( {v_n^2 - {{\left( {{v_n} + {\Delta }{v_s}} \right)}^2}} \right)\) For very small \({\Delta }{v_s}\) linearization leads to \(\omega \ {\Delta }{v_s}\). Due to the cosine modulating filter function, the NSE instrument measures the cosine transform of S(Q,ω). The detector output of an ideal NSE instrument at exact symmetry is therefore
where the time parameter \(t = J{\lambda^3}\gamma \frac{m^2}{{2\pi {h^2}}}\). The time parameter depends on J—the integral of the magnetic induction along the flight path of the neutron from the π/2-flipper to the sample and γ = 1.83033 × 108 rad/s T. J is easily modified by varying the main solenoid current to which J is proportional. Note that the maximum achievable time depends linearly on the maximum field and on the third power of the neutron wavelength. The \(S\left( Q \right)\) is a static scattering function at t = 0 measured by SANS. The fact that the instrument output represents the intermediate scattering function S(Q,t) directly and not S(Q,ω) makes it particularly useful for scattering from a sample featuring relaxation dynamics [25].
Introductory educational material and a list of neutron scattering facilities is available e.g. at www.neutronsources.org.
3 Examples highlighting the potentials of QENS
3.1 Diffusion of liquid and gases in nano- and mesoporous materials
Details of adsorption process and diffusion of liquid and gas phases in nano- and mesoporous materials are crucial in many industrial processes, e.g. catalysis, separation [26], development of novel materials for hydrogen storage [27].
Since the adsorption process is a diffusion-related phenomenon, measurement of adsorption isotherms and macroscopic uptake are often performed in combination with microscopic diffusion measurements covering the displacements of molecular species inside the nanoporous solid. In particular, how diffusivities of guest molecules depend on the size, shape and interaction of the molecules with the host surface, on the temperature, on the concentration and composition of the sorbent phase, and on the geometry and topology of the porous channel system has been the focus of investigations over many years. Considering that the guest molecules are typically organic compounds, methods sensitive to the presence of adsorbed hydrogen are able to provide valuable microscopic information [28,29,30]. QENS extended the time and length scale of investigations down to 10−3–500 ns and 0.1–100 nm respectively, thus making it possible to observe hitherto unobservable details of local dynamics of guest molecules and of even collective dynamical effects.
In earlier times, many experimental studies were focused on zeolites and other microporous solids. They included experimental methods as well as computer simulations. They were notably promoted by the application of QENS following Hervé Jobic’s pioneering work in the field [31, 32], with special emphasis on the understanding of the dynamics of specific zeolite/sorbate systems [28]. Those systems are encountered in significant industrial processes such as hydrocarbon separations, fluid catalytic cracking and catalytic dewaxing. At the same time, they serve as model systems, e.g. to study the dependence on the alkane chain length. Mobility of n-alkanes in silicalite-1 and in MFI zeolites was found to be strongly dependent on the carbon atoms per alkane molecule (Fig. 2). It was found that the diffusivity values obtained by QENS are in reasonable agreement with MD simulations. On the other side, the diffusion of n-alkanes in Na-ZSM-5 is much slower than in silicalite-1 demonstrating importance of Na counterions in transport processes.

Self-diffusion coefficient, obtained at 300 K by different techniques, for n-alkanes in silicalite-1: (○) MD simulations (●) hierarchical simulations, (□) QENS in Na-ZSM-5, (\(\boxdot\)) QENS in silicalite-1, (\(\vartriangle\)) PFG NMR. Asterisked symbols correspond to extrapolation to 300 K. [33] Copyright 2006 ACS Publishing
Combination of PFG NMR and QENS provided a new insight into the study of mesoporous zeolite materials [34]. Due to the vast differences in the range of molecular displacements (from nanometers probed by QENS to micrometers by the PFG NMR), both methods are able to explain different details of mesoporous structure. In addition to the regular pore structure, one may expect several transport resistances acting in the interior of zeolite crystallites and this has been confirmed by PFG NMR. The details of local mobilities influenced by intra-crystalline morphology and surface interactions are to be clarified by QENS. Indeed, the diffusivities of n-alkanes in NaCaA zeolites found by PFG NMR demonstrate almost no dependence on the number of carbon atoms (long time scale) beyond n = 3. The diffusivities obtained by the QENS are much more dispersed indicating the importance of the zeolite morphology on the local scale (see Fig. 3, curves (e) and (d)).

n-alkane chain length dependence of self-diffusivities in different zeolites: a silicalite-1 determined by PFG NMR at 423 K and b Na-ZSM-5 determined by QENS at 475 K [35] (the dotted squares correspond to the newer measurements in silicalite-1) and comparison with previous PFG NMR measurements in (c) NaX at 475 K [36] and in (d) NaCaA at 443 K [37] and e NSE collective diffusion measurements in NaCaA at 475 K [38]. (see Ref. [34].) Copyright 2006 Elsevier
As a significant difference between QENS and PFG NMR studies, transport resistances within the individual microporous crystals with mutual spacings in between the measuring ranges of the techniques, remain invisible by QENS but are of immediate influence on the NMR data. In this way, comparative QENS and PFG NMR studies provided direct evidence of the existence of intracrystralline transport resistances which could, moreover, be identified by structural analysis via transmission electron microscopy [39].
In 2010 Jobic et al. reported the first application of in situ quasielastic neutron scattering (QENS) of zeolite-guest systems subjected to microwave (MW) irradiation, for comparison with MW-driven molecular simulations, providing an unprecedented picture of selective heating and athermal effects in these systems [40]: selective heating of methanol in silicalite, but little to no heating of benzene in silicalite. The hydrogen atoms of a methanol molecule experience several molecular motions: translation, rotation, and vibration, which occur on different time scales and can be nicely discriminated on the time/length scale provided by the TOF instrument. It was found that the mobility modes are differently influenced by the MW irradiation, with athermal effects causing increased rotational and translational diffusivity of the methanol molecules. The QENS results were supported by the MD computer simulations. Such progress helps guide new ways to selectively heat heterogeneous material which is extremely important for tuning the selectivity of adsorption in zeolites.
Understanding the behavior of hydrogen in clay rock systems opens new approaches to underground hydrogen storage and is also relevant for deep underground nuclear waste repository technology. Clays (such as bentonites) and clay rocks are presently considered respectively as engineered barriers and host rocks preventing the migration of radionuclides released form the wastes. In particular, hydrogen is expected to develop from anaerobic corrosion of the waste steel canisters [41,42,43]. The development of reliable methods to estimate hydrogen diffusion coefficients in cap rock layers and their hydrogen absorption capacity at temperatures and pressures of importance is crucial for the estimation of hydrogen leakage rates and thus for assessment of the overall performance, safety and efficiency of storage facilities. Adsorption and diffusion of hydrogen gas was investigated in model smectite at high pressure by QENS and volumetric techniques at the temperature and pressure of practical interest [44]. In particular, QENS revealed that hydrogen diffuses inside the clay porous network according to Fick's law (continuous diffusion), while jump diffusion cannot be excluded at distances lower than 6.3 Å, i.e. less than the one between two Na+ exchangeable ions.
QENS was used by Silverwood et al. [45] to study the diffusion of propane in zeolite ZSM-5 under static and macroscopic flow conditions. It was found that the rate of self-diffusion in the adsorbed phase was slightly faster under the flow conditions studied. Control measurements showed that the broadening seen in the quasielastic signal must result from propane condensed in the zeolitic pores and not from gas, either in the bulk or inter-particle voids. Due to the highly penetrating nature of the neutron probe, this study provides the possibility of direct diffusion measurements in zeolites under realistic operating conditions for a wide range of applications such as gas separation and storage, pressure swing adsorption and catalysis.
Some of the self-diffusional processes which are important for adsorption and catalysis applications can be monitored by the solid state NMR Kolokolov et al. [46]. In particular, deuterium (2H) NMR has the advantage of tracking down the angular displacement of the target deuteron chemical bond. In addition to the dipolar broadening of common solid-state NMR spectra, for solid-state 2H NMR spectra the so called first-order quadrupolar broadening is observed [47, 48]. 2H NMR provides information about the geometry of the reorientation of the chemical bond and its characteristic time. Details of the local dynamics of n-butane adsorbed in H-ZSM-5 studied by Kolokolov et al. [49] were found to be in agreement with the QENS experiment. In particular, the activation energy of translational diffusion related to the diffusion on the length scale of a zeolite channel found by QENS and 2H NMR are equivalent [50]. In addition to translational motion, local conformational isomerization and jump exchange between the nanoporous channels were detected by the 2H NMR. Metal–Organic Frameworks (MOF) materials with 3D interconnecting porous system are characterized by hierarchical dynamics over a length scale from one to tens of nanometers. In addition to the benzene ring rotation detected by QENS, 2H NMR reveals the rotational barriers and activation energy for the anisotropic rotation [51].
Processes taking place at the solid/aqueous solution interface have a strong impact on the evolution of materials in the fields of construction, environment, geochemistry, membranes, catalysis, and nuclear wastes. Since most of them are partially or completely porous, the processes and chemical reactions occurring in this nanoconfinement have a major impact on the evolution of materials at the macroscopic scale. Baum et al. studied the dynamics of water confined in highly ordered mesoporous silica with various pore sizes in the presence of ions of different kosmotropic properties [52]. Using QENS they determined the critical pore size (2.6 nm) and the electrolyte concentration at which the influence of the ion nature becomes the main factor affecting the water properties. This opens new perspectives regarding the underlying mechanisms of the dissolution and precipitation of minerals in nanoconfinement.
3.2 Hydrogen dynamics in high temperature polymer electrolyte fuel cells (HT-PEFCs)
Conception, design and optimization of fuel cells in general requires a good knowledge of the underlying physical processes at all length scales. In Fig. 4 the schematic setup of a fuel cell is presented, showing how microscopic techniques can be related to macroscopic properties of the fuel cell. The central part of the HT-PEFC is a membrane-electrode assembly (MEA), which consists of two catalytic layers separated by a proton exchange membrane. Water-free proton conducting membranes, polybenzimidazole-type (PBI) polymer films doped with phosphoric acid (PA), which has the highest reported proton conductivity are mostly used in HT-PEFC and have been studied intensively during the last decade [53, 54]. In particular, it has been shown that optimizing the thermal and chemical stability of the polymer, acid management, and the integral interface between the electrode and membrane essentially improve the proton conductivity and cell performance. Therefore, investigation of proton mobility in the MEA on a broad time/length scale is crucial for further fuel cell development.

Schematic picture of a HT-PEFC with proton conducting polymer membrane surrounded by the electrode layers. The zoom on the right shows a sketch of the electrode layer consisting of Pt catalyst particles on a carbon-PTFE support with phosphoric acid in the remaining space [55]
Different kinds of fuel cells have been investigated by macroscopic, microscopic and scattering techniques, in order to gain insight into the local structure [56] or proton conduction mechanisms [57]. Computer simulations shed light from another perspective on local processes [58]. High Temperature Polymer Electrolyte Fuel Cells (HT-PEFCs) [59] based on a proton conducting membrane such as poly2,2′-m-(phenylene)-5,5′-bibenzimidazole (PBI) doped with phosphoric acid (PA) are promising electrochemical energy converters. Because of the high operation temperature of 160–180 °C a HT-PEFC has a high CO tolerance. In contrast to the standard perfluorosulphonic acid (PFSA) based low-temperature fuel cells (LT-PEFCs) water management is not a critical issue of HT-PEFC [60]. One draw-back of this fuel cell type is the lower performance compared to standard low temperature fuel cells due to the activation losses caused by phosphoric acid [59].
Hopfenmüller et al. studied proton diffusion in PBI membranes using neutron scattering techniques [61]. Time of flight (TOF) spectroscopy and backscattering (BS) spectroscopy have been combined to cover a broad dynamic range from 1 ps to about 5 ns. In order to selectively observe the diffusion of protons potentially contributing to the ion conductivity, two samples were prepared, where in one of the samples the phosphoric acid was used with hydrogen replaced by deuterium. The scattering data from the two samples were subtracted in a suitable way after measurement. Thereby, the protons have been found to undergo subdiffusion, this means, their mean square displacement was observed to increase following the relation
with a time exponent \(\kappa < 1.\) The time exponent is correlated with the fractal dimension \({d_w}\) (“fractal exponent”) of the trajectories (see, e.g., Sect. 2.6.3 of Ref. [8]).
The fractal exponent for the PBI membranes was thus found to be dw = 2.46, notably differing from dw = 2 for ordinary diffusion. Dependence of the relaxation times on the wave vector \(\tau \left( Q \right)\) shows a transition from normal to fractal behavior at \(Q\) = 1.1–1.4 Å−1, resulting in a characteristic distance of 5 Å. This distance was attributed to the lower border of the fractal regime. The study was supported by PFG NMR studies that reveal the fractal like diffusion over a length scale of tens nanometer [62].
Khaneft et al. investigated the structure of the catalytic layer with different Pt loadings starting from carbon supported catalyst [63]. Using small angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) it was shown that the structure of the catalytic layer is mainly defined by the structure of the original catalytic powder. At the same time the preparation process of the electrodes does not have a strong influence on the structural properties in the investigated \(Q\)-range. Using QENS the proton dynamics of phosphoric acid (PA) in the catalytic layer depending on Pt content was investigated over a broad temperature range. Mean squared displacement of PA protons as a function of temperature can be determined as described in Ref. [64] and shown in Fig. 5. The mean squared displacement (msd) is constant at low temperatures where the dynamics are supposed to be frozen but one can clearly see the difference in the behavior of the samples containing different amounts of Pt. The shift of the step on elastic scans (Fig. 5, insert) and subsequently on msd curves to the high temperatures is related to the structure of the catalytic layer and particularly depends on the amount of Pt particles in the layer. The authors assume that PA is trapped around Pt particles and this effect is stronger for higher local Pt concentration.

Mean squared displacement (msd) of the PA in the electrode (Fig. 4) obtained by fitting the \(Q\)-dependence of the intensity in the range 0.2 Å−1 < Q < 0.7 Å−1. In the insert: intensity of the elastic scattering of the PA in the electrode at Q = 0.6 Å−1, normalized to the intensity at the lowest temperature where dynamics is frozen. Decrease of the intensity at high temperatures indicates the change in dynamical process [63] Copyright 2016 WILEY–VCH Verlag GmbH
3.3 Dynamics of polymers in nanopores: effect of interaction with the surface
During the last several years a dramatic increase in the synthesis of new nanoporous materials has been observed [65]. They have found many unique applications in nanotechnology, chemistry and biochemistry. From an application perspective, the dominance of surfaces in nanoporous systems makes them attractive as matrices for gas storage, gas separation, catalysis and energy harvesting. Furthermore, nanoporous materials can be used as a step in the fabrication of multifunctional nanostructures or as membranes in separation processes [66]. In particular, nanostructured materials based on organic polymers led to the development of new miniature devices that exhibit unique capabilities not available in larger scale devices [67]. The majority of the applications are related to the structural changes and transport properties of the polymer embedded in the nanopores.
Polymer chain dynamics is commonly described in terms of the Rouse and the reptation model [25]. The relaxation of the Rouse modes, determined by a balance of viscous and entropic forces, only depends on the monomeric friction. In addition, long polymers heavily interpenetrate each other and mutually restrict their motions at long times in forming topological constraints (‘‘entanglements’’) [68]. The important question that is addressed by computer simulations [69, 70] as well as by a variety of experiments on a macroscopic level [71,72,73] is how the dynamics in the Rouse and reptation regime change under confinement.
3.3.1 Model nanoporous structures
Self-ordered nanoporous Anodic aluminum oxide (AAO), which is accessible by a two-step anodization process introduced by Masuda and Fukuda [74], contains arrays of aligned cylindrical nanopores with a pore diameter Dp from about 20 nm up to a few hundred nm, with a narrow pore diameter distribution and uniform pore depth, which can be adjusted between about 1 μm and several 100 μm. Since a few decades these nanoporous materials have been used as a tool (i.e. hard templates) for fabrication of polymer nanostructured materials (tubes, rods, fibers etc.) [75]. It is easily possible to tailor the diameter and the aspect ratio of the obtained nanostructures. However, the most important advantage is the possibility to control mesoscopic structure formation processes inside the pores. There are relatively few limitations regarding the materials that can be infiltrated into nanotubes via hard templates. Mixtures, sols, semi-crystalline and liquid-crystalline polymers, thermoplastics, as well as block copolymers, are eligible for this approach. The inner surface of Alumina nanopores is covered by hydroxyl groups resulting in a hydrophilic surface [76]. In the focus of this review are linear polymer melts infiltrated in nanoporous AAO.
The high surface-to-volume ratio of the nanoporous AAO hard templates makes it possible to observe and control influence of the surface on polymer chain properties. It can be reached by either using polymers with chemically different segmental structure or adjusting the properties of the pore walls (pore surface functionalization).
The porous silicon is produced employing an electrochemical anodic etching process of Si (100) wafers (thickness ∼0.5 mm). The pores are mostly linear and oriented along the Si crystallographic direction < 100 > (perpendicular to the membrane surface [77]) with a length of ∼ 0.3 mm. After a special treatment with hydrofluoric acid and hydrogen peroxide the pores have a roughly circular cross-section with a mean diameter ∼10 nm and their walls are covered with silicon rendering them highly hydrophilic. The porosity of the porous layer is about 0.55 in volume fraction. In general, the porosity is higher, but the pores are not as well ordered as for nanoporous AAO.
3.3.2 Infiltration of polymers in AAO
Infiltration of the polymer is performed by different methods [67] depending on polymer composition and nanopore size. For most of the experiments presented here, self-ordered AAO with a pore diameter of 20–40 nm, a nearest-neighbor distance of 55 nm, and a pore depth of 100 µm were used (Fig. 6). Those Alumina nanoporous templates were prepared following a procedure reported by Jessensky et al. [78]. The AAO layers were attached to underlying 400 µm thick Al layers so that the AAO pore bottoms were closed. For NSE experiment it is important to avoid the strong small angle scattering of the templates. Therefore, the infiltrated polymers need to have the same scattering length density as the AAO template. In blending deuterated and protonated polymers appropriately matching can be achieved (see e.g. [79].).

a, b Electron micrograph (SEM) of anionic aluminum oxide (AAO) nanopores, c model interaction of a polydimethylsiloxane (PDMS) chain with the AAO surface OH groups via H-bonds
3.3.3 Chain dynamics of polydimethylsiloxane (PDMS) confined in AAO nanopores
Microscopic studies of the dynamics at the chain level are mainly available through simulations, while only a few experimental studies on chain dynamics under confinement can be found. In particular, physical adsorption of polymer chains on the surface of nanoparticles was reported, leading to the formation of a glassy layer on the particle [80]. The simultaneous slowing down of Rouse modes, i.e. increasing of the friction coefficient, was found for unentangled polymer melts [69]. The combined effects of increased effective friction and entanglement mesh sizes were even found to lead to a non-monotonic behavior of the chain relaxation with confinement size [70]. The strong confinement effect (so-called corset effect) was concluded from NMR relaxometry studies performed on poly(ethylene oxide) confined in solid methacrylate matrix. A reduction of the effective tube diameter of the Doi-Edwards reptation model in confined polymer by about one order of magnitude as compared to the bulk has been reported [81]. These results have been suggested to be the consequence of a number of combined effects: polymer-surface interaction, the mutual uncrossability of polymer chains (entanglements) and confinement geometry.
Dynamics of PDMS on the chain level was studied by the neutron spin echo (NSE) spectroscopy [82]. Figure 6c schematically displays the system indicating hydrogen bonds with the OH-groups at the AAO surface.
PDMS has an entanglement molecular weight of Me = 12 kg/mol leading to an entanglement spacing of about 8 nm. The polymers under investigation had a molecular weight of 17.4 kg/mol and were basically non-entangled (see Fig. 7 representing bulk PDMS measured as a reference).

The NSE data obtained at 373 K show a significantly slowed down chain dynamics compared to the bulk (Fig. 7). Proper inspection of the plateaus measured at different values of the momentum transfer Q (some data are not presented here) at long times made it possible to exclude the existence of an immobilized (glassy) layer, since the plateaus are Q–dependent. Note, that this result agrees with our previous data on PEO confined in Alumina nanopores [21] and with a recent neutron scattering study [84]. Furthermore, the observed Q–dependence of the plateaus cannot be described in terms of a simple confined polymer, i.e. one-phase system, because fitting such a model to the data yields a Q-dependent confinement length.
Therefore, a new model was derived containing (i) a fraction of free Rouse-like chains, since many chains far away from the surface are not affected and (ii) a second fraction of chains, which is close to the surface and assumed to be effectively confined. The respective dynamic structure factor reads:
where \({A_{{\text{bulk}}}}\) and \({A_{{\text{conf}}}} = 1 - {A_{{\text{bulk}}}}\) are the fractions of the bulk and confined phases, respectively; \({S_{{\text{bulk}}}}\left( {Q,t} \right)\;\) is the dynamic structure factor of the bulk chain defined by the Rouse model [85] and \({S_{{\text{conf}}}}\left( {Q,t} \right)\) is the dynamic structure factor of the confined chain. This model contains three variable parameters: fraction \({A_{{\text{bulk}}}}\) (or \({A_{{\text{conf}}}} = 1 - {A_{{\text{bulk}}}}\)), the number of suppressed Rouse modes psupp reflecting the effective confinement in \({S_{{\text{conf}}}}\left( {Q,t} \right)\) and an additional parameter Δp, the width of a cut off function describing the transition from active to suppressed modes. Δp accounts for a variety of anchoring and confinement conditions. By fitting all Q simultaneously \({A_{{\text{bulk}}}}\) was found to be 25% of the total amount of polymer in the pores, psupp = 5 and Δp = 1 (Fig. 8). From the longest active mode in the confined phase one can calculate the effective confinement length \(d =\) 3.4 nm.

Schematic representation of the surface-induced entanglements in the confined polymer melt in cylindrical nanopore (a) and perpendicular to the pore surface (b). The chains adsorbed on the surface (anchored chains) are shown by black colour, and the entangled chains in the confined phase (interphase) are represented by red colour [83]
The fractions \({A_{{\text{bulk}}}}\) and \({A_{{\text{conf}}}}\) obtained can be transferred to a corresponding layer thickness which was found to be rconf = rbulk = 6.5 nm (Fig. 8). The fact that the size of the PDMS polymer chain considered here in the bulk is 3.5 nm and should even be reduced for a multiple adsorbed chain, clearly indicates that the layer with a thickness of 6.5 nm is made up not only of chains adsorbed at the surface. The polymer chains anchored to the surface may form loops; neighboring chains can then interpenetrate these loops (Fig. 8). These penetrating chains can be considered as an interphase between the polymers adsorbed on the surface and the bulk polymer phase.
Formation of loops and also trains and tails by the polymer chains on the attractive surface was observed by computer simulations [86] and theory [87, 88]. In particular, it was found that the loop distribution follows a power law relationship.
In general, the confined phase composed of the anchored chains and the chains in the interphase is internally highly mobile and not glassy as has so far been claimed frequently in the literature. These results are inferred from the space–time dependent chain dynamics that is observed in terms of the single chain dynamic structure factor and represent the first direct and quantitative observation of the interphase [82].
A similar situation was observed for (PEO) melts confined in porous silicon with narrower pores (diameter up to 13 nm) [89, 90]. NSE spectra were successfully analyzed in terms of a two-state model, where 19–36% of chains are adsorbed to the pore walls and exhibit much slower large scale internal dynamics than in the bulk and their centers-of-mass do not move, while free chains have bulk-like internal dynamics and diffuse within the cylinder in the center of the pore. For PEO with a molecular weight (Mw) 3 kg/mol the radius of this cylinder was found to be 1.4 nm; with this the thickness of the adsorbed layer was found to be approximately equal to the radius of gyration Rg. For larger PEO chains with a molecular weight of 10 kg/mol that exhibit a Rg of order of the pore diameter, the center of mass diffusion could not be discerned.
For small oligomeric n-Hexatriacontane C36H74 in porous silicon at 364 K an adsorbed bilayer and at 435 K a monolayer (in both cases, the long axis of the molecules is oriented parallel to the surface) was found [89]. There is no indication of an adsorbed layer at 512 K. The results support the existence of a sticky boundary layer inferred from capillary filling experiments. In contrast to longer PEO chains, the diffusion of the C36H74 is isotropic. In addition, the segments close to the surface order in parallel direction. Thus, a preferential orientation of the oligomeric alkanes was found.
In general, for attractive surfaces like nanoporous AAO and nanoporous Silicon the segmental dynamics is slowed down in direction perpendicular to the surface, while in the parallel direction such a slowing down could not be observed. The dynamic neutron scattering results demonstrate that the polymer surface layer is not glassy, but mobile with increased segmental relaxation times compared to the bulk. In addition, the presence of interphase results in drastic property changes of the confined polymer.
4 Conclusions
Neutron scattering is one of the unique methods allowing to measure dynamics of molecules over a few orders of magnitudes in both time and length scale. We have presented some representative results on the diffusion of liquid and gases in nanoporous materials, hydrogen dynamics in energy related materials for fuel cells and polymer dynamics in well-ordered nanoporous material. A few examples demonstrating the power of the QENS method were considered. In particular, we focused on (i) investigation of the diffusion of liquid and gases in micro- and mesoporous solid materials like zeolites; (ii) diffusion of hydrogen in high temperature polymer electrolyte fuel cell (HT-PEFC) and energy relevant materials; and (iii) dynamics of macromolecules in well-ordered nanopores: effect of interaction with the surface.
Abbreviations
- \({A_{{\text{bulk}}}},{A_{{\text{conf}}}}\) [–]:
-
Fractions of the bulk and confined phases
- b [m]:
-
Scattering length
- \(d\) [m]:
-
Effective confinement length
- \({d_w}\) [–]:
-
Fractal dimension
- D t [m2 s− 1]:
-
Transport diffusion coefficient
- \(\Delta E\) [J]:
-
Energy transfer
- \({E_i},\;{E_f}\) [J]:
-
Energy of incident and scattered neutrons
- G [m− 1]:
-
Reciprocal lattice vector
- \({G_{pair}}\left( {r,t} \right)\) [–]:
-
Van Hove pair correlation function
- \(h\) [J s]:
-
Planck constant
- \(\hbar\) [J s]:
-
Reduced Planck constant
- \({I_{eff}}\) [–]:
-
Effective intensity
- J [T]:
-
Integral of the magnetic induction
- \({\vec k_i}\), \({\vec k_f}\) [m−1]:
-
Wave vectors of the incoming and outgoing (scattered) neutrons
- \(m\) [kg]:
-
Mass of neutron
- M w [kg mol−1]:
-
Molecular weight of the polymer
- N [–]:
-
Number of scatterers
- Δp [–]:
-
Width of a cut off function
- p supp [–]:
-
Number of suppressed Rouse modes
- \(Q\) [m−1]:
-
Momentum transfer
- \(\vec Q\) [m−1]:
-
Scattering wave vector
- r bulk, r conf [m]:
-
Bulk and confined layer thickness
- R g [m]:
-
Gyration radius of polymer chain
- \({r_n}\left( t \right)\) [m]:
-
Position vector of an atom “n” at time t
- \(\langle r_n^2\left( t \right)\rangle\) [m2]:
-
Mean-square displacement of atom “n”
- \(S\left( {Q,t} \right)\) [–]:
-
Intermediate scattering function
- \(S\left( {Q,\omega } \right)\)[–]:
-
Dynamic structure factor
- \({S_{self}}\left( {Q,t} \right)\) [–]:
-
Incoherent scattering function
- γ [rad s− 1 T− 1]:
-
Gyromagnetic ratio of neutron
- θ [rad]:
-
Scattering angle
- \(\kappa\) [–]:
-
Time exponent
- \({\lambda_i},\;{\lambda_f}\) [m]:
-
Wavelengths of the incoming and outgoing (scattered) neutrons
- \({\nu_i},\;{\nu_f}\) [m s−1]:
-
Velocity of the incoming and outgoing (scattered) neutrons
- ρ [m− 2]:
-
Scattering length density
- Δρ [m− 2]:
-
Contrast
- σ [–]:
-
Scattering cross section
- \(\tau\) [s]:
-
Relaxation time
- \(\omega\) [rad s−1]:
-
Angular frequency
- \(\Omega\) [sr]:
-
Solid angle
References
Slade, R.C.T., Hirst, P.R., West, B.C.: Ammonium-ion motions in the hexagonal tungsten trioxide framework: a neutron scattering study of the bronze (NH4)0.22WO3 and of [(NH4)2o]0.085WO3. J. Mater. Chem. 1, 281–288 (1991). https://doi.org/10.1039/JM9910100281
Slade, R.C.T., Pressman, H.A.: Neutron scattering investigation of hydrogenic species in the ammonium molybdenum bronze (NH4)0.24H003MoO3. J. Mater. Chem. 4, 501–508 (1994). https://doi.org/10.1039/jm9940400501
Biehl, R., Richter, D.: Slow internal protein dynamics in solution. J. Phys. (2014). https://doi.org/10.1088/0953-8984/26/50/503103
Richter, D., Monkenbusch, M., Arbe, A., Colmenero, J.: Neutron Spin Echo in Polymer Systems. Springer, Berlin (2005)
Kolokolov, D.I., Lim, D.W., Kitagawa, H.: Characterization of proton dynamics for the understanding of conduction mechanism in proton conductive metal-organic frameworks. Chem. Rec. (2020). https://doi.org/10.1002/tcr.202000072
Lim, D.W., Kitagawa, H.: Proton Transport in Metal-Organic Frameworks (2020)
Kärger, J., Avramovska, M., Freude, D., Haase, J., Hwang, S., Valiullin, R.: Pulsed field gradient NMR diffusion measurement in nanoporous materials. Adsorption 1, 3 (2021). https://doi.org/10.1007/s10450-020-00290-9
Kärger, J., Ruthven, D.M., Theodorou, D.N.: Diffusion in Nanoporous Materials. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim (2012)
Jobic, H.: Diffusion in zeolites measured by neutron scattering techniques. In: Fluid Transport in Nanoporous Materials, pp. 333–352. Kluwer Academic Publishers (2006)
Jobic, H., Makrodimitris, K., Papadopoulos, G.K., Schober, H., Theodorou, D.N.: Diffusivities of CO2 and N2 in silicalite, comparison between quasi-elastic neutron scattering and molecular simulations. In: Studies in Surface Science and Catalysis, pp. 2056–2061. Elsevier Inc. (2004)
Mondelli, C., González, M.A., Albergamo, F., Carbajo, C., Torralvo, M.J., Enciso, E., Bermejo, F.J., Fernández-Perea, R., Cabrillo, C., Leon, V., Saboungi, M.L.: Collective excitations in liquid D2 confined within the mesoscopic pores of a MCM-41 molecular sieve. Phys. Rev. B. (2006). https://doi.org/10.1103/PhysRevB.73.094206
Motokawa, R., Kobayashi, T., Endo, H., Mu, J., Williams, C.D., Masters, A.J., Antonio, M.R., Heller, W.T., Nagao, M.: A telescoping view of solute architectures in a complex fluid system. ACS Cent. Sci. 5, 85–96 (2019). https://doi.org/10.1021/acscentsci.8b00669
Novikov, V.N., Schweizer, K.S., Sokolov, A.P.: Coherent neutron scattering and collective dynamics on mesoscale. J. Chem. Phys. (2013). https://doi.org/10.1063/1.4802771
Schleger, P., Farago, B., Lartigue, C., Kollmar, A., Richter, D.: Clear evidence of reptation in polyethylene from neutron spin-echo spectroscopy. Phys. Rev. Lett. 81, 124–127 (1998). https://doi.org/10.1103/PhysRevLett.81.124
Oeser, R., Ewen, B., Richter, D., Farago, B.: Dynamic fluctuations of crosslinks in a rubber: a neutron-spin-echo study. Phys. Rev. Lett. 60, 1041–1044 (1988). https://doi.org/10.1103/PhysRevLett.60.1041
Zamponi, M., Wischnewski, A., Monkenbusch, M., Willner, L., Richter, D., Likhtman, A.E., Kali, G., Farago, B.: Molecular observation of constraint release in polymer melts. Phys. Rev. Lett. 96, 238302 (2006). https://doi.org/10.1103/PhysRevLett.96.238302
Radulescu, A., Székely, N.K., Polachowski, S., Leyendecker, M., Amann, M., Buitenhuis, J., Drochner, M., Engels, R., Hanslik, R., Kemmerling, G., Lindner, P., Papagiannopoulos, A., Pipich, V., Willner, L., Frielinghaus, H., Richter, D.: Tuning the instrument resolution using chopper and time of flight at the small-angle neutron scattering diffractometer KWS-2. J. Appl. Crystallogr. 48, 1849–1859 (2015). https://doi.org/10.1107/S1600576715019019
Lohstroh, W., Evenson, Z.: TOFTOF: cold neutron time-of-flight spectrometer. J. Large-Scale Res. Facil. JLSRF. 1, 15 (2015). https://doi.org/10.17815/jlsrf-1-40
Zamponi, M., Khaneft, M.: SPHERES: backscattering spectrometer. J. Large-Scale Res. Facil. JLSRF. 1, 30 (2015). https://doi.org/10.17815/jlsrf-1-38
Holderer, O., Ivanova, O., Hopfenmüller, B., Zamponi, M., Maier, W., Majerus, A., Lehnert, W., Monkenbusch, M., Zorn, R.: Observing proton motion on the nanoscale in polymeric electrolyte membranes with quasielastic neutron scattering. Int. J. Hydrogen Energy. 39, 21657–21662 (2014). https://doi.org/10.1016/j.ijhydene.2014.09.018
Krutyeva, M., Martin, J., Arbe, A., Colmenero, J., Mijangos, C., Schneider, G.J., Unruh, T., Su, Y., Richter, D.: Neutron scattering study of the dynamics of a polymer melt under nanoscopic confinement. J. Chem. Phys. 131, 174901 (2009). https://doi.org/10.1063/1.3258329
Pasini, S., Holderer, O., Kozielewski, T., Richter, D., Monkenbusch, M.: J-NSE-Phoenix, a neutron spin-echo spectrometer with optimized superconducting precession coils at the MLZ in Garching. Rev. Sci. Instrum. 90, 043107 (2019). https://doi.org/10.1063/1.5084303
Monkenbusch, M., Schätzler, R., Richter, D.: The Jülich neutron spin-echo spectrometer—design and performance. Nucl. Instruments Methods Phys. Res. Sect. A 399, 301–323 (1997). https://doi.org/10.1016/S0168-9002(97)00956-X
Hahn, E.L.: Spin echoes. Phys. Rev. 80, 580–594 (1950). https://doi.org/10.1103/PhysRev.80.580
Richter, D., Monkenbusch, M., Arbe, A., Colmenero, J.: Neutron spin echo in polymer systems. Adv. Polym. Sci. 174, 1 (2005). https://doi.org/10.1007/b106578
Krishna, R.: The Maxwell-Stefan description of mixture diffusion in nanoporous crystalline materials. Microporous Mesoporous Mater. 185, 30–50 (2014). https://doi.org/10.1016/j.micromeso.2013.10.026
Härmas, R., Palm, R., Russina, M., Kurig, H., Grzimek, V., Härk, E., Koppel, M., Tallo, I., Paalo, M., Oll, O., Embs, J., Lust, E.: Transport properties of H2 confined in carbide-derived carbons with different pore shapes and sizes. Carbon N. Y. 155, 122–128 (2019). https://doi.org/10.1016/j.carbon.2019.08.041
Jobic, H., Theodorou, D.N.: Quasi-elastic neutron scattering and molecular dynamics simulation as complementary techniques for studying diffusion in zeolites (2007)
Kärger, J., Freude, D., Haase, J.: Diffusion in nanoporous materials: Novel insights by combining MAS and PFG NMR (2018)
Schneider, D., Kondrashova, D., Valiullin, R., Kärger, J., Bunde, A.: Mesopore-promoted transport in microporous materials. Chem. Ingenieur Technik (2015). https://doi.org/10.1002/cite.201500037
Jobic, H.: Applications of neutron scattering to catalysis. In: Catalyst Characterization: Physical Techniques for Solid Materials, pp. 347–375 (1994)
Jobic, H.: Diffusion studies using quasi-elastic neutron scattering. In: Kannelopoulos, N. (ed.) Recent Advances in Gas Separation by Microporous Ceramic Membranes (2000)
Jobic, H., Theodorou, D.N.: Diffusion of long n-alkanes in silicalite. A comparison between neutron scattering experiments and hierarchical simulation results. J. Phys. Chem. B. 110, 1964–1967 (2006). https://doi.org/10.1021/jp056924w
Jobic, H., Schmidt, W., Krause, C.B., Kärger, J.: PFG NMR and QENS diffusion study of n-alkane homologues in MFI-type zeolites. Microporous Mesoporous Mater. 90, 299–306 (2006). https://doi.org/10.1016/j.micromeso.2005.10.020
Jobic, H.: Diffusion of linear and branched alkanes in ZSM-5. A quasi-elastic neutron scattering study. In: Journal of Molecular Catalysis A: Chemical, pp. 135–142. Elsevier (2000)
Kärger, J., Pfeifer, H., Rauscher, M., Walter, A.: Self-diffusion of n-paraffins in NaX zeolite. J. Chem. Soc. Faraday Trans. 76, 717–737 (1980). https://doi.org/10.1039/F19807600717
Jobic, H., Kärger, J., Krause, C., Brandani, S., Gunadi, A., Methivier, A., Ehlers, G., Farago, B., Haeussler, W., Ruthven, D.M.: Diffusivities of n-alkanes in 5A zeolite measured by Neutron spin echo, pulsed-field gradient NMR, and zero length column techniques. In: Adsorption, pp. 403–407. Springer (2005)
Jobic, H., Méthivier, A., Ehlers, G., Farago, B., Haeussler, W.: Accelerated diffusion of long-chain alkanes between nanosized cavities. Angew. Chem. Int. Ed. 43, 364–366 (2004). https://doi.org/10.1002/anie.200352691
Feldhoff, A., Caro, J., Jobic, H., Ollivier, J., Krause, C.B., Galvosas, P., Kärger, J.: Intracrystalline transport resistances in nanoporous zeolite X. ChemPhysChem 10, 2429–2433 (2009). https://doi.org/10.1002/cphc.200900279
Jobic, H., Santander, J.E., Conner, W.C., Wittaker, G., Giriat, G., Harrison, A., Ollivier, J., Auerbach, S.M.: Experimental evidence of selective heating of molecules adsorbed in nanopores under microwave radiation. Phys. Rev. Lett. 106, 157401 (2011). https://doi.org/10.1103/PhysRevLett.106.157401
Ortiz, L., Volckaert, G., Mallants, D.: Gas generation and migration in Boom Clay, a potential host rock formation for nuclear waste storage. Eng. Geol. 64, 287–296 (2002). https://doi.org/10.1016/S0013-7952(01)00107-7
Senger, R., Marschall, P., Finsterle, S.: Investigation of two-phase flow phenomena associated with corrosion in an SF/HLW repository in Opalinus Clay, Switzerland. Phys. Chem. Earth. 33, S317–S326 (2008). https://doi.org/10.1016/j.pce.2008.10.034
Xu, T., Senger, R., Finsterle, S.: Corrosion-induced gas generation in a nuclear waste repository: reactive geochemistry and multiphase flow effects. Appl. Geochem. 23, 3423–3433 (2008). https://doi.org/10.1016/j.apgeochem.2008.07.012
Mondelli, C., Bardelli, F., Vitillo, J.G., Didier, M., Brendle, J., Cavicchia, D.R., Robinet, J.-C., Charlet, L.: Hydrogen adsorption and diffusion in synthetic Na-montmorillonites at high pressures and temperature. Int. J. Hydrogen Energy. 40, 2698–2709 (2015). https://doi.org/10.1016/j.ijhydene.2014.12.038
Silverwood, I.P., Sakai, V.G.: Propane diffusion in ZSM-5 pores measured by quasielastic neutron scattering under macroscopic flow. Chem. Eng. Sci. 186, 116–121 (2018). https://doi.org/10.1016/j.ces.2018.04.051
Kolokolov, D.I., Freude, D., Stepanov, A.G.: Dynamics in nanoporous materials probed by 2h solid state nmr. estimation of self- diffusion coefficients. Adsorpt.-J. Int. Adsorpt. Soc.
Freude, D., Haase, J.: Quadrupole effects in solid-state NMR. Basic principles and experimental techniques for nuclei with half-integer spins. http://www.quad-nmr.de/
Harris, R.K., Becker, E.D., Cabral De Menezes, S.M., Goodfellow, R., Granger, P.: NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001). Pure Appl. Chem. 73, 1795–1818 (2001). https://doi.org/10.1351/pac200173111795
Kolokolov, D.I., Jobic, H., Stepanov, A.G.: Mobility of n-butane in ZSM-5 zeolite studied by 2H NMR. J. Phys. Chem. C. 114, 2958–2966 (2010). https://doi.org/10.1021/jp908464f
Jobic, H., Bee, M., Caro, J.: Translational mobility of n-butane and n-hexane in ZSM-5 measured by quasi-elastic neutron SCATTERING. In: Proceedings from the Ninth International Zeolite Conference, pp. 121–128. Elsevier (1993)
Kolokolov, D.I., Stepanov, A.G., Guillerm, V., Serre, C., Frick, B., Jobic, H.: Probing the dynamics of the porous Zr terephthalate UiO-66 framework using 2H NMR and neutron scattering. J. Phys. Chem. C. 116, 12131–12136 (2012). https://doi.org/10.1021/jp3029193
Baum, M., Rieutord, F., Juranyi, F., Rey, C., Rébiscoul, D.: Dynamical and structural properties of water in silica nanoconfinement: impact of pore size, ion nature, and electrolyte concentration. Langmuir 35, 10780–10794 (2019). https://doi.org/10.1021/acs.langmuir.9b01434
Asensio, J.A., Sánchez, E.M., Romero, P.G.: Proton-conducting membranes based on benzimidazole polymers for high-temperature PEM fuel cells. A chemical quest. . Chem. Soc. Rev. 39, 3210–3239 (2010). https://doi.org/10.1039/b922650h
Bose, S., Kuila, T., Nguyen, T.X.H., Kim, N.H., Lau, K., Lee, J.H.: Polymer membranes for high temperature proton exchange membrane fuel cell: recent advances and challenges. Prog. Polym. Sci. 36, 813–843 (2011). https://doi.org/10.1016/j.progpolymsci.2011.01.003
Holderer, O., Khaneft, M., Lin, Y., Liu, S., Feoktystov, A., Kruteva, M., Zorn, R., Lehnert, W.: Nanostructure of HT-PEFC electrodes investigated with scattering methods. ECS Trans. 80, 19–25 (2017). https://doi.org/10.1149/08008.0019ecst
Schmidt-Rohr, K., Chen, Q.: Parallel cylindrical water nanochannels in Nafion fuel-cell membranes. Nat. Mater. 7, 75–83 (2008). https://doi.org/10.1038/nmat2074
Ueki, T., Watanabe, M.: Macromolecules in ionic liquids: progress, challenges, and opportunities. https://pubs.acs.org/sharingguidelines (2008)
Vilčiauskas, L., Tuckerman, M.E., Bester, G., Paddison, S.J., Kreuer, K.D.: The mechanism of proton conduction in phosphoric acid. Nat. Chem. 4, 461–466 (2012). https://doi.org/10.1038/nchem.1329
Lehnert, W., Wannek, C., Zeis, R.: Chapter 3. Trends in High-Temperature Polymer Electrolyte Fuel Cells. Presented at the October 18 (2010)
Li, Q., He, R., Jensen, J.O., Bjerrum, N.J.: PBI-based polymer membranes for high temperature fuel cells—preparation, characterization and fuel cell demonstration. https://doi.org/10.1002/fuce.200400020 (2004)
Hopfenmüller, B., Zorn, R., Holderer, O., Ivanova, O., Lehnert, W., Lüke, W., Ehlers, G., Jalarvo, N., Schneider, G.J., Monkenbusch, M., Richter, D.: Fractal diffusion in high temperature polymer electrolyte fuel cell membranes. J. Chem. Phys. 148, 204906 (2018). https://doi.org/10.1063/1.5018717
Ivanova, O., Lüke, W., Majerus, A., Krutyeva, M., Szekely, N.K., Pyckhout-hintzen, W., Appavou, M., Monkenbusch, M., Zorn, R., Lehnert, W., Holderer, O.: In fl uence of morphology on physical properties of poly (2, 5-benzimidazole ) membranes. J. Membr. Sci. 533, 342–350 (2017). https://doi.org/10.1016/j.memsci.2017.03.015
Khaneft, M., Holderer, O., Ivanova, O., Lüke, W., Kentzinger, E., Appavou, M.S., Zorn, R., Lehnert, W.: Structure and proton dynamics in catalytic layer of HT-PEFC. Fuel Cells. 16, 406–413 (2016). https://doi.org/10.1002/fuce.201500167
Zorn, R.: On the evaluation of neutron scattering elastic scan data. Nucl. Instrum. Methods Phys. Res. Sect. A. 603, 439–445 (2009). https://doi.org/10.1016/j.nima.2009.02.040
Kanellopoulos, N. (ed.): Nanoporous Materials: Advanced Techniques for Characterization, Modelling and Processing. CRC Press Taylor & Francis Group, Boca Raton (2017)
Stroeve, P., Ileri, N.: Biotechnical and other applications of nanoporous membranes. Trends Biotechnol. 29(6), 259–266 (2011). https://doi.org/10.1016/j.tibtech.2011.02.002
Martín, J., Mijangos, C.: Tailored polymer-based nanofibers and nanotubes by means of different infiltration methods into alumina nanopores. Langmuir 25, 1181–1187 (2009). https://doi.org/10.1021/la803127w
Doi, M., Edwards, S.F.: The Theory of Polymer Dynamics. Clarendon press Oxford, Oxford (1986)
Dionne, P.J., Ozisik, R., Picu, C.R.: Structure and dynamics of polyethylene nanocomposites. Macromolecules 38, 9351–9358 (2005). https://doi.org/10.1021/ma051037c
Li, Y., Wei, D., Han, C.C., Liao, Q.: Dynamics of polymer melts confined by smooth walls: crossover from nonentangled region to entangled region. J. Chem. Phys. 126, 204907 (2007). https://doi.org/10.1063/1.2737044
Bäumchen, O., Fetzer, R., Jacobs, K.: Reduced interfacial entanglement density affects the boundary conditions of polymer flow. Phys. Rev. Lett. 103, 247801 (2009). https://doi.org/10.1103/PhysRevLett.103.247801
Nusser, K., Schneider, G.J., Pyckhout-Hintzen, W., Richter, D.: Viscosity decrease and reinforcement in polymer à silsesquioxane composites. Macromolecules 44, 7820–7830 (2011)
Si, L., Massa, M.V., Dalnoki-Veress, K., Brown, H.R., Jones, R.A.L.: Chain entanglement in thin freestanding polymer films. Phys. Rev. Lett. 94, 127801 (2005). https://doi.org/10.1103/PhysRevLett.94.127801
Masuda, H., Fukuda, K.: Ordered metal nanohole arrays made by a two-step replication of honeycomb structures of anodic alumina. Science 268, 1466–1468 (1995). https://doi.org/10.1126/science.268.5216.1466
Steinhart, M.: Supramolecular organization of polymeric materials in nanoporous hard templates. Adv. Polym. Sci. 220, 123–127 (2008). https://doi.org/10.1007/12-2008-142
Digne, M., Sautet, P., Raybaud, P., Euzen, P., Toulhoat, H.: Hydroxyl groups on γ-alumina surfaces: a DFT study. J. Catal. 211, 1–5 (2002). https://doi.org/10.1006/JCAT.2002.3741
Christophersen, M., Carstensen, J., Rönnebeck, S., Jäger, C., Jäger, W., Föll, H.: Crystal orientation dependence and anisotropic properties of macropore formation of p- and n-type silicon. J. Electrochem. Soc. 148, E267 (2001). https://doi.org/10.1149/1.1369378
Jessensky, O., Müller, F., Gösele, U.: Self-organized formation of hexagonal pore arrays in anodic alumina. Appl. Phys. Lett. 72, 1173–1175 (1998). https://doi.org/10.1063/1.121004
Martín, J., Krutyeva, M., Monkenbusch, M., Arbe, A., Allgaier, J., Radulescu, A., Falus, P., Maiz, J., Mijangos, C., Colmenero, J., Richter, D.: Direct observation of confined single chain dynamics by neutron scattering. Phys. Rev. Lett. 104, 197801 (2010). https://doi.org/10.1103/PhysRevLett.104.197801
Kim, S.Y., Meyer, H.W., Saalwächter, K., Zukoski, C.F.: Polymer dynamics in PEG-silica nanocomposites: effects of polymer molecular weight, temperature and solvent dilution. Macromolecules 45, 4225–4237 (2012). https://doi.org/10.1021/ma300439k
Fatkullin, N., Kimmich, R., Fischer, E., Mattea, C., Beginn, U., Kroutieva, M.: The confined-to-bulk dynamics transition of polymer melts in nanoscopic pores of solid matrices with varying pore diameter. New J. Phys. 6, 1–14 (2004). https://doi.org/10.1088/1367-2630/6/1/046
Krutyeva, M., Wischnewski, A., Monkenbusch, M., Willner, L., Maiz, J., Mijangos, C., Arbe, A., Colmenero, J., Radulescu, A., Holderer, O., Ohl, M., Richter, D.: Effect of nanoconfinement on polymer dynamics: surface layers and interphases. Phys. Rev. Lett. 110, 108303 (2013). https://doi.org/10.1103/PhysRevLett.110.108303
Krutyeva, M., Wischnewski, A., Richter, D.: Polymer dynamics in nanoconfinement: interfaces and interphases. EPJ Web Conf. 83, 02009 (2015). https://doi.org/10.1051/epjconf/20158302009
Glomann, T., Schneider, G.J., Allgaier, J., Radulescu, A., Lohstroh, W., Farago, B., Richter, D.: Microscopic dynamics of polyethylene glycol chains interacting with silica nanoparticles. Phys. Rev. Lett. 110, 178001 (2013). https://doi.org/10.1103/PhysRevLett.110.178001
Rouse, P.E.: A theory of the linear viscoelastic properties of dilute solutions of coiling polymers. J. Chem. Phys. 21, 1272–1280 (1953). https://doi.org/10.1063/1.1699180
De Virgiliis, A., Milchev, A., Rostiashvili, V.G., Vilgis, T.A.: Structure and dynamics of a polymer melt at an attractive surface. Eur. Phys. J. E. 35, 97 (2012). https://doi.org/10.1140/epje/i2012-12097-6
Hoeve, C.A.J., Dimarzio, E.A., Peyser, P.: Adsorption of polymer molecules at low surface coverage. J. Chem. Phys. 42, 2558–2563 (1965). https://doi.org/10.1063/1.1696332
Scheutjens, J.M.H.M., Fleer, G.J.: Statistical theory of the adsorption of interacting chain molecules. 2. Train, loop, and tail size distribution. J. Phys. Chem. 84, 178–190 (1980). https://doi.org/10.1021/j100439a011
Kusmin, A., Gruener, S., Henschel, A., Holderer, O., Allgaier, J., Richter, D., Huber, P.: Evidence of a sticky boundary layer in nanochannels: a neutron spin echo study of n -hexatriacontane and poly(ethylene oxide) confined in porous silicon. J. Phys. Chem. Lett. 1, 3116–3121 (2010). https://doi.org/10.1021/jz1012406
Kusmin, A., Gruener, S., Henschel, A., de Souza, N., Allgaier, J., Richter, D., Huber, P.: Polymer dynamics in nanochannels of porous silicon: a neutron spin echo study. Macromolecules 43, 8162–8169 (2010). https://doi.org/10.1021/ma1004925
Acknowledgements
The author thanks Prof. Jörg Kärger and Prof. Douglas M. Ruthven for fruitful discussions and suggestions.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kruteva, M. Dynamics studied by Quasielastic Neutron Scattering (QENS). Adsorption 27, 875–889 (2021). https://doi.org/10.1007/s10450-020-00295-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-020-00295-4