
Abstract
Nanoporous solids, including microporous, mesoporous and hierarchically structured porous materials, are of scientific and technological interest because of their high surface-to-volume ratio and ability to impose shape- and size-selectivity on molecules diffusing through them. Enormous efforts have been put in the mechanistic understanding of diffusion–reaction relationships of nanoporous solids, with the ultimate goal of developing materials with improved catalytic performance. Single-molecule localization microscopy can be used to explore the pore space via the trajectories of individual molecules. This ensemble-free perspective directly reveals heterogeneities in diffusion and diffusion-related reactivity of individual molecules, which would have been obscured in bulk measurements. In this article, we review developments in the spatial and temporal characterization of nanoporous solids using single-molecule localization microscopy. We illustrate various aspects of this approach, and showcase how it can be used to follow molecular diffusion and reaction behaviors in nanoporous solids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Nanoporous solids are omnipresent in adsorption, molecular separation, and catalytic conversion technologies [1,2,3,4,5,6,7], and diffusion of guest molecules in these functional materials is among the rate-limiting processes in their technological application. The economic feasibility of these technologies often depends on the diffusivity of the guest molecules, therefore understanding of the underlying processes is not only of fundamental, but also of industrial and societal importance [8,9,10,11]. However, as the pore size of nanoporous solids is comparable to the size of the diffusant, a theoretical framework explaining this diffusion is particularly complex. Frequent interactions between guest (molecule) and host (nanoporous solid) strongly affect diffusion and adsorption behavior giving rise to complex heterogeneous motion [8,9,10,11]. Investigations at the single-molecule level can reveal these heterogeneities and thus provide deeper insight into such guest–host interactions and their effect on mass transport [4, 12,13,14,15,16,17].
Before the advent of single-molecule tracking studies in nanoporous solids, in particular in silica sol–gel films, diffusion was characterized with ensemble techniques such as pulsed-field gradient nuclear magnetic resonance (PFG NMR), quasi-elastic neutron scattering (QENS), vibrational spectroscopy, and fluorescence recovery after photobleaching (FRAP). Average diffusion constants were obtained with these techniques, but spatial heterogeneity could not be inferred [4, 18]. It was known from transmission electron microscopy (TEM) images that the pore size and topology were not uniform within the studied materials and that structural defects might close off pore domains [19]. To better understand the functioning of these porous materials, a microscopic understanding of the local pore geometry and the functionality correlated with molecular motion was required. This has driven the search for techniques that are able to measure local variations in mass transport at the single-molecule level.
Laser-induced fluorescence emission can be detected with high sensitivity, which makes it an excellent tool to probe local heterogeneities at the single-molecule level. In fluorescence correlation spectroscopy (FCS), the diffusion constant is inferred from temporal intensity fluctuations caused by single fluorescent emitters passing through a small (∼1 fL) detection volume. The strength and duration of these intensity fluctuations are quantified by a correlation analysis, which provides information about the underlying processes responsible for the fluctuations. The choice of an appropriate model to describe this result requires prior knowledge about the local environment that interacts with the moving fluorescent molecule, such as adsorption sites. The fitted parameters are still a (local) ensemble average of the molecules diffusing through the detection volume, and the spatial resolution of ∼200 nm is limited by the size of this volume [18, 20].
Single-molecule localization and tracking is a more direct way to visualize and quantify molecular motion. Locations of single molecules can be extracted with nanometer precision from time-lapse videos recorded with super-resolution fluorescence microscopy. Diffusion is then quantified from the displacements of a molecule within a trajectory [21, 22]. This allows a spatial and temporal mapping of the heterogeneities without a priori knowledge of the diffusion process. It was shown that the diffusion constants obtained from single-molecule trajectories, that is the time-average, and the ensemble value measured with PFG NMR are in perfect agreement as predicted by the ergodicity theorem [23, 24]. This demonstrates the consistency between the two conceptually different approaches in equilibrium conditions and validates the interpretation of diffusion constants obtained from single-molecule trajectories. Alternatively, single-molecule localization can be employed to record single reaction events using fluorogenic molecules, which directly maps local reactivity and provides indirect information about mass transport to the active reaction sites. These two single-molecule localization microscopy (SMLM) techniques are the topic of the following discussion.
The aim of this article is to review the insights into diffusion and chemical conversion that have been obtained with SMLM. This technique is unique since it can directly reconstruct the paths of individual molecules, thus giving an exclusive view on the heterogeneities that often remain unresolvable with ensemble techniques. We first discuss the fundamentals of SMLM, trajectory data analysis, and the most popular fluorescent probes for the investigation of nanoporous solids in Sect. 2. In Sect. 3, we discuss how motion heterogeneities are studied from trajectories of non-reactive fluorophores. Using the example of mesoporous silica model systems, we discuss how diffusion is correlated to the host topology and how these insights are translated to other nanoporous materials. In Sect. 4, we discuss heterogeneities in chemical conversion in a selection of nanoporous solids, including zeolites, mesoporous silica-coated metal nanoparticles, and metal–organic frameworks (MOFs). The application of correlative SMLM approaches for establishing the exact site–reactivity relationships is reviewed as well. We conclude with our perspective on the current developments in the field.
2 Single-molecule localization microscopy
2.1 Overcoming the diffraction limit: single-molecule localization and tracking
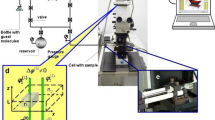
The resolution in optical microscopy is fundamentally limited by the diffraction limit. Diffraction is well demonstrated by a ‘single-slit’ experiment, in which a coherent light source is imaged onto a plane via a circular aperture. The interference of light then generates the well-known (interference) pattern consisting of a bright center surrounded by alternating dark and bright concentric rings, which is better known as the Airy disk. In an optical microscope, the objective lens has a finite angle under which it can collect light from the sample (Fig. 1a). It therefore effectively acts as an aperture that gives rise to the same Airy-disk diffraction pattern. The pattern is now denoted as the point spread function (PSF) as it describes the microscope’s response to a point emitter. The full width at half maximum (FWHM) of the PSF is often used as a short-hand resolution limit for conventional light microscopy, and results in a maximum resolution of ∼200 nm [22, 25].

Adapted from Manzo and Garcia-Parajo [21] (Color figure online)
Schematic representation of single-molecule localization and tracking. a A typical fluorescence microscope set-up consists of a laser excitation source, a dichroic mirror, an objective lens, and a detector. The dichroic mirror reflects the excitation light (green) via the objective onto the sample, but lets the light emitted by the sample (yellow) pass to the detector. A parallel beam can be used to illuminate the sample over the field of view (i.e. wide-field microscopy) or a focused spot can be scanned over the sample (i.e. confocal microscopy). The fluorescent emission of the excited fluorophores is recorded by the detector creating a micrograph. b When tracking individual emitters, the center of the diffraction-limited spots in the time-lapse movie are fitted, and the individual localizations are grouped together in a trajectory. These tracks can be interpreted in a final step.
Super-resolution fluorescence microscopy is a hypernym for techniques that bypass the diffraction limit [22]. As the location of the fluorescent molecule is at the center of the PSF, the position of a single fluorescent molecule can be determined with a precision much higher than the diffraction limit by fitting the PSF’s center position. Eric Betzig recognized that a prerequisite for sub-diffraction localization is sparsity in the molecules’ diffraction-limited spots [26], that is, overlapping PSF’s need to be avoided. Then, by selective excitation of subpopulations of fluorophores, a super-resolution image with a high fluorophore density can be reconstructed. Two years later, a new piece of the puzzle was added: pioneering in the field of single-molecule spectroscopy, William E. Moerner reported a specific mutant of the green fluorescent protein (GFP) with strong intermittent fluorescence, so-called blinking, before going to a stable dark state. The blinking behavior could be recovered with a burst of UV-light [27], giving exactly the fluorophore properties and control over emission Betzig was looking for. Using Moerner’s discovery, Betzig in collaboration with the biologist Jennifer Lippincott-Schwartz, demonstrated super-resolution imaging of labelled intracellular proteins using SMLM [28]. After this first demonstration, SMLM quickly became a standard imaging tool in the fields of biology and chemistry, and it should not come as a surprise that a share of the 2014 Nobel Prize for chemistry was awarded to Eric Betzig and William E. Moerner for their pioneering work in the field.
Diffusion of individual fluorescent molecules can be followed as a function of time using SMLM. The pore space of the porous host is explored by the moving fluorogenic probes, and sparsity is automatically achieved when a low fluorophore density is employed. A typical analysis consists of localization, tracking, and interpretation (Fig. 1b) [21]. First, individual emitters are localized in each individual frame. The PSF is often approximated by a Gaussian intensity distribution, which can be fitted to individual emitters. In this way, the center coordinates of an isolated PSF are retrieved with sub-pixel precision, i.e. down to ∼2 nm [21, 22, 29, 30]. During tracking, the localizations are grouped into tracks, which represent the path of a single fluorophore. Popular algorithms require cut-off values to prevent unphysical linking, these values represent the maximum displacement between localizations and the maximum number of consecutive frames the molecule is allowed to be in an ‘off-state’ [21, 31]. Finally, the obtained tracks can be interpreted using motion analysis or counted to determine the local reactivity in a porous material.
Fluorogenic substrates are used to map the reactivity of a working catalyst. Here, a non-fluorescent reactant is converted into a fluorescent product over the active sites of the catalyst, which allows the detection of single catalytic turnovers. Each turnover is represented by a single track, that is the formation of a product molecule, from which catalytically active zones in the material can be reconstructed with super-resolution. Because reaction events occur stochastically on the catalysts’ active sites, these tracks are spatially and temporally separated and therefore can be resolved individually for each turnover. Moreover, fluorescent products photo bleach quickly under the imaging conditions or rapidly diffuse away from the active site, which renders them invisible due to their fast motion, and prevents overlap of different tracks. This concept forms the basis of a method named nanometer accuracy by stochastic chemical reactions (NASCA) microscopy and is a common approach to image the reactivity of a catalyst at work [13, 14, 17, 32, 33].
Methods to estimate the diffusion constant from single-fluorophore tracks come in varying degrees of complexity. The mean-squared displacement (MSD) analysis is one of the most common tools to do this, because of its visual interpretability [29, 34, 35]. The shape of the MSD curve is an indication of the type of motion and aids in selecting the appropriate diffusion model. It is computed from a time series of positions \({{\varvec{x}}}_{0},{{\varvec{x}}}_{1},\ldots,{{\varvec{x}}}_{N}\) for a single trajectory as
with the lag time \({t}_{n}=n\Delta t\) for \(n=\mathrm{1,2},\ldots,N\) and the time between frames \(\Delta t\) [34,35,36,37,38]. Argued by Albert Einstein in his 1905 seminal work, the mean displacement recorded for a particle suspended in a liquid or gas medium is not proportional to the lag time, and thus the mean velocity cannot be inferred from this value. However, the mean-squared displacement (MSD) is proportional to the lag time and the motion can be quantified with a proportionality constant known as the diffusion constant \(D\) [39]. Hence for normal diffusion, the diffusion constant can be estimated from the MSD via
for a dimensionality \(n\). However, scaling of the MSD with the lag time is not always linear and deviation from normal Brownian diffusion occurs. An important scenario is when the host provides a heterogeneous environment for the guest molecules over the probed length and times scales or when the guests interact with the host. The resulting motion is called anomalous diffusion and is described by a power-law scaling
with a generalized diffusion constant \(K\) and an anomalous exponent \(\alpha \). The motion is called sub-diffusive when \(\alpha <1\) and super-diffusive for \(\alpha >1\), and there are many examples of this type of motion in biological systems [21]. These models can be expanded to account for localization noise and motion blur [34, 35].
Tracks with many localizations are required for reliable diffusion constant estimation, particularly with MSD analysis [38]. This is often not possible as molecules rapidly diffuse out of focus or photo bleach too quickly, which in turn has driven the search for alternative measures. A maximum likelihood estimator (MLE) has been derived for free diffusion [34, 35] considering blinking [40], motion blur, and variable localization error [41]. However, one of the challenges is that both the MSD and MLE diffusion constant estimates are biased for tracks typically obtained in experiments. The unbiased covariance-based estimator (CVE) was therefore derived as an alternative to MSD and MLE analysis, however it does not perform well in cases where the displacement per step is short with respect to the localization error [38, 42]. A promising way to increase the precision of diffusion constant estimation is to quantify the localization error and include this in the model for the MLE or CVE [38, 43]. When multiple underlying diffusional states are present, the model becomes more complex, and other approaches have been developed for this task, which are often based on Bayesian statistics [44,45,46,47]. Considering these recent developments, motion analysis of multistate diffusion seems to come within reach, but there is no golden standard yet, and the optimal analysis method still has to be determined on a case-by-case basis.
2.2 Fluorescent molecular probes for imaging diffusion and catalytic conversions
The optimal fluorophore is a trade-off between many competing factors, and good fluorophore selection is the fundament of any SMLM experiment. In material science we cannot directly copy the toolkit developed for SMLM in biological systems, and there are multiple reasons for this: (1) as biological systems are often water-based, the solvent and the material have a very similar refractive index. In nanoporous solids, the refractive index of the host material is less homogeneous and can be much higher than that of the solvent. In that case, light scattering of the host material, due to the refractive index mismatch with the solvent, results in a high background hindering or even preventing localization. A careful choice of the solvent, i.e. index matching, minimizes this effect. (2) As inorganic materials of interest are often non-polar, compatibility with polar dyes used in biological media is limited, because their fluorescence and solubility is often low in non-polar media [48]. (3) The typical conditions of interest are often completely different for biological systems and functional materials. Rigorously air- and moisture-free conditions and non-ambient temperature and pressure are not common in biological systems, but can be, for example, in heterogeneous catalysis [49]. (4) To probe diffusion in nanoporous materials, the size of the diffusing probe is another important factor closely linked to the research question of the study. Preferably, the probe size is comparable to that of the relevant species, such as the reactant and product in a nanoporous catalyst, but strong light scattering of the host might require very bright fluorophores. Altogether, the selection of the optimal fluorophore is paramount for experimental success.
For optimal diffusion coefficient estimation from single-molecule tracks, a fluorophore with minimum blinking, limited photobleaching, and a high photon-emission rate is desired, as this results in long tracks [50]. Blinking and photobleaching can be controlled in aqueous systems by buffering the solution with oxygen scavengers or oxidator–reductor couples [51, 52]. Such protocols would be desirable for non-polar solvents as well [49]. Nevertheless, enhanced photostability inside p-terphenyl crystals was observed without oxygen scavengers due to shielding from oxidative species directly by the host [53]. For a high photon emission rate, the fluorophore needs to possess a large absorption cross section and high quantum yield. This is an intrinsic property of the fluorophore and can be highly dependent on the solvent. Rylene dyes, such as perylene diimide (PDI) and terylene diimide (TDI) derivatives, are particularly popular for tracking studies [54,55,56,57,58,59,60] owing to their high photon emission rate, photostability, and modifiability (Fig. 2a) [61, 62]. It is worth mentioning that nanoparticles, in particular quantum dots, have already been proven to be excellent probes to study mass transport in porous solids because of their even higher photon emission rate and excellent photostability [63, 64]. However, their larger size in comparison to fluorescent molecules limits the application in many micro- and mesoporous systems (pore diameters ≤ 50 nm). Here, nanographenes hold great potential for SMLM as they are the happy medium between the small size of molecules and favorable photoemission properties of quantum dots [65].

Comprehensive overview of fluorescent molecules to probe diffusion (a), reactivity in zeolites (b–d), and reactivity in other nanoporous solids (e–g). (a) Rylene dyes perylene diimide (n = 1) and terylene diimide (n = 2), (b) oligomerization of furfuryl alcohol, (c) dimerization of styrene derivatives, (d) thiophene oligomerization, (e) reduction of resazurin into resorufin (left) and oxidation of Amplex Red (right), (f) phenylbutadienyl-BODIPY epoxidation, and (g) fluorescein diacetate ester hydrolysis into fluorescein over a metal–organic framework (MOF)
The number of reported reactive fluorophores is limited, as their design, synthesis, and purification are not straightforward. In zeolites (pore diameters < 2 nm) used in catalysis, the oligomerization products of furfuryl alcohol, thiophene, and styrene have been successfully reported as fluorescent single-molecule probes by the groups of Roeffaers, Hofkens, and Weckhuysen (Fig. 2b–d) [33, 66, 67]. Their small size, determined by carbon 5- and 6-rings, allows these reactants to enter the micropores of zeolites and oligomerize at its active sites. Oligomerization is an acid catalyzed process and happens readily over the Brønsted acid sites in the zeolite pores. The conversion rate of styrene and thiophene reactants is strongly dependent on their side groups, because it alters the stability of the carbocation in the fluorescent product and limits transport into the porous catalyst. Systematic study of these substituent effects have been proven valuable on the single-particle and single-molecule level [67,68,69,70,71]. A challenge in the analysis of these single-molecule events is the strong intrinsic blinking due to proton transfer from the acid sites [67, 70, 72]. To discriminate between the formation of a new oligomer, which is an indication of mass transport of the reactant to the active site, and proton transfer, careful experimental design and analysis are required.
In meso- (2–50 nm) and macropores (> 50 nm), well-defined probes are preferred, which allows quantitative interpretation of the results (Fig. 2e–g). Here, the modification of highly fluorescent and photostable dyes with a reactive functional group is a common approach. This requires that the fluorescent domain is chemically stable or reacts in a controlled manner. However, free electron-pairs present in many popular fluorophores can result in undesired reactivity. BODIPY dyes deserve special attention as they are chemically inert, relatively nonpolar, and electrically neutral [49]. They have been successfully applied by de Cremer et al. to study the epoxidation over mesoporous Ti-MCM-41 (Fig. 2f) [73, 74]. Modification of the conjugated π-system due to a reaction event causes a spectral shift in the fluorescence emission, which potentially allows the monitoring of reactant and product diffusion as well as the reaction event. An opposed approach is to use the reactive lone pair to report a reaction event, since oxidation of the free electron pair can induce a spectral shift or quenching of the fluorescence [49]. The fluorescence is then recovered after reduction of the molecule, which is exemplified by the catalytic conversion of resazurin into resorufin as shown in the left reaction in Fig. 2e. Resorufin is among the most popular reactive dyes and has been widely used in the group of Chen to study the reaction kinetics of individual metal nanoparticles [32, 75]. Its small size makes it particularly interesting to investigate mass transport in the mesopore regime [76]. However, larger fluorophores with good photophysical properties, such as rhodamine, coumarin, and perylene, can be modified in the same manner to produce reactive dyes [77, 78].
3 Visualizing diffusion of single molecules in nanoporous solids
3.1 Molecular transport in silica films
Mesoporous silica makes an excellent model system to study single-molecule diffusion due to the high degree of control over both pore size and chemical environment inside the pores. Such control can be achieved by tuning the precursor solution before application on a cover slip substrate. The structure-directing agent forms micelles in the silica precursor solution, which templates the pores in the 2–30 nm range [15, 79]. Hexagonal, lamellar, and cubic geometries can be obtained via tuning of the silica/structure-directing agent ratio [58]. Addition of a functionalized-silica source to the precursor solution allows direct chemical modification of the pore walls [59]. A wide range of differently-sized fluorophores can be incorporated inside the pores [79, 80]. Exposure to solvent vapor is required to complete the formation of the cylindrical mesopores [81] and to mobilize the dye molecules for tracking studies [57]. All these components have their unique interaction with the diffusing dye, leading to complex motion behavior.
The first observations of single-molecule trajectories in mesoporous silica revealed strong heterogeneities in molecular motion [18, 55, 82, 83]. Inter- and intra-trajectory heterogeneities were observed in both non-ordered glassy and ordered templated mesoporous silicas with pore sizes of respectively 3 and 22 nm. In the non-ordered glassy sample, trajectory segments were observed in which the molecule appeared to be trapped for some time (Fig. 3a). The authors speculated that this could either be an adsorption event by temporary bonding to the silica surface or confinement e.g. in a narrow pore leading to strongly hindered motion [18]. In the ordered mesoporous silica, no temporary trapping was observed, but the motion could not be described by a single diffusion constant (Fig. 3b, d) [55]. These different diffusion regimes were not spatially separated within a single trajectory and indicated variations in the microenvironment of the dye [55, 58]. A final remarkable observation was that the dye diffusion appeared to follow a specific direction instead of moving randomly as would be expected for normal diffusion (Fig. 3c) [55]. The pore geometry and local obstructions could confine the dye into a pore domain. Transient trapping of the fluorescent dye, multiple diffusion regimes, and apparent confinement to the pore geometries turned out to be key observations and have led to two decades of single-molecule studies of motion heterogeneity in mesoporous silica.

Adapted from Hellriegel et al. [55]
Examples of anomalous diffusion behavior in mesoporous silica as observed in early days of single-molecule tracking in nanoporous solids. a Single-molecule trajectories displaying trapping of the molecule in the sol–gel, b anomalous diffusion, and c diffusion directed along the pore direction. d Step length histogram and fit (thick solid line) of the trajectory displayed in b. The full step length distribution can only be explained by the contribution of two different diffusion constants (thin solid lines) indicating at least two different underlying diffusion regimes.
3.1.1 Confinement imposed by the pores
Comparison of a known pore geometry and single-molecule trajectories revealed how probes can infer the topology of mesoporous silica. Samples with pure hexagonal, pure lamellar and mixed geometries were compared by Kirstein et al. (Fig. 4a, b) [58]. The motion of the molecules in these geometries was qualitatively and quantitatively different (Fig. 4c). Molecular motion resembled random Brownian motion in the lamellar phase, while motion appeared correlated in the hexagonal phase as if the molecule was probing the domain boundaries. The shape of these domains did not in any way resemble structures on the outer surfaces, which were mapped with atomic force microscopy, and the authors concluded that the probed domains were located inside the mesoporous silica film. The mixed phase was more defect-rich, and even escape from the bulk to the surface was recognized followed by re-entrance at a different location in the structure. Here, switching between diffusion modes corresponding to lamellar, hexagonal, and surface domains was observed. TEM images showed that these domains coexisted, being stacked on top, and confirmed the topology inferred from the trajectories [58]. Optical microscopy cannot directly map the underlying pore structure, and correlation with other microscopy techniques is required for direct correlation with the local pore geometry. Zürner et al. correlated TEM images with the single-molecule tracks in hexagonal mesoporous silica (Fig. 4d–i) [60]. The motion of the molecules followed the direction of the linear and curved pores almost perfectly. However, defects and grain boundaries of the hexagonal domains stopped the path of the molecules, and ‘leaky’ channels allowed lateral travel. This has shown that single-molecule trajectories can provide a direct observation of the nanoscale-connectivity and accessibility in mesoporous samples [58, 60]. The location of the molecules could be determined with a precision of about five to ten parallel channels, and a higher resolution was required to study the exact mechanism of lateral travel [60].

a Schematic representation of lamellar and hexagonal mesoporous silica with the order of the structure-directing agent shown inside the pores. b Schematic diagram of the diverse diffusion modes and the transitions between them in the mixed phase. c The MSD curves for the populations depicted in b. Individual tracks are plotted only for the phase mixture, and the thick lines represent the average values for the pure hexagonal and pure lamellar phases. Reproduced from Kirstein et al. [58] with permission from Springer Nature. d Overlay of single-molecule trajectories and TEM micrographs of the pore space. The orientation of the solid black lines indicates the orientation of the pores, and the blue boxes along the trajectories illustrate the localization error. The molecules explore domains with parallel channels and follow strongly curved regions and domain boundaries. e Magnified area from d showing the domain boundary at which the molecule turns back. f Trajectory of another molecule, which follows an S shape corresponding to the underlying pore direction. The different colors correspond to different sections of the track that were due to blinking of the molecule. g The yellow trajectory shows lateral motion between ‘leaky’ channels. h Obstruction of molecular motion at a less ordered region and i domain boundary. Adapted from Zürner et al. [60] with permission from Springer Nature (Color figure online)
At high excitation intensity, the molecules could be localized with high resolution, and Jung et al. found discrete jumps in the trajectories perpendicular to the dominant diffusion direction (Fig. 5a, b) [57]. The localization error was smaller than these jumps, which therefore reflected the structure of the material itself. The mean jump distance was found to be 5–6 nm (Fig. 5c), which is only slightly larger than the pore-to-pore distance obtained with X-ray diffraction (XRD). This showed direct evidence of diffusion through defects in the material. Comparison with random walk simulations in a similar geometry indicated that the defect density of the wall is low, in the order of 0.1% permeability. In some cases, even further parallel pores could be explored, which was observed for a molecule moving to a parallel pore about six channels away. This indicated the presence of both nanodefects in the pore wall and less ordered domains, which allowed for single and multichannel hopping [57, 60]. The single-molecule paths follow directly the pore geometry and topology and revealed the origin of both domain-size and local motion heterogeneities.

Reproduced from Jung et al. [57]. Copyright 2008 American Chemical Society (Color figure online)
Diffusion through defects in pore walls of hexagonal mesoporous silica. a Projected x and y coordinates as a function of time for a single molecule. Discrete jumps in the position indicate hopping to a parallel pore marked by the transition from black to green at 103 s. b Trajectory of the molecule in a. c Histograms of the projected y coordinate for the black and green time domain with green and black striped bars, respectively. The two maxima are separated by 5–6 nm.
3.1.2 Transient trapping of probe molecules
Temporary trapping of single molecules is another remarkable motion heterogeneity observed in the early days of single-molecule tracking in mesoporous silica. When this was reported for the first time, authors could only speculate about the origin. It was suggested that it could either be an adsorption event by temporary bonding to the silica surface or confinement in a narrow pore or at a defect site leading to strongly hindered motion [18]. However, correlation with the pore topology did not reveal any trap sites, not even at defects [60]. To study a trapping event at the single-molecule level, the orientation and emission wavelength were mapped in the work of Jung et al. (Fig. 6) [84, 85]. Small changes in interaction between the host and the guest were detectable in the fluorescence emission spectra, and the length of the adsorption events was suggested to be related to the strength of the adsorption sites. Seconds-long time periods were observed in which the emission wavelength maximum and orientation angle remained unchanged, which suggested strong adsorption of the probe. After about a minute, the emission spectrum and orientation angle of the probe started to vary a lot, which indicated a rapidly changing environment, that is, the presence of various environments probed by the now mobile molecule. The duration of the adsorption events followed a broad distribution with adsorption times up to minutes. Adsorption could be explained by chemical bonding to the silica walls and electrostatic interactions with the cationic template as well as interactions at defect sites [84, 85]. Due to the various factors involved in an adsorption event, strong heterogeneity in the effective strength of adsorption sites was expected and in line with experimental observations.

Reproduced from Jung et al. [84] with permission from Wiley
The angular and spectral behavior of a single molecule during a trapping event in hexagonal mesoporous silica. a The simultaneous fluorescence emission spectra, b the wavelength maximum of the emission spectra, and c molecular orientation of the probe molecule as a function of time. The gray areas indicate time periods in which the molecule was non-emissive.
3.1.3 Anomalous diffusion
To understand how the fluorophore moves through pores, it is important to understand how it interacts with the chemical species in the pore. This was studied in detail via the quantification of rotational motion of rod-shaped PDI-based fluorophores (Fig. 2a) around the mesopore axis, so called wobbling, using single-molecule emission dichroism (SMED, Fig. 7a). The ratio of the vertically versus horizontally polarized light obtained with SMED was used to compute the maximum wobbling angle and mean accessible pore space [80, 86]. Wobbling of PDI-based dyes with various carbon chain lengths in cetyl trimethyl ammonium bromide (CTAB)-templated hexagonal mesoporous silica was investigated. The mean accessible cavity diameter was about one third of the pore diameter [80]. Alignment of the dye with the pore direction indicated constrained rotational freedom due to interaction with the pore-void constituents [57, 80, 86]. A spread in the maximum wobbling angle was observed and indicated a 28% relative standard deviation in the pore sizes. This was much larger than the 4% relative standard deviation found from X-ray scattering, where the silica pore itself was probed. Thus, it was concluded that the effective pore size as experienced locally by single molecules is smaller and more heterogeneous than the physical pore size of the mesoporous silica [80].

Single-molecule wobbling and partitioning in hexagonal mesoporous silica. a Schematic representation of the wobbling of a Nile Red molecule around the pore axis of a templated mesopore. The maximum wobbling angle θ and effective pore diameter d are depicted. b Single-frame emission ratios and Clausius–Mossotti (CM) factors for 0D (immobile), 1D, and 2D diffusing Nile Red molecules. A larger CM factor indicates a more polar microenvironment. The solid black lines show fits of Gaussian functions to the main populations. Adapted from Kumarasinghe et al. [87]. Copyright 2016 American Chemical Society. c Schematic of TDI-based dye molecules immobilized at the pore wall in air (top) and mobilized in the presence of solvent molecules (bottom). The mobile molecules partition more towards the center of the pore. Reproduced from Jung et al. [57]. Copyright 2008 American Chemical Society (Color figure online)
An important implication of this is that the microenvironment of the molecules is not only dependent on the mesopore itself, but also on the location in the mesopore, since they only probe a fraction of the pore diameter. The polarity is a good indicator for this location, as the microenvironment becomes more polar near the pore wall due to the presence of charged silica and surfactant functional groups. The local polarity was probed with the solvatochromic dye Nile Red (Fig. 7a, b) utilizing the fact that the intensity ratio between two emission bands reflects the local polarity around single Nile Red molecules [87]. A large fraction of the molecules was found stuck, and these probes were either in nonpolar or highly polar microenvironments. This suggested that some dyes were immobile because they were trapped inside the silica matrix or on the silica surface. Molecules with 1D motion were assigned to the hexagonal mesopores. The local polarity in the mesopores was similar to that of n-hexane, but the microenvironment of molecules that exhibited 1D diffusion was several percent more polar than the non-polar microenvironments of the immobile molecules. Even though the overall non-polar microenvironment showed that the mobile molecules were not in very close proximity to the pore wall, some interaction with polar moieties was possible [87]. In general, the dye appears to partition in the core region of the micelle, but the mobile molecules appear to have some other interactions as well.
Anomalous diffusion was observed in a few cases for both neutral and charged dyes, also indicating local variations in the microenvironment of the dye [55, 58, 88]. It was hypothesized that oxygen atoms of the fluorescent probe can form hydrogen bonds with the exposed silanol groups of the mesoporous host, which results in intermittent adsorption events. Shielding of the exposed silanols by e.g. solvent molecules’ functional groups would then increase the dye mobility (Fig. 7c) [57, 59, 85]. A wide range of adsorption times on time scales much shorter than the observation time could account for the observed motion heterogeneity [21, 37]. Alternatively, Kumarasinghe et al. attributed the motion heterogeneity in microenvironments to an inhomogeneous distribution of solvent molecules [87]. Local variations in the concentration of solvent molecules could result in a variable shielding, which might account for the observed motion heterogeneity. Both hypotheses are in line with the observation that the 1D mobile molecules experience a slightly more polar microenvironment than the 0D and 2D molecules (Fig. 7b), and it remains an open question whether the observed motion heterogeneity originates from short adsorption events or local variations of solvent molecule concentration [59, 88].
The packing of the structure-directing agent in the micelle and guest–host interactions are other factors to consider. Interestingly, a strong increase in mobility was observed with increasing density of C1–C3 alkyl surface groups. An increasing number of functional groups was found to introduce additional defects in the packing of the structuring agent, which was observed with nuclear magnetic resonance (NMR) spectroscopy, and this provided a more open structure through which the dyes could move more rapidly. Remarkably, an opposite trend was observed for phenyl-functionalized mesoporous silica. Attractive π–π interactions between the phenyl and the aromatic groups of the dye were found to be dominant in this case. Even though it was concluded in other work that the dye partitioned in the non-polar hydrophobic part of the micelle template, interactions with the pore wall dominated its mobility. Altogether, a trade-off between the shielding of the silanol groups, packing rigidity of structure-directing agent, and additional guest–host attractions make up the motion of molecules through mesoporous silica [59].
Combination of single-molecule tracking and SMED allowed the correlation of translational motion and mean accessible pore space for both neutral and charged dyes. The accessible pore size was found to be constant for the C4–C11 functionalized PDI-based dyes, which had all a similar charge and polarity. The diffusion constant was also the same for these dyes, which indicated that the accessible pore diameter rather than the size of the diffusing probe determined the mobility along the pore axis [80]. However, the accessible pore diameter was dependent on the polarity and electrostatic interaction of charged molecule [80, 88]. Positively and negatively charged PDI-based dyes were found to have a similar mean effective pore size as the neutral dyes, but had a reduced mobility with respect to these fluorophores [88]. The cationic dye had an almost two times lower mobility than the neutral dyes and was one and a half times less mobile than the negatively charged dye. Even though the mean effective pore size of the probe was similar to the neutral ones, a larger spread in the obtained effective pore size was observed, which points to a stronger and more variable interaction with its environment [88]. Indeed, the charged dyes could more easily approach the pore walls and move through defects, which was attributed to the more polar molecule tips and electrostatic interaction of the dye with the negatively charged silica and the positively charged CTAB head groups [88, 89]. Altogether, the mobility of charged probes was found to decrease significantly, likely due to a stronger interaction with the pore-void constituents.
In summary, these findings sketch a picture where the micelle core acts as the highway for dye molecules and their degree of rotational freedom and affinity for the pore wall sets the speed limit. An open structure allows for even higher speed limits via the packing density of the structure-directing agent. Incorporation of functional groups at the pore wall decreases the packing density and affinity of the dye for the pore wall. Electrostatic interactions also allow for increased interaction of the dye with its environment, which in turn slows down diffusion in the discussed silica films. Control over mobility is of paramount importance for the development of e.g. drug-delivery systems and chromatographic particles [90, 91]. Finetuning of host-diffusant interaction resulted in a successful gradual release of the anticancer drug Doxorubicin hydrochloride [91]. Local variations in the microenvironment of the diffusant have been discussed at the single-molecule level and could help us to better understand mass transport in these systems.
3.2 Mass transport in other nanoporous solids
Fundamental insights about mass transport in mesoporous silica can be translated to other nanoporous solids that have been studied at the single-molecule level. Diffusion of Nile Red fluorophores was studied in crystalline microporous MOFs by Liao et al. [92] revealing a strong correlation between the pore structure of the MOFs and diffusion heterogeneity. The trajectories in the MOFs with 1D linear micropores were qualitatively similar to the ones in mesoporous silica. Structural defects were observed indirectly as the molecule appeared to hit a ‘dead end’ and was confined to this region before finding a way out. Lateral diffusion was more frequent than in mesoporous silica, which might indicate that it is a more defect-rich material or could be due to the lack of a structure-directing agent in the pores [92].
The step to less well-defined systems with a broad distribution in pore- and/or probe size requires a different approach, which is being developed in our group [56, 93, 94]. In such systems, complex motion patterns arise and the trajectories first have to be classified based on their properties, for example via machine learning. This yields populations of trajectories with similar motion behavior, and the diffusion constant can then be inferred from individual tracks or from the population mean in the case of short trajectories [94]. This approach was used for the first time to study single-molecule diffusion of a PDI-containing dye in fluid catalytic cracking (FCC) particles, an important industrial catalyst, by Hendriks et al. (Fig. 8a, b) [56]. These catalyst particles are not only heterogeneous in material composition, but also contain a hierarchical pore network with pore sizes in the micro-, meso-, and macroporous regime [95]. Even though the large PDI-based dye could not probe the microporous zeolite domains, it is no surprise that single-molecule diffusion in a single FCC particle revealed a large range of diffusion constants spanning at least three orders of magnitude. Moreover, heterogeneity in motion patterns was observed, and the trajectories were segmented into mobile, immobile, and hybrid categories using a hierarchical decision tree (Fig. 8c–g). The mobile and hybrid tracks were spatially homogeneously distributed, and the pore network could not be resolved directly from the tracks mainly due to the limited track lengths obtained (Fig. 8h). The spread in diffusion constants (Fig. 8i was attributed to the large pore size distribution; however, the heterogeneous material composition could also have contributed via intermittent physisorption events. The obtained diffusion constant was similar to that of similarly-sized vacuum gas oil feedstock molecules measured with adsorption experiments [56]. Intraparticle diffusion has been found to be the limiting factor in the conversion at operating conditions [96, 97], which is discussed in detail by Kärger et al. in their PFG NMR-themed contribution in this issue [10]. Indeed, the Thiele modulus was approximated for the cracking of cetane (C16) to octane (C8) and was in line with previously reported values, validating the single-molecule approach to study mass transport in these systems [56].

Adapted from Hendriks et al. [56] with permission from American Chemical Society (Color figure online)
Single-molecule trajectory classification and motion analysis of PDI-containing dye molecules in a single FCC catalyst particle. a Schematic of the imaging plane at the middle cross section in the FCC particle. b Schematic representation of the hierarchical pore structure of an FCC particle, where light gray represents the matrix and the darker gray squares the embedded zeolite active sites. c Color-coded map of each recorded track within the FCC particle, showing d immobile (red), e hybrid (green) and f mobile tracks (blue). g Trajectory of a dye molecule immobilized in a polystyrene thin film, which has a similar radius as the immobile track in d indicating a correct classification. h Voronoi diagram showing a spatial map of single trajectory diffusion coefficients. The center of mass of the tracks are indicated by a dot, which is surrounded by an area that is closer to that track than to any other. The color of each area indicates the diffusion coefficient, with areas around immobile tracks being white. i The diffusion coefficients of all trajectories; the gray box indicates the values which fall within the localization inaccuracy. The inset Voronoi diagram shows the spatial distribution of each track type.
Clever design of model systems can resolve diffusion anisotropy. Direct comparison of single-molecule diffusion in the sinusoidal and straight channels of ZSM-5 was performed by Fu et al. [93]. A thin-film model system was synthesized with either straight or sinusoidal pores in the observation plane. As small dyes are required for diffusion studies in microporous crystals such as zeolites, oligomerization products of furfuryl alcohol were used as fluorophore. Qualitatively similar heterogeneity in motion patterns was observed as in FCC particles, and the trajectories were again pooled into mobile, immobile and hybrid categories by adapting the hierarchical decision tree from Hendriks et al. [56, 93]. An order of magnitude faster average diffusion was observed in the straight than in the sinusoidal channels, which is in line with PFG NMR measurements [98]. This was explained directly with steric effects imposed by the pore geometry. Introduction of mesopores in the sample resulted mainly in one order of magnitude faster diffusion in the sinusoidal pores, while the mobility in the straight pores remained roughly the same. This was attributed to a selective etching in the plane of the sinusoidal pores facilitating faster diffusion in this direction [93]. Altogether, we can conclude that motion in nanoporous materials is heterogeneous, and that single-molecule microscopy has a pivotal role in the elucidation of these heterogeneities.
4 Mapping single-turnover catalytic conversions in nanoporous solids
4.1 Single-molecule study of structure–reactivity relationships in nanoporous solids
SMLM has been demonstrated to be a powerful technique for the visualization of reaction behaviors in nanoporous materials, in particular zeolite-type materials and mesoporous silica [33, 70, 73, 74, 99,100,101,102]. Roeffaers and co-workers were among the first to use single-molecule fluorescence to investigate zeolite chemistry in ZSM-22 and ZSM-5 zeolites [33]. They demonstrated that the acid-catalyzed furfuryl alcohol oligomerization reaction (Fig. 2b) could produce pore-entrapped products with sufficiently bright fluorescence to localize the individual product molecules [103]. Using the NASCA approach, they were able to record super-resolution reactivity maps of individual ZSM-22 particles with a diameter of ca. 100 nm (Fig. 9a). The reaction turnovers were accumulated into 20 × 20 nm2 bins and revealed zones with different activity within a single ZSM-22 rod, which has a 1D pore geometry. Three zones with a higher activity spaced along the long axis of the rod were observed (Fig. 9b), which is in line with the notion of aggregative growth of ZSM-22 [104]. As the 1D pores of the oriented crystals are aligned along the long axis, molecules could enter the channels in the center of the aggregates at the grain boundaries due to imperfect fusion during crystal growth, resulting in the observed reactivity distribution [105]. Zeolite ZSM-5 with known intergrown structures was studied using the same fluorogenic reaction. A remarkable heterogeneity in catalytic activity with a high reaction rate near the border between two intergrown crystal components was observed (Fig. 9c). This was attributed to the differences in accessibility in this area as a result of different orientations of the 10-membered rings of the zeolite material. Thus, defects at grain boundaries between zeolite aggregates appear to have a strong effect on the local reactivity.

Adapted from Roeffaers et al. [33] with permission from American Chemical Society
Inhomogeneous reactivity in zeolites revealed with NASCA microscopy. a Scatter plot of all the spatially resolved catalytic conversions of furfuryl alcohol on an isolated zeolite ZSM-22. Insert shows the needle-like morphology of zeolite ZSM-22 as observed with scanning electron microscopy (SEM). b Super-resolution reaction map calculated with a 20 × 20 nm2 bin size of the same sample as in a. The inset shows the local reaction rate as measured along the white line in b. c SEM image of three intergrown coffin-shaped ZSM-5 crystals (top), and a super-resolution reactivity map of a similar sample (bottom). The reconstructed fluorescence map clearly shows the location of the active intergrowth region.
In 2015, Ristanović et al. demonstrated that single-molecule fluorescence experiments allowed mapping of the catalytic activity in a more complex, industrially applied catalyst, i.e. FCC particles as discussed above (Fig. 10a) [99]. These catalysts are hierarchically structured, multi-component particles, which contain acidic zeolite Y or ZSM-5 embedded in a matrix of clay, silica, and alumina [95]. As the fluorescent products are formed exclusively at Brønsted acid sites, their fluorescence can be used for the 3D localization of zeolite ZSM-5 domains embedded within the matrix material of a single FCC particle. Due to the high structural complexity and intrinsic background fluorescence, super-resolution optical fluctuation imaging (SOFI) was selected as a complementary method to the NASCA approach. SOFI is a post-processing method, which maps out regions with fluctuating fluorescent emitters and allows super-resolution imaging in experiments with a low signal-to-noise ratio [106]. From binarized SOFI images, zeolite clusters could be identified, and the size distribution of the domains was computed (Fig. 10b, c). The results showed that the zeolite domains were well-dispersed throughout the imaging volume and the individual domains were about 500 nm in diameter. This is consistent with the size distribution of zeolite particles in FCC catalysts obtained via scanning transmission X-ray microscopy using a 100 nm pixel size [107] and confocal laser scanning microscopy [108]. Analysis of reactivity using NASCA microscopy showed that the average turnover rate of highly active zeolite domains was around 5 µm−2 s−1, which is about an order of magnitude higher than the least reactive zeolite ZSM-5 domains (Fig. 10d). It was speculated that this could be a result of differences in framework aluminum content and/or accessibility.

Adapted from Ristanović et al. [99] with permission from Wiley
Zeolite domain size quantification in an FCC particle. a Schematic of the single-molecule fluorescence approach used in the study of a single FCC catalyst particle, containing zeolite ZSM-5 domains depicted in red (top), and the geometry of the analyzed focal slices at various focal depths (bottom). The inner regions of the FCC particles (depicted in gray) were not included in the data analysis due to the attenuation of fluorescent light and mass transfer limitations. b SOFI images of zeolite ZSM-5 domains size within a single FCC catalyst particle at focal depths Z = 0 (left), Z = 1 (middle) and Z = 2 μm (right). c The corresponding histograms of size distributions of the zeolite domains; the insets denote single clusters larger than 0.4 μm2. d Catalytic turnover rate from NASCA microscopy as a function of average brightness in the SOFI image, calculated for 65 individual zeolite domains.
MOFs are another class of nanoporous materials that have attracted great attention in the catalysis field [109,110,111]. Their chemical properties are highly tunable as the metal cation and the organic linkers can be varied independently, which offers unique opportunities for the development of e.g. bi-functional acid–base catalysts. Therefore, it is of great interest to identify the location and properties of catalytically active sites in MOF crystals. Valvekens et al. mapped fluorescein diacetate (FDA) hydrolysis (Fig. 2g) over Ba2(BTC)(NO3) MOFs using the NASCA approach [112]. It was shown that the catalytic activity of the MOFs associated with defect sites generated via an activation process, which exclusively occurred in the hexagonal prism-shaped crystals. Even though the pore size was only slightly larger than FDA, the fluorogenic molecules could diffuse through the 1D pores of the MOF. A homogeneous reactivity throughout the hexagonal prism-shaped crystals was observed, indicating that the active sites were well dispersed (Fig. 11a). Kubarev et al. investigated the catalytic activity distribution of single ZIF-8 crystals (Fig. 11b) using FDA [113]. The fluorogenic FDA molecule is ∼4 times larger than the pore aperture size of 3.4 Å and cannot diffuse freely through the crystalline domains. The NASCA results showed a homogeneous activity at the outer surface of ZIF-8 (Fig. 11c), while the inside of the crystals was free of catalytic events (Fig. 11d). However, some crystals showed catalytic activity in well-defined domains inside the particle indicating a relation between catalytic activity, accessibility, and defects in these materials (Fig. 11e). This was further corroborated by mapping the catalytic events over mesoporous ZIF-8 crystals generated by oleic acid etching. As shown in Fig. 11f, the introduction of additional extra framework porosity allowed catalysis to occur deeper inside ZIF-8 crystals. However, an overall loss of catalytic activity was observed, which was ascribed to poisoning of the acid sites by the oleic acid [113].

FDA hydrolysis catalyzed by MOFs as studied with NASCA microscopy. a Reactivity map of a Ba2(BTC)(NO3) hexagonal prism-shaped MOF crystal. Adapted from Valvekens et al. [112] with permission from The Royal Society of Chemistry. b A scanning electron microscopy image of a representative ZIF-8 MOF crystal and c–f NASCA reactivity maps of a parent (c–e) and oleic acid etched ZIF-8 crystal (f). Note that the reactivity maps were recorded at the bottom surface of a crystal (c) and inside of several different crystals (d–f). Maps were reconstructed from events during 150 s (a), 150 s with a 50 × 50 nm2 bin size (c–e), and 500 s with a 100 × 100 nm2 bin size (f). False color shows the observed reaction rate on the logarithmic scale from 2.2 × 10−9 to 6 × 10−7 M s−1 (c), 2.2 × 10−9 to 7.4 × 10−7 M s−1(d, e), 2.9 × 10−10 to 3.0 × 10−8 M s−1 (f). Adapted from Kubarev and Roeffaers [113] with permission from The Royal Society of Chemistry
It is known that heterogeneous catalytic performance in porous materials is determined by a synergistic interplay between molecular transport and reaction kinetics [114]. In many zeolite catalysts, the pore diameters are only slightly larger than the molecular diameter of the reactants, leading to slow diffusion and reduced performance. Among various strategies to overcome diffusion limitations, the introduction of mesopores in the microporous crystal bulk phase via dealumination has attracted special attention [4, 114]. However, the post-synthesis introduced mesopores can create new geometrical constraints that will ultimately alter the catalytic performance of zeolites due to a different shape-selectivity [114,115,116,117]. Therefore, it is critical to study the effect of dealumination on the diffusion–reactivity interplay at the nanoscale. This was investigated by Ristanović et al. by following real-time changes in the catalytic turnovers of the furfuryl alcohol oligomerization in parent, mildly, and severely steamed large zeolite H-ZSM-5 crystals (Fig. 12a) [72]. The 3D reactivity of both the parent zeolite H-ZSM-5-P crystals, mildly steamed H-ZSM-5-MT, and severely steamed H-ZSM-5-ST zeolite crystals were obtained (Fig. 12b, c). Mass transfer limitations were observed in both parent and mildly steamed crystals with most of the fluorescent events detected within the 500 nm near-surface layers of the single crystals. Mildly steamed zeolites showed a four times higher reactivity in similar regions than those of the parent crystals. Moreover, the surface reactivity had become very inhomogeneous on the micrometer length scale. Therefore, mild steaming could notably enhance both accessibility and reactivity, while simultaneously causing a highly heterogeneous distribution of accessible acid sites at the macroscopic level due to migration of extra-framework aluminum species. Severe steaming at 700 °C led to a significant loss of Brønsted acidity and a two orders of magnitude lower average turnover frequency. Thus, dealumination has a large effect on both mass transport and reactivity in zeolite ZSM-5 catalysts.

Adapted from Ristanović et al. [72] with permission from American Chemical Society
a Schematic of the single-molecule fluorescence approach applied for 3D reactivity mapping of a single H-ZSM-5 crystal with intergrowth structures. The Brønsted acid site catalyzed furfuryl alcohol oligomerization was used as the probe reaction. Channel orientations in different subunits are indicated (top), and the estimated analyzed crystalline volume is shown below (bottom). The localization precision in the Z-direction was estimated to be ~500 nm. b Single-molecule reactivity maps for H-ZSM-5-P (parent), H-ZSM-5-MT (mildly steamed), and H-ZSM-5-ST (severely steamed) crystals recorded at three different focal depths [Z = 0 (surface), 2, and 4 μm]. Color bar: turnovers per 200 × 200 nm2. c Normalized turnover rates of zeolites H-ZSM-5-P, H-ZSM-5-MT, and H-ZSM-5-ST in a 5.75 mM solution of furfuryl alcohol plotted as a function of time and focal depth Z.
The authors further demonstrated that not only the reactivity and diffusion properties were changed as a result of the introduction of a secondary pore structure, but also the reaction intermediates could be altered by the defect-chemistry [67]. From single-particle ensemble measurements in ZSM-5 zeolites, it was known that the formed styrene oligomerization reaction products are dependent on the local pore geometry. Linear products were primarily formed in the straight channels, while cyclic products were found at intersections between the straight and sinusoidal channels and close to the catalyst’s surface. The authors could not assign the observed absorption bands to trimeric products with certainty and therefore did not include this reaction product in the discussion [118]. By using the NASCA approach, dimeric and trimeric oligomers of 4-methoxystyrene (Fig. 13a) could be resolved based on their different photostability [67]. These fluorescent probes were used to map the accessibility and reaction intermediates in parent and mildly steamed large zeolite ZSM-5 crystals. The highly crystalline parent zeolite H-ZSM-5 crystals typically have a silicon-rich layer (Si/Al ratio of ∼160) of up to 100 nm in thickness. This layer also seemed to act as a shape-selective reaction barrier hindering the formation of trimeric styrene, the bulkier fluorescent product, as shown in Fig. 13b, c. In the mildly steamed crystals, the diffusion properties were drastically changed with the formation of larger mesoporous defects, which can facilitate the formation of the bulkier oligomers with high photostability (Fig. 13d, e). Furthermore, the developed approach was also demonstrated to be efficient for the detection of mesoporosity. The high-resolution maps of a steamed H-ZSM-5 crystal measured at four different focal depths (Fig. 13f) showed that the photostable reaction products were formed in a 2–3 μm thick surface layer of the zeolite crystal, suggesting that the mesopores were primarily formed on the outer surface. This is consistent with the reported mesopore distribution as recorded with high resolution SEM [119].

Adapted from Ristanović et al. [67] with permission from American Chemical Society (Color figure online)
a Simplified reaction pathway of Brønsted acid catalyzed oligomerization of 4-methoxystyrene and 4-fluorostyrene as well as their corresponding absorbance wavelength. b–e Probing the accessibility of b, c parent and d, e steamed zeolite H-ZSM-5 crystals. The red circles in b, d indicate the efficiency of the localization algorithm to detect individual fluorescent events. c, e Cartoons illustrate the formation of dimeric (blue dots) and trimeric (red dots) fluorescent species in parent (c) and steamed (e) zeolite H-ZSM-5 crystals. f Accumulated images of photoactivity for different focal depths denoted in the top left corner, recorded after ∼1 h from the start of the reaction.
NASCA microscopy was also used to provide direct experimental evidence and achieve quantitative understanding of the synergistic effect of molecular transport and reaction kinetics at the single-molecule and single-particle level. De Cremer et al. demonstrated that intraparticle diffusion during the epoxidation of phenylbutadienyl (PBD)-BODIPY (1.8 × 0.5 nm) with tBuOOH over Ti-MCM-41 (2.7 nm pore diameter, Fig. 2f) could be directly measured (Fig. 14). This allowed not only a microscopic quantification of diffusion, but also macroscopic parameters that identify mass transfer limitations to be calculated at the single-particle level [33, 73]. In the epoxidation of cholesterol and cyclohexene, catalytic testing showed an increase in selectivity and reaction rate for catalyst particles with a size down to 100 nm, which indicates the presence of diffusion limitations [120, 121]. Interestingly, the distribution of reaction events in the epoxidation of PBD-BODIPY in a micrometer-sized particle revealed that product formation only occurred in the outer 300 nm shell (Fig. 14a, b). A comparable Si/Ti ratio was observed from X-ray photoelectron spectroscopy (XPS) measurements and bulk chemical analysis, demonstrating homogenous distribution of titanium species. Thus, the enrichment of active sites at the surface was excluded as the cause of the peripheral active zone. Moreover, excess reactant in absence of the oxidant showed that it was able to access the full pore volume and that no physical barriers were present (Fig. 14c), excluding the hypothesis that pore discontinuities are the origin of the observed catalytic zoning effects. Taken this together, intraparticle diffusion must be the limiting factor in case of Ti-MCM-41 catalyzed epoxidation of diluted reactants [73, 74]. This was corroborated by the calculation of Thiele modulus and effectiveness factor, two dimensionless parameters commonly used in heterogeneous catalysis to identify diffusion limitations at the bulk scale. The value of the Thiele modulus was estimated to be about 3, corresponding to an effectiveness factor of only 30% [73].

Monitoring individual epoxidation events in the individual Ti-MCM-41 catalysts. a The transmission optical images (grayscale) are overlaid with a scatter plot (red dots) of the fitted positions of fluorescent spots originating from individual product molecules, which were formed by epoxidation of PBD-BODIPY with tBuOOH. Two catalysts particles can be distinguished. b Zoom-in image of the rectangle region highlighted in panel a. The positions of product formation are distributed over the outer 200 to 300 nm of the particle. Adapted from De Cremer et al. [74] with permission from Wiley. c Fluorescence image of the middle of Ti-MCM-41 particles after adsorption of excess PBD-BODIPY in the absence of the oxidant t-BuOOH. Confocal microscopy was used to record a thin optical slice through the sample without background signal from the out-of-focus regions. Adapted from De Cremer et al. [73], Copyright 2010, with permission from Elsevier (Color figure online)
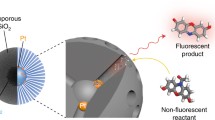
Dong et al. designed a well-defined model system (Fig. 15a) with platinum nanoparticles sandwiched between an optically transparent solid silica core and a mesoporous silica shell with aligned nanopores (∼2 nm pore size) [76]. This model system allowed them to decouple mass transport factors from reaction kinetics, which enabled the quantitative analysis of their interplay at the single-molecule level. From the single-molecule trajectories and NASCA analysis of the Amplex Red oxidation reaction (Fig. 2e), the concentration dependent diffusion rate and reaction rate were calculated (Fig. 15b, c). A comparable diffusion rate (0.03 s−1) and reaction rate (0.02 s−1) was observed at 0.02 µM Amplex Red concentration (Fig. 15d). Therefore, it was concluded that the catalytic conversion was diffusion limited at the low concentration range. Conversely, saturation was achieved when the concentration of Amplex Red was much higher, and the reaction rate was limited solely by the chemical conversion rate. Mass transport of Amplex Red played a negligible role in the reaction rate when nanoconfinement was not present, i.e. without a mesoporous silica shell (Fig. 15e). Further analysis revealed a promotion of the reaction rate constant (keff) by six times in the nanoconfined material, compared to that of the bare platinum nanoparticles. The enhanced activity could be a result of a different adsorption strength or higher effective concentration of the reactant when the platinum nanoparticles are encapsulated by mesoporous silica.

Adapted from Dong et al. [76] with permission from Springer Nature
a Schematic representation of the nanocatalyst; it consists of 5 nm platinum nanoparticles (NPs) sandwiched between a solid 100 nm silica core and a mesoporous 120 nm mesoporous silica shell. b A typical fluorescence intensity trajectory from a single NP at a temporal resolution of 50 ms. τon and τoff correspond to the resident time of resorufin in nanopores and the interval time between two consecutive catalytic events, respectively. c Single-molecule trajectories over many particles during a short imaging period. d and e Ratio of the measured reaction rate over the diffusion rate over d NPs encapsulated by mesoporous silica shell and e bare NPs, respectively.
4.2 Correlative approaches
Although SMLM has made it possible to study single catalytic turnovers with unprecedent spatial resolution, exact site–reactivity relationships are still missing. Therefore, different correlation approaches were developed to integrate or combine complementary techniques with SMLM.
Liu et al. has combined SMLM with stimulated Raman scattering (SRS) microscopy (Fig. 16a) to probe reactivity, acid site distribution, and site accessibility in individual mordenite (H-MOR) zeolites [122]. NASCA analysis revealed that the catalytic performance of dealuminated acid mordenites shows a remarkable heterogeneity at the intercrystal and subcrystal level (Fig. 16b). Using benzonitrile (PhCN) as the probe molecule, the acid site distribution and accessibility of H-MOR crystals at different stages of the dealumination process were imaged with SRS microscopy (Fig. 16c). Initially, the acid site density was homogeneous throughout the crystal and the sites inside the crystal were inaccessible to the aromatic PhCN probe, limiting the oligomerization of furfuryl alcohol to the surface of the fresh catalysts. Slight dealumination allowed the regions near the outer surface to become partially accessible to aromatics while the core remained inaccessible to PhCN, which correlated to the catalytic results. Further dealumination resulted in an overall increase in accessibility accompanied by a decrease in active site density in the crystallite outer shell. Due to the good overall accessibility, the local catalytic performance became largely determined by the local acid site density, which was higher in the center of the crystal, resulting in a core–shell reactivity distribution. When the whole crystal was fully accessible to the reactants after extensive dealumination (i.e., later stage dealuminated MD-MOR and SD-MOR), a strongly reduced acid site density was also observed in the center, leading to a homogeneous reactivity map with a relatively low turnover rate. A similar volcano-like trend in reactivity as function of dealumination has also been observed for ZSM-5 on the single-particle and single-molecule level [72, 118].

Adapted from Liu et al. [122] with permission from American Chemical Society
a Schematic representation of the used optical micro-(spectro)scopic assay of NASCA (top) and stimulated Raman scattering (SRS) microscopy. b Quantitative analysis of NASCA reactivity maps obtained for 500 × 500 × 800 nm3. (A) Parent H-MOR crystal; (B) severely dealuminated H-MOR crystal; (C–E) mildly dealuminated MOR crystals at different stages of dealumination. c SRS micro-(spectro)scopic mapping (2249 cm−1) of PhCN adsorbed in H-MORs from (A) early, (B) intermediate and (C) late stages of dealumination as well as (D) severely dealuminated H-MOR. Scale bar 3 μm. Note that the color bars are enlarged from original figures for clarification.
Zeolite activity and selectivity is determined by the host–guest chemistry between inorganic zeolite and organic guest molecules, which often involves proton and hydrogen-transfer reactions [67]. It has been shown at both the ensemble and single-molecule level that a change of substituent on the styrene moiety as well as the solvent medium alters the turnover rate of proton-transfer processes and reactivities of reactants [67, 69, 123]. However, an accurate structural elucidation of the guest molecule was missing, which is essential to understand the guest–host chemistry. Therefore, we have further combined SMLM, solid-state NMR spectroscopy, and density functional theory (DFT) calculations [70] to establish molecular understanding of host–guest chemistry in detail. A large variation in blinking dynamics was observed in the SMLM measurements for styrene-derived emitters, suggesting a heterogeneous microenvironment of the fluorescent molecules. This was corroborated by NMR spectroscopy, as both line broadening and multiple resonances were observed for the reactant. Based on the observed reaction products with NMR spectroscopy, a reaction pathway was proposed. When a styrene molecule is chemisorbed, it is prone to an attack from a second styrene molecule resulting in a fluorescent carbocationic product. Its fluorescence is quenched after proton transfer to the zeolite Brønsted acid site, and a neutral non-fluorescent dimer is formed. DFT calculations showed that this proton transfer is reversible, and the switching between these two states is largely depended on the relative stability of the products and the local position of the Brønsted acid site within the zeolite micropore. The equilibrium between carbocation and neutral states was significantly shifted by changing solvents and substituents on the styrene. This combined approach showed that the blinking dynamics as observed with SMLM strongly depended on the local chemical environment imposed by zeolite framework and guest solvent molecules.
Super-resolution fluorescence microscopy has also been integrated with electron microscopies, i.e. SEM and TEM, which enables the direct correlation of catalyst structure with its performance at the nanoscale [66, 105, 124]. Van Loon et al. used an integrated fluorescence and electron microscope (iFlEM, Fig. 17a) to investigate the effect of nanocrystallite structure on the catalytic performance of industrial MOR zeolites [124]. Figure 17b displays the SEM micrograph and optical transmission image of a H-MOR aggregate from an industrial sample. A direct comparison clearly demonstrates that the structural complexity of the small particles in typical zeolite samples cannot be captured by diffraction-limited optical imaging. The crystal aggregates consisted of intact crystals together with other protruding fragments. Morphology analysis showed that the two main crystals are crystallographically perpendicular to each other. Overlay of the NASCA and SEM images revealed significant activity at all the interfaces between crystals, suggesting that intercrystalline intergrowth structures in H-MOR aggregates have a major contribution to the overall activity. The developed iFlEM approach was also applied to investigate the effects of acid leaching (Fig. 17c). The correlated SEM micrograph and quantitative NASCA activity map showed that the catalytic activity in the acid leached particle still predominantly resided at the edges of the orange crystal and the intergrowth (Fig. 17c). Recently, the catalytic hotspots in H-ZSM-22 as discussed above were verified by the iFlEM approach [105]. Another application of the integrated light electron microscopy (iLEM) was reported by Hendriks et al. for the study of structure–activity relationships in complex, hierarchically structured FCC catalysts with the combination of NASCA and TEM [66]. The thiophene oligomerization reaction product was used as fluorescent reporter (Fig. 2d) and revealed regions with high catalytic activity (Fig. 17d). The correlation between reactivity and structural features was explored by quantitative analysis of a region of interest (ROI) within a catalyst thin section of 20 × 20 μm2. Based on the structural features from the TEM image, the region of interest (ROI) was then manually classified into zeolite material and non-zeolitic matrix material (Fig. 17e). The zeolite material was found to be homogeneously distributed in this ROI, covering 38% of the surface of the thin section. A further 46% was covered by matrix material, while 16% contained no material. As discussed before, Ristanović et al. found that the reactivity found with NASCA microscopy correlated directly with the SOFI intensity [99]. Because the number of detected single turnovers was low, the SOFI intensity of each of these categories was evaluated as a measure of reactivity, as shown in Fig. 17f. A significantly higher reactivity was found for the zeolite phase compared to the matrix material, showing that the zeolite material has most catalytic activity within the catalyst for the thiophene oligomerization probe reaction.

a Schematic representation of the iFlEM. The insets show an inside view of the electron beam–sample–objective lens configuration for taking correlated SEM (top) and SMF images (bottom). b and c Overlay images of the detected individual fluorescent products (cyan dots) on the SEM micrographs (left) and the corresponding schematic representations indicating the different crystallographic axes (right) for b parent and c acid leached MOR zeolites, respectively. Adapted from Van Loon et al. [124] with permission from American Chemical Society. d Schematic of sample preparation and the analysis of an FCC catalyst particle thin section using the iLEM. FCC particles embedded in epoxy resin (yellow) are microtomed into thin sections and deposited onto a SiN membrane (left). The thin section was then analyzed by TEM (middle), NASCA microscopy, and SOFI analysis of thiophene-oligomerization reaction products (right). The red dots indicate single turnovers and are manually overlaid on the SOFI reactivity map. Because the sample was slightly out of focus in the top right corner, fewer turnovers are observed in this region. e TEM images show different types of ultrastructure including zeolite crystals (red), non-zeolitic matrix material (blue) and no material (gray). f Box plots of the SOFI intensity in the areas of each type of material classified. Adapted from Hendriks et al. [66] with permission from Wiley (Color figure online)
5 Conclusions and outlook
Single-molecule measurements have uncovered fascinating heterogeneous motion behavior and reactivity in nanoporous solids. To date, most studied materials have been well-defined or ordered materials, such as mesoporous silica and zeolites, and single-molecule microscopy has proven to be very sensitive towards defects in these materials. Therefore, it would be particularly interesting to study how mass transport and reactivity are affected by defects in materials such as MOFs via correlation with material properties and surface topology obtained with atomic force microscopy–infrared microscopy (AFM–IR) and scanning tunneling microscopy (STM). High resolution spectral mapping of the fluorescent reaction products using tip-enhanced fluorescence (TEFL) microscopy could provide additional information about the reaction products formed at high concentration [125]. Such correlative approaches will also be highly beneficial for the study of structure–performance relationships in less-ordered materials such as complex heterogeneous catalysts, for example FCC particles and extrudates.
Correlation of SMLM with other microscopy techniques that are capable of capturing the pore space has great value [101] with high potential for the future. By overlay SMLM images with TEM micrographs, Zürner et al. have provided the first direct evidence that single-molecule trajectories follow the pore-connectivity and accessibility in mesoporous silica [60]. In more complex, heterogeneous pore spaces such as FCC particles, mass transport was dominated by transient trapping of the molecules [56]. The origin of these traps remains an open question. Correlation of the single-molecule trajectories with the pore space and composition imaged with e.g. X-ray tomography techniques or focused ion beam–SEM could answer this question [126, 127]. Recently, sequential two-dimensional correlative imaging of a microtomed FCC particle was demonstrated by Hendriks et al., which allowed correlation of reactivity with the material morphology and elemental composition as shown in Fig. 18 [128]. However, alignment of the micrographs obtained by different microscopy techniques is challenging, especially when they are recorded consecutively and in three dimensions [101, 129]. The combination of super-resolution imaging correlated with SMLM appears to be a promising new route. A recently developed super-resolution technique that specifically targets interfaces is a promising approach to obtain fast and reliable information on the accessible pore space and would allow correlation in a single experiment [130].

Adapted from Hendriks [128] (Color figure online)
a Schematic representation of a correlative microscopy approach showcased on a sliced FCC particle (yellow circles). Three steps were performed sequentially: (1) fluorescence imaging of the thiophene-stained sample to quantify the reactivity using SOFI analysis, (2) X-ray fluorescence micro-spectroscopy to collect the elemental distribution, and (3) SEM imaging after platinum coating to capture the material morphology. b Experimental results following the experiment described in a of an FCC particle microtomed into 100 nm thin slices. Three different regions of interest are shown, and the SEM, SOFI, and elemental distribution maps are given for each of these regions. The yellow region has a different morphology than the other regions, which is reflected in the different SOFI reactivity and elemental distribution.
Ultimately, one would like to be able to predict mass transport properties based on a material’s pore space and vice versa. This would be a big step towards a rational design of functional nanoporous materials, such as catalysts and membranes. A promising way to do this is using machine learning (ML), that is, by training a computer how a single-molecule trajectory ‘looks’ when a probe moves in a specific pore environment [94] complemented with the known fundamentals of diffusion [4, 11]. For ML, a large and diverse (i.e. unbiased) training set is required, which ‘knows’ both the trajectories and the pore space. Here, high-throughput correlative microscopy is crucial to record these data sets, and poses an interesting experimental challenge. Moreover, ML could also significantly speed up localization and tracking analysis in such large data sets, as deep learning methods have already been demonstrated to speed up the image-reconstruction in SMLM [131].
In less complex systems, that is, well-defined pore systems, imaging of the pore space is not always necessary. Such systems allow the measurement of diffusion along specific axes of the material, which allows direct correlation of mass transport with the pore structure. As discussed, materials with large ordered porous domains have made such correlation possible in mesoporous silica [57, 58, 60] and MOFs [92]. Model systems with uniform pore alignments are particularly interesting for materials with an anisotropic pore structure. Fu et al. demonstrated that this allowed differentiation between diffusion in straight and sinusoidal pores in ZSM-5 zeolites, which governs the differences in reaction performance [93, 132]. Altogether, this is a powerful approach and holds great potential to be applied with other ordered microporous materials as well [133].
Reactive fluorophores have been shown to provide a detailed insight into the spatial and temporal conversion behavior of nanoporous catalysts; however, mass transport is always probed indirectly. An experiment in which both diffusion of the reactant and product as well as the reaction event are recorded would allow direct observation of the relation between mass transport and reactivity. Up to date, such experiment has not been reported in nanoporous catalysts, probably due to challenging fluorophore design. The emission of the BODIPY-dye developed by de Cremer et al. undergoes a spectral shift upon epoxidation (Fig. 2e), which would allow such measurements, however only reaction events were reported in their work [73, 74]. An alternative approach is to use energy transfer from an acceptor fluorophore to a donor fluorophore, which are connected by a cleavable linker, to identify and track the individual components. Such a reactive fluorescent probe represents cleaving processes of large organic species found in e.g. biomass and vacuum gas oil. Energy transfer in dyads and dendrimeric structures of rylene dyes have a high efficiency (> 90%), but upon cleaving at an active site of the catalyst, energy transfer becomes readily inefficient due to an increased distance between the fluorophores [134, 135]. By alternating excitation of the fluorophore moieties, the uncleaved dye can be differentiated from its cleaved subunits and tracked individually. It was demonstrated that the trajectories of a photocleavable PDI–TDI-based dyad allowed tracking of the ‘reaction’ event as well as diffusion of the dyad and subunits [135]. Moreover, the strong distance dependence of the energy-transfer efficiency allowed the authors to measure low diffusion constants in the order of D < 10−19 m2/s.
Single-molecule trajectories contain a wealth of information about the interplay between mass transport properties and pore space in nanoporous solids. While mapping those trajectories is a common tool to study transport in biological systems, it is still a niche subject in materials science [21]. Nevertheless, the potential is enormous despite the many more experimental challenges that need to be overcome when studying solid materials in comparison to the systems studied in biology. Many nanoporous solids have an inhomogeneous refractive index and strong absorption of visible light, however, both advanced model system and experiment design can tackle these challenges: (1) development and application of fluorophores operating in the infrared allows deeper imaging into the sample due to reduced light scattering. This comes at the expense of a lower resolution, because of the longer wavelength of infrared than visible light. (2) One could for example design optically transparent model systems with pore spaces representative for real pore systems, which increases the signal-to-noise and allows imaging deep into the sample. The mass transport behaviors could then be directly translated to the nanoporous solid of interest. (3) For materials with a heterogeneous matrix, the signal-to-noise ratio can be drastically improved by slicing the nanoporous solid into slabs [66]. Reconstruction of mass transport through the pore space will, however, still be a highly complex task if the accessibility of the full pore space has to be considered. Here, (correlative) microscopy approaches using 3D imaging methods that are able to capture the whole pore space will be needed.
In conclusion, SMLM has established itself as a powerful method to reveal heterogeneities in mass transport and reactivity in nanoporous solids at the single-molecule level. The origin of these heterogeneities has been partially unraveled, but many open questions remain—particularly in complex, hierarchical pore systems. We see a bright future for SMLM in materials science and believe that many of the open questions can be addressed with the methods outlined in this work.
Abbreviations
- MSD (m2):
-
Mean-squared displacement
- \({\varvec{x}}\) (m):
-
Particle position
- \({t}_{n}\) (s):
-
Lag time
- \(\Delta t\) (s):
-
Time between frames
- \(n\) (–):
-
Dimensionality
- \(K\) (m2/s−α):
-
Generalized diffusion constant
- \(\alpha \) (–):
-
Anomalous exponent
- AFM–IR:
-
Atomic force microscopy–infrared microscopy
- CM:
-
Clausius–Mossotti
- CTAB:
-
Cetyl trimethyl ammonium bromide
- CVE:
-
Covariance-based estimator
- DFT:
-
Density functional theory
- FCC:
-
Fluid catalytic cracking
- FCS:
-
Fluorescence correlation spectroscopy
- FDA:
-
Fluorescein diacetate
- FRAP:
-
Fluorescence recovery after photobleaching
- FWHM:
-
Full width at half maximum
- GFP:
-
Green fluorescent protein
- iFlEM:
-
Integrated fluorescence and electron microscope
- iLEM:
-
Integrated light electron microscopy
- ML:
-
Machine learning
- MLE:
-
Maximum likelihood estimator
- MOF:
-
Metal–organic framework
- MSD:
-
Mean-squared displacement
- NASCA:
-
Nanometer accuracy by stochastic chemical reactions
- NMR:
-
Nuclear magnetic resonance
- NP:
-
Nanoparticle
- PDI:
-
Perylene diimide
- PFG NMR:
-
Pulsed-field gradient nuclear magnetic resonance
- PBD:
-
Phenylbutadienyl
- PhCN:
-
Benzonitrile
- PSF:
-
Point spread function
- QENS:
-
Quasi-elastic neutron scattering
- ROI:
-
Region of interest
- SEM:
-
Scanning electron microscopy
- SMED:
-
Single-molecule emission dichroism
- SMLM:
-
Single-molecule localization microscopy
- SOFI:
-
Super-resolution optical fluctuation imaging
- SRS:
-
Stimulated Raman scattering
- STM:
-
Scanning tunneling microscopy
- TDI:
-
Terylene diimide
- TEFL:
-
Tip-enhanced fluorescence
- TEM:
-
Transmission electron microscopy
- XPS:
-
X-ray photoelectron spectroscopy
- XRD:
-
X-ray diffraction
References
Čejka, J., Corma, A., Zones, S. (eds.): Zeolites and Catalysis: Synthesis, Reactions and Applications. Wiley, Weinheim (2010)
Davis, M.E.: Ordered porous materials for emerging applications. Nature 417, 813–821 (2002). https://doi.org/10.1038/nature00785
Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J. (eds.): Handbook of Heterogeneous Catalysis. Wiley, Weinheim (2008)
Kärger, J., Ruthven, D.M., Theodorou, D.N.: Diffusion in Nanoporous Materials. Wiley, Weinheim (2012)
Kulprathipanja, S. (ed.): Zeolites in Industrial Separation and Catalysis. Wiley, Weinheim (2010)
Morris, R.E., Wheatley, P.S.: Gas storage in nanoporous materials. Angew. Chem. Int. Ed. 47, 4966–4981 (2008). https://doi.org/10.1002/anie.200703934
Vogt, E.T.C., Whiting, G.T., Dutta Chowdhury, A., Weckhuysen, B.M.: Zeolites and zeotypes for oil and gas conversion. In: Advances in Catalysis, pp. 143–314. Elsevier, Amsterdam (2015)
Chmelik, C., Heinke, L., Valiullin, R., Kärger, J.: A new view of diffusion in nanoporous materials. Chem. Ing. Tech. 82, 779–804 (2010). https://doi.org/10.1002/cite.201000038
Chmelik, C., Kärger, J.: In situ study on molecular diffusion phenomena in nanoporous catalytic solids. Chem. Soc. Rev. 39, 4864–4884 (2010). https://doi.org/10.1039/C0CS00100G
Kärger, J., Avramovska, M., Freude, D., et al.: Pulsed field gradient NMR diffusion measurement in nanoporous materials. Adsorption (2021). https://doi.org/10.1007/s10450-020-00290-9
Kärger, J., Ruthven, D.M.: Diffusion in nanoporous materials: fundamental principles, insights and challenges. N. J. Chem. 40, 4027–4048 (2016). https://doi.org/10.1039/C5NJ02836A
Buurmans, I.L.C., Weckhuysen, B.M.: Heterogeneities of individual catalyst particles in space and time as monitored by spectroscopy. Nat. Chem. 4, 873–886 (2012). https://doi.org/10.1038/nchem.1478
De Cremer, G., Sels, B.F., Vos, D.E.D., Hofkens, J., Roeffaers, M.B.J.: Fluorescence micro(spectro)scopy as a tool to study catalytic materials in action. Chem. Soc. Rev. 39, 4703–4717 (2010). https://doi.org/10.1039/C0CS00047G
Janssen, K.P.F., Cremer, G.D., Neely, R.K., Kubarev, A.V., Loon, J.V., Martens, J.A., Vos, D.E.D., Roeffaers, M.B.J., Hofkens, J.: Single molecule methods for the study of catalysis: from enzymes to heterogeneous catalysts. Chem. Soc. Rev. 43, 990–1006 (2014). https://doi.org/10.1039/C3CS60245A
Michaelis, J., Bräuchle, C.: Reporters in the nanoworld: diffusion of single molecules in mesoporous materials. Chem. Soc. Rev. 39, 4731–4740 (2010). https://doi.org/10.1039/C0CS00107D
Tachikawa, T., Majima, T.: Single-molecule, single-particle fluorescence imaging of TiO2-based photocatalytic reactions. Chem. Soc. Rev. 39, 4802–4819 (2010). https://doi.org/10.1039/b919698f
Weckhuysen, B.M.: Chemical imaging of spatial heterogeneities in catalytic solids at different length and time scales. Angew. Chem. Int. Ed. 48, 4910–4943 (2009). https://doi.org/10.1002/anie.200900339
Hellriegel, C., Kirstein, J., Bräuchle, C., Latour, V., Pigot, T., Olivier, R., Lacombe, S., Brown, R., Guieu, V., Payrastre, C., Izquierdo, A., Mocho, P.: Diffusion of single streptocyanine molecules in the nanoporous network of sol–gel glasses. J. Phys. Chem. B 108, 14699–14709 (2004). https://doi.org/10.1021/jp049412a
Brinker, C.J., Scherer, G.W.: Sol–Gel Science: The Physics and Chemistry of Sol–Gel Processing. Academic, Boston (2013)
Naredi‐Rainer, N., Prescher, J., Hartschuh, A., Lamb, D.C.: Confocal microscopy. In: Fluorescence Microscopy, pp. 165–202. Wiley, Weinheim (2017)
Manzo, C., Garcia-Parajo, M.F.: A review of progress in single particle tracking: from methods to biophysical insights. Rep. Prog. Phys. 78, 124601 (2015). https://doi.org/10.1088/0034-4885/78/12/124601
Vangindertael, J., Camacho, R., Sempels, W., Mizuno, H., Dedecker, P., Janssen, K.P.F.: An introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl. Fluoresc. 6, 022003 (2018). https://doi.org/10.1088/2050-6120/aaae0c
Birkhoff, G.D.: Proof of a recurrence theorem for strongly transitive systems. Proc. Natl Acad. Sci. U S A 17, 650–655 (1931)
Feil, F., Naumov, S., Michaelis, J., Valiullin, R., Enke, D., Kärger, J., Bräuchle, C.: Single-particle and ensemble diffusivities—test of ergodicity. Angew. Chem. Int. Ed. 51, 1152–1155 (2012). https://doi.org/10.1002/anie.201105388
Kubitscheck, U.: Principles of light microscopy. In: Fluorescence Microscopy, pp. 23–83. Wiley, Chichester (2017)
Betzig, E.: Proposed method for molecular optical imaging. Opt. Lett. 20, 237–239 (1995). https://doi.org/10.1364/ol.20.000237
Dickson, R.M., Cubitt, A.B., Tsien, R.Y., Moerner, W.E.: On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 388, 355–358 (1997). https://doi.org/10.1038/41048
Betzig, E., Patterson, G.H., Sougrat, R., Lindwasser, O.W., Olenych, S., Bonifacino, J.S., Davidson, M.W., Lippincott-Schwartz, J., Hess, H.F.: Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006)
Deschout, H., Zanacchi, F.C., Mlodzianoski, M., Diaspro, A., Bewersdorf, J., Hess, S.T., Braeckmans, K.: Precisely and accurately localizing single emitters in fluorescence microscopy. Nat. Methods 11, 253–266 (2014). https://doi.org/10.1038/nmeth.2843
Small, A., Stahlheber, S.: Fluorophore localization algorithms for super-resolution microscopy. Nat. Methods 11, 267–279 (2014). https://doi.org/10.1038/nmeth.2844
Crocker, J.C., Grier, D.G.: Methods of digital video microscopy for colloidal studies. J. Colloid Interface Sci. 179, 298–310 (1996). https://doi.org/10.1006/jcis.1996.0217
Chen, P., Zhou, X., Shen, H., Andoy, N.M., Choudhary, E., Han, K.-S., Liu, G., Meng, W.: Single-molecule fluorescence imaging of nanocatalytic processes. Chem. Soc. Rev. 39, 4560–4570 (2010). https://doi.org/10.1039/B909052P
Roeffaers, M.B.J., De Cremer, G., Libeert, J., Ameloot, R., Dedecker, P., Bons, A.-J., Bückins, M., Martens, J.A., Sels, B.F., De Vos, D.E., Hofkens, J.: Super-resolution reactivity mapping of nanostructured catalyst particles. Angew. Chem. Int. Ed. 48, 9285–9289 (2009). https://doi.org/10.1002/anie.200904944
Berglund, A.J.: Statistics of camera-based single-particle tracking. Phys. Rev. E 82, 011917 (2010). https://doi.org/10.1103/PhysRevE.82.011917
Michalet, X., Berglund, A.J.: Optimal diffusion coefficient estimation in single-particle tracking. Phys. Rev. E 85, 061916 (2012). https://doi.org/10.1103/PhysRevE.85.061916
Qian, H., Sheetz, M.P., Elson, E.L.: Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 60, 910–921 (1991). https://doi.org/10.1016/S0006-3495(91)82125-7
Saxton, M.J.: Modeling 2D and 3D diffusion. In: Dopico, A.M. (ed.) Methods in Membrane Lipids, pp. 295–321. Humana Press, Totowa (2007)
Vestergaard, C.L., Blainey, P.C., Flyvbjerg, H.: Optimal estimation of diffusion coefficients from single-particle trajectories. Phys. Rev. E 89, 022726 (2014). https://doi.org/10.1103/PhysRevE.89.022726
Einstein, A.: Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. Ann. Phys. 322, 549–560 (1905). https://doi.org/10.1002/andp.19053220806
Shuang, B., Byers, C.P., Kisley, L., Wang, L.-Y., Zhao, J., Morimura, H., Link, S., Landes, C.F.: Improved analysis for determining diffusion coefficients from short, single-molecule trajectories with photoblinking. Langmuir 29, 228–234 (2013). https://doi.org/10.1021/la304063j
Relich, P.K., Olah, M.J., Cutler, P.J., Lidke, K.A.: Estimation of the diffusion constant from intermittent trajectories with variable position uncertainties. Phys. Rev. E 93, 042401 (2016). https://doi.org/10.1103/PhysRevE.93.042401
Vestergaard, C.L., Pedersen, J.N., Mortensen, K.I., Flyvbjerg, H.: Estimation of motility parameters from trajectory data. Eur. Phys. J. Spec. Top. 224, 1151–1168 (2015). https://doi.org/10.1140/epjst/e2015-02452-5
Lindén, M., Ćurić, V., Amselem, E., Elf, J.: Pointwise error estimates in localization microscopy. Nat. Commun. 8, 15115 (2017). https://doi.org/10.1038/ncomms15115
Karslake, J.D., Donarski, E.D., Shelby, S.A., Demey, L.M., DiRita, V.J., Veatch, S.L., Biteen, J.S.: SMAUG: analyzing single-molecule tracks with nonparametric Bayesian statistics. Methods (2020). https://doi.org/10.1016/j.ymeth.2020.03.008
Lindén, M., Elf, J.: Variational algorithms for analyzing noisy multistate diffusion trajectories. Biophys. J. 115, 276–282 (2018). https://doi.org/10.1016/j.bpj.2018.05.027
Persson, F., Lindén, M., Unoson, C., Elf, J.: Extracting intracellular diffusive states and transition rates from single-molecule tracking data. Nat. Methods 10, 265–269 (2013). https://doi.org/10.1038/nmeth.2367
Rowland, D.J., Biteen, J.S.: Measuring molecular motions inside single cells with improved analysis of single-particle trajectories. Chem. Phys. Lett. 674, 173–178 (2017). https://doi.org/10.1016/j.cplett.2017.02.052
Wöll, D., Flors, C.: Super-resolution fluorescence imaging for materials science. Small Methods 1, 1700191 (2017). https://doi.org/10.1002/smtd.201700191
Cordes, T., Blum, S.A.: Opportunities and challenges in single-molecule and single-particle fluorescence microscopy for mechanistic studies of chemical reactions. Nat. Chem. 5, 993–999 (2013). https://doi.org/10.1038/nchem.1800
Vestergaard, C.L.: Optimizing experimental parameters for tracking of diffusing particles. Phys. Rev. E 94, 022401 (2016). https://doi.org/10.1103/PhysRevE.94.022401
Aitken, C.E., Marshall, R.A., Puglisi, J.D.: An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 94, 1826–1835 (2008). https://doi.org/10.1529/biophysj.107.117689
Vogelsang, J., Kasper, R., Steinhauer, C., Person, B., Heilemann, M., Sauer, M., Tinnefeld, P.: A reducing and oxidizing system minimizes photobleaching and blinking of fluorescent dyes. Angew. Chem. Int. Ed. 47, 5465–5469 (2008). https://doi.org/10.1002/anie.200801518
Werley, C.A., Moerner, W.E.: Single-molecule nanoprobes explore defects in spin-grown crystals. J. Phys. Chem. B 110, 18939–18944 (2006). https://doi.org/10.1021/jp057570b
Feil, F., Cauda, V., Bein, T., Bräuchle, C.: Direct visualization of dye and oligonucleotide diffusion in silica filaments with collinear mesopores. Nano Lett. 12, 1354–1361 (2012). https://doi.org/10.1021/nl2039474
Hellriegel, C., Kirstein, J., Bräuchle, C.: Tracking of single molecules as a powerful method to characterize diffusivity of organic species in mesoporous materials. N. J. Phys. 7, 23 (2005). https://doi.org/10.1088/1367-2630/7/1/023
Hendriks, F.C., Meirer, F., Kubarev, A.V., Ristanović, Z., Roeffaers, M.B.J., Vogt, E.T.C., Bruijnincx, P.C.A., Weckhuysen, B.M.: Single-molecule fluorescence microscopy reveals local diffusion coefficients in the pore network of an individual catalyst particle. J. Am. Chem. Soc. 139, 13632–13635 (2017). https://doi.org/10.1021/jacs.7b07139
Jung, C., Kirstein, J., Platschek, B., Bein, T., Budde, M., Frank, I., Müllen, K., Michaelis, J., Bräuchle, C.: Diffusion of oriented single molecules with switchable mobility in networks of long unidimensional nanochannels. J. Am. Chem. Soc. 130, 1638–1648 (2008). https://doi.org/10.1021/ja075927e
Kirstein, J., Platschek, B., Jung, C., Brown, R., Bein, T., Bräuchle, C.: Exploration of nanostructured channel systems with single-molecule probes. Nat. Mater. 6, 303–310 (2007). https://doi.org/10.1038/nmat1861
Lebold, T., Mühlstein, L.A., Blechinger, J., Riederer, M., Amenitsch, H., Köhn, R., Peneva, K., Müllen, K., Michaelis, J., Bräuchle, C., Bein, T.: Tuning single-molecule dynamics in functionalized mesoporous silica. Chem. Eur. J. 15, 1661–1672 (2009). https://doi.org/10.1002/chem.200801380
Zürner, A., Kirstein, J., Döblinger, M., Bräuchle, C., Bein, T.: Visualizing single-molecule diffusion in mesoporous materials. Nature 450, 705–708 (2007). https://doi.org/10.1038/nature06398
Avlasevich, Y., Li, C., Müllen, K.: Synthesis and applications of core-enlarged perylene dyes. J. Mater. Chem. 20, 3814–3826 (2010). https://doi.org/10.1039/C000137F
Weil, T., Vosch, T., Hofkens, J., Peneva, K., Müllen, K.: The rylene colorant family-tailored nanoemitters for photonics research and applications. Angew. Chem. Int. Ed. 49, 9068–9093 (2010). https://doi.org/10.1002/anie.200902532
Hoang, H.T., Segers-Nolten, I.M., Tas, N.R., van Honschoten, J.W., Subramaniam, V., Elwenspoek, M.C.: Analysis of single quantum-dot mobility inside 1D nanochannel devices. Nanotechnology 22, 275201 (2011). https://doi.org/10.1088/0957-4484/22/27/275201
Lee, C.H., Crosby, A.J., Emrick, T., Hayward, R.C.: Characterization of heterogeneous polyacrylamide hydrogels by tracking of single quantum dots. Macromolecules 47, 741–749 (2014). https://doi.org/10.1021/ma402373s
Liu, X., Chen, S.-Y., Chen, Q., Yao, X., Gelléri, M., Ritz, S., Kumar, S., Cremer, C., Landfester, K., Müllen, K., Parekh, S.H., Narita, A., Bonn, M.: Nanographenes: ultrastable, switchable, and bright probes for super-resolution microscopy. Angew. Chem. Int. Ed. 59, 496–502 (2020). https://doi.org/10.1002/anie.201909220
Hendriks, F.C., Mohammadian, S., Ristanović, Z., Kalirai, S., Meirer, F., Vogt, E.T.C., Bruijnincx, P.C.A., Gerritsen, H.C., Weckhuysen, B.M.: Integrated transmission electron and single-molecule fluorescence microscopy correlates reactivity with ultrastructure in a single catalyst particle. Angew. Chem. Int. Ed. 57, 257–261 (2018). https://doi.org/10.1002/anie.201709723
Ristanović, Z., Kubarev, A.V., Hofkens, J., Roeffaers, M.B.J., Weckhuysen, B.M.: Single molecule nanospectroscopy visualizes proton-transfer processes within a zeolite crystal. J. Am. Chem. Soc. 138, 13586–13596 (2016). https://doi.org/10.1021/jacs.6b06083
Kox, M.H.F., Mijovilovich, A., Sättler, J.J.H.B., Stavitski, E., Weckhuysen, B.M.: The catalytic conversion of thiophenes over large H-ZSM-5 crystals: an X-ray, UV/Vis, and fluorescence microspectroscopic study. ChemCatChem 2, 564–571 (2010). https://doi.org/10.1002/cctc.200900329
Kox, M.H.F., Stavitski, E., Weckhuysen, B.M.: Nonuniform catalytic behavior of zeolite crystals as revealed by in situ optical microspectroscopy. Angew. Chem. Int. Ed. 46, 3652–3655 (2007). https://doi.org/10.1002/anie.200700246
Ristanović, Z., Chowdhury, A.D., Brogaard, R.Y., Houben, K., Baldus, M., Hofkens, J., Roeffaers, M.B.J., Weckhuysen, B.M.: Reversible and site-dependent proton-transfer in zeolites uncovered at the single-molecule level. J. Am. Chem. Soc. 140, 14195–14205 (2018). https://doi.org/10.1021/jacs.8b08041
Stavitski, E., Kox, M.H.F., Weckhuysen, B.M.: Revealing shape selectivity and catalytic activity trends within the pores of H-ZSM-5 crystals by time- and space-resolved optical and fluorescence microspectroscopy. Chem. Eur. J. 13, 7057–7065 (2007). https://doi.org/10.1002/chem.200700568
Ristanović, Z., Hofmann, J.P., De Cremer, G., Kubarev, A.V., Rohnke, M., Meirer, F., Hofkens, J., Roeffaers, M.B.J., Weckhuysen, B.M.: Quantitative 3D fluorescence imaging of single catalytic turnovers reveals spatiotemporal gradients in reactivity of zeolite H-ZSM-5 crystals upon steaming. J. Am. Chem. Soc. 137, 6559–6568 (2015). https://doi.org/10.1021/jacs.5b01698
De Cremer, G., Bartholomeeusen, E., Pescarmona, P.P., Lin, K., De Vos, D.E., Hofkens, J., Roeffaers, M.B.J., Sels, B.F.: The influence of diffusion phenomena on catalysis: a study at the single particle level using fluorescence microscopy. Catal. Today 157, 236–242 (2010). https://doi.org/10.1016/j.cattod.2010.04.035
De Cremer, G., Roeffaers, M.B.J., Bartholomeeusen, E., Lin, K., Dedecker, P., Pescarmona, P.P., Jacobs, P.A., De Vos, D.E., Hofkens, J., Sels, B.F.: High-resolution single-turnover mapping reveals intraparticle diffusion limitation in Ti-MCM-41-catalyzed epoxidation. Angew. Chem. Int. Ed. 49, 908–911 (2010). https://doi.org/10.1002/anie.200905039
Chen, P., Zhou, X., Andoy, N.M., Han, K.-S., Choudhary, E., Zou, N., Chen, G., Shen, H.: Spatiotemporal catalytic dynamics within single nanocatalysts revealed by single-molecule microscopy. Chem. Soc. Rev. 43, 1107–1117 (2014). https://doi.org/10.1039/C3CS60215J
Dong, B., Pei, Y., Zhao, F., Goh, T.W., Qi, Z., Xiao, C., Chen, K., Huang, W., Fang, N.: In situ quantitative single-molecule study of dynamic catalytic processes in nanoconfinement. Nat. Catal. 1, 135–140 (2018). https://doi.org/10.1038/s41929-017-0021-1
Ameloot, R., Roeffaers, M., Baruah, M., Cremer, G.D., Sels, B., Vos, D.D., Hofkens, J.: Towards direct monitoring of discrete events in a catalytic cycle at the single molecule level. Photochem. Photobiol. Sci. 8, 453–456 (2009). https://doi.org/10.1039/B821657F
Hirayama, T., Tsuboi, H., Niwa, M., Miki, A., Kadota, S., Ikeshita, Y., Okuda, K., Nagasawa, H.: A universal fluorogenic switch for Fe(II) ion based on N-oxide chemistry permits the visualization of intracellular redox equilibrium shift towards labile iron in hypoxic tumor cells. Chem. Sci. 8, 4858–4866 (2017). https://doi.org/10.1039/C6SC05457A
Lebold, T., Michaelis, J., Bräuchle, C.: The complexity of mesoporous silica nanomaterials unravelled by single molecule microscopy. Phys. Chem. Chem. Phys. 13, 5017–5033 (2011). https://doi.org/10.1039/C0CP02210A
Pramanik, R., Ito, T., Higgins, D.A.: Molecular length dependence of single molecule wobbling within surfactant- and solvent-filled silica mesopores. J. Phys. Chem. C 117, 15438–15446 (2013). https://doi.org/10.1021/jp404991m
Jung, C., Schwaderer, P., Dethlefsen, M., Köhn, R., Michaelis, J., Bräuchle, C.: Visualization of the self-assembly of silica nanochannels reveals growth mechanism. Nat. Nanotechnol. 6, 87–92 (2011). https://doi.org/10.1038/nnano.2010.258
McCain, K.S., Hanley, D.C., Harris, J.M.: Single-molecule fluorescence trajectories for investigating molecular transport in thin silica sol–gel films. Anal. Chem. 75, 4351–4359 (2003). https://doi.org/10.1021/ac0345289
Seebacher, C., Hellriegel, C., Deeg, F.-W., Bräuchle, C., Altmaier, S., Behrens, P., Müllen, K.: Observation of translational diffusion of single terrylenediimide molecules in a mesostructured molecular sieve. J. Phys. Chem. B. 106, 5591–5595 (2002). https://doi.org/10.1021/jp013198y
Jung, C., Hellriegel, C., Michaelis, J., Bräuchle, C.: Single-molecule traffic in mesoporous materials: translational, orientational, and spectral dynamics. Adv. Mater. 19, 956–960 (2007). https://doi.org/10.1002/adma.200602173
Jung, C., Hellriegel, C., Platschek, B., Wöhrle, D., Bein, T., Michaelis, J., Bräuchle, C.: Simultaneous measurement of orientational and spectral dynamics of single molecules in nanostructured host–guest materials. J. Am. Chem. Soc. 129, 5570–5579 (2007). https://doi.org/10.1021/ja0684850
Pramanik, R., Ito, T., Higgins, D.A.: Single molecule wobbling in cylindrical mesopores. J. Phys. Chem. C 117, 3668–3673 (2013). https://doi.org/10.1021/jp400479w
Kumarasinghe, R., Higgins, E.D., Ito, T., Higgins, D.A.: Spectroscopic and polarization-dependent single-molecule tracking reveal the one-dimensional diffusion pathways in surfactant-templated mesoporous silica. J. Phys. Chem. C 120, 715–723 (2016). https://doi.org/10.1021/acs.jpcc.5b10152
Kumarasinghe, R., Ito, T., Higgins, D.A.: Nanoconfinement and mass transport in silica mesopores: the role of charge at the single molecule and single pore levels. Anal. Chem. 92, 1416–1423 (2020). https://doi.org/10.1021/acs.analchem.9b04589
Park, S.C., Ito, T., Higgins, D.A.: Dimensionality of diffusion in flow-aligned surfactant-templated mesoporous silica: a single molecule tracking study of pore wall permeability. J. Phys. Chem. C 119, 26101–26110 (2015). https://doi.org/10.1021/acs.jpcc.5b06835
Cooper, J.T., Peterson, E.M., Harris, J.M.: Fluorescence imaging of single-molecule retention trajectories in reversed-phase chromatographic particles. Anal. Chem. 85, 9363–9370 (2013). https://doi.org/10.1021/ac402251r
Lebold, T., Jung, C., Michaelis, J., Bräuchle, C.: Nanostructured silica materials as drug-delivery systems for doxorubicin: single molecule and cellular studies. Nano Lett. 9, 2877–2883 (2009). https://doi.org/10.1021/nl9011112
Liao, Y., Yang, S.K., Koh, K., Matzger, A.J., Biteen, J.S.: Heterogeneous single-molecule diffusion in one-, two-, and three-dimensional microporous coordination polymers: directional, trapped, and immobile guests. Nano Lett. 12, 3080–3085 (2012). https://doi.org/10.1021/nl300971t
Fu, D., Maris, J.J.E., Stanciakova, K., Nikolopoulos, N., van der Heijden, O., Siemons, M.E., Salas Pastene, D., Kapitein, L.C., Rabouw, F.T., Meirer, F., Weckhuysen, B.M.: Nanospectroscopic Quantification of Molecular Diffusion within Single-Oriented Zeolite Channels (2021)
Maris, J.J.E., Rabouw, F.T., Weckhuysen, B.M., Meirer, F.: Segmentation-based motion analysis of trajectories using Diffusion Lab (2021)
Vogt, E.T.C., Weckhuysen, B.M.: Fluid catalytic cracking: recent developments on the grand old lady of zeolite catalysis. Chem. Soc. Rev. 44, 7342–7370 (2015). https://doi.org/10.1039/C5CS00376H
Kortunov, P., Vasenkov, S., Kärger, J., Fé Elía, M., Perez, M., Stöcker, M., Papadopoulos, G.K., Theodorou, D., Drescher, B., McElhiney, G., Bernauer, B., Krystl, V., Kočiřík, M., Zikánová, A., Jirglová, H., Berger, C., Gläser, R., Weitkamp, J., Hansen, E.W.: Diffusion in fluid catalytic cracking catalysts on various displacement scales and its role in catalytic performance. Chem. Mater. 17, 2466–2474 (2005). https://doi.org/10.1021/cm050031z
Kortunov, P., Vasenkov, S., Kärger, J., Fé Elía, M., Perez, M., Stöcker, M., Papadopoulos, G.K., Theodorou, D., Drescher, B., McElhiney, G., Bernauer, B., Krystl, V., Kocirik, M., Zikanova, A., Jirglova, H., Berger, C., Gläser, R., Weitkamp, J., Hansen, E.W.: Pulsed-field gradient nuclear magnetic resonance study of transport properties of fluid catalytic cracking catalysts. Magn. Reson. Imaging 23, 233–237 (2005). https://doi.org/10.1016/j.mri.2004.11.016
Zeng, S., Xu, S., Gao, S., Gao, M., Zhang, W., Wei, Y., Liu, Z.: Differentiating diffusivity in different channels of ZSM-5 zeolite by pulsed field gradient (PFG) NMR. ChemCatChem 12, 463–468 (2020). https://doi.org/10.1002/cctc.201901689
Ristanović, Z., Kerssens, M.M., Kubarev, A.V., Hendriks, F.C., Dedecker, P., Hofkens, J., Roeffaers, M.B.J., Weckhuysen, B.M.: High-resolution single-molecule fluorescence imaging of zeolite aggregates within real-life fluid catalytic cracking particles. Angew. Chem. Int. Ed. 54, 1836–1840 (2015). https://doi.org/10.1002/anie.201410236
Roeffaers, M.B.J., Sels, B.F., Uji-i, H., De Schryver, F.C., Jacobs, P.A., De Vos, D.E., Hofkens, J.: Spatially resolved observation of crystal-face-dependent catalysis by single turnover counting. Nature 439, 572–575 (2006). https://doi.org/10.1038/nature04502
Van Loon, J., Kubarev, A.V., Roeffaers, M.B.J.: Correlating catalyst structure and activity at the nanoscale. ChemNanoMat 4, 6–14 (2018). https://doi.org/10.1002/cnma.201700301
Zhang, J., Chen, C., Chen, S., Hu, Q., Gao, Z., Li, Y., Qin, Y.: Highly dispersed Pt nanoparticles supported on carbon nanotubes produced by atomic layer deposition for hydrogen generation from hydrolysis of ammonia borane. Catal. Sci. Technol. 7, 322–329 (2017). https://doi.org/10.1039/C6CY01960A
Choura, M., Belgacem, N.M., Gandini, A.: Acid-catalyzed polycondensation of furfuryl alcohol: mechanisms of chromophore formation and cross-linking. Macromolecules 29, 3839–3850 (1996). https://doi.org/10.1021/ma951522f
Hayasaka, K., Liang, D., Huybrechts, W., De Waele, B.R., Houthoofd, K.J., Eloy, P., Gaigneaux, E.M., van Tendeloo, G., Thybaut, J.W., Marin, G.B., Denayer, J.F.M., Baron, G.V., Jacobs, P.A., Kirschhock, C.E.A., Martens, J.A.: Formation of ZSM-22 zeolite catalytic particles by fusion of elementary nanorods. Chem. Eur. J. 13, 10070–10077 (2007). https://doi.org/10.1002/chem.200700967
Layek, A., Loon, J.V., Roeffaers, M.B.J., Kubarev, A.V.: Correlated super-resolution fluorescence and electron microscopy reveals the catalytically active nanorods within individual H-ZSM-22 zeolite particles. Catal. Sci. Technol. 9, 4645–4650 (2019). https://doi.org/10.1039/C9CY00948E
Dertinger, T., Colyer, R., Iyer, G., Weiss, S., Enderlein, J.: Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc. Natl Acad. Sci. U S A 106, 22287–22292 (2009). https://doi.org/10.1073/pnas.0907866106
Kalirai, S., Paalanen, P.P., Wang, J., Meirer, F., Weckhuysen, B.M.: Visualizing dealumination of a single zeolite domain in a real-life catalytic cracking particle. Angew. Chem. Int. Ed. 55, 11134–11138 (2016). https://doi.org/10.1002/anie.201605215
Sprung, C., Weckhuysen, B.M.: Dispersion and orientation of zeolite ZSM-5 crystallites within a fluid catalytic cracking catalyst particle. Chem. Eur. J. 20, 3667–3677 (2014). https://doi.org/10.1002/chem.201303549
Chughtai, A.H., Ahmad, N., Younus, H.A., Laypkov, A., Verpoort, F.: Metal–organic frameworks: versatile heterogeneous catalysts for efficient catalytic organic transformations. Chem. Soc. Rev. 44, 6804–6849 (2015). https://doi.org/10.1039/C4CS00395K
Rogge, S.M.J., Bavykina, A., Hajek, J., Garcia, H., Olivos-Suarez, A.I., Sepúlveda-Escribano, A., Vimont, A., Clet, G., Bazin, P., Kapteijn, F., Daturi, M., Ramos-Fernandez, E.V., Xamena, F.X.L.I., Van Speybroeck, V., Gascon, J.: Metal–organic and covalent organic frameworks as single-site catalysts. Chem. Soc. Rev. 46, 3134–3184 (2017). https://doi.org/10.1039/C7CS00033B
Rivera-Torrente, M., Pletcher, P.D., Jongkind, M.K., Nikolopoulos, N., Weckhuysen, B.M.: Ethylene polymerization over metal–organic framework crystallites and the influence of linkers on their fracturing process. ACS Catal. 9, 3059–3069 (2019). https://doi.org/10.1021/acscatal.9b00150
Valvekens, P., Jonckheere, D., Baerdemaeker, T.D., Kubarev, A.V., Vandichel, M., Hemelsoet, K., Waroquier, M., Speybroeck, V.V., Smolders, E., Depla, D., Roeffaers, M.B.J., Vos, D.D.: Base catalytic activity of alkaline earth MOFs: a (micro)spectroscopic study of active site formation by the controlled transformation of structural anions. Chem. Sci. 5, 4517–4524 (2014). https://doi.org/10.1039/C4SC01731E
Kubarev, A.V., Roeffaers, M.B.J.: Surface acid–base catalytic activity of ZIF-8 revealed by super-resolution fluorescence microscopy. CrystEngComm 19, 4162–4165 (2017). https://doi.org/10.1039/C7CE00074J
van den Bergh, J., Gascon, J., Kapteijn, F.: Diffusion in zeolites—impact on catalysis. In: Zeolites and Catalysis, pp. 361–387. Wiley, Hoboken (2010)
Campbell, S.M., Bibby, D.M., Coddington, J.M., Howe, R.F., Meinhold, R.H.: Dealumination of HZSM-5 zeolites: I. Calcination and hydrothermal treatment. J. Catal. 161, 338–349 (1996). https://doi.org/10.1006/jcat.1996.0191
De Lucas, A., Canizares, P., Durán, A., Carrero, A.: Dealumination of HZSM-5 zeolites: effect of steaming on acidity and aromatization activity. Appl. Catal. Gen. 154, 221–240 (1997). https://doi.org/10.1016/S0926-860X(96)00367-5
Ong, L.H., Dömök, M., Olindo, R., van Veen, A.C., Lercher, J.A.: Dealumination of HZSM-5 via steam-treatment. Microporous Mesoporous Mater. 164, 9–20 (2012). https://doi.org/10.1016/j.micromeso.2012.07.033
Sprung, C., Weckhuysen, B.M.: Differences in the location of guest molecules within zeolite pores as revealed by multilaser excitation confocal fluorescence microscopy: which molecule is where? J. Am. Chem. Soc. 137, 1916–1928 (2015). https://doi.org/10.1021/ja511381f
Karwacki, L., de Winter, D.A.M., Aramburo, L.R., Lebbink, M.N., Post, J.A., Drury, M.R., Weckhuysen, B.M.: Architecture-dependent distribution of mesopores in steamed zeolite crystals as visualized by FIB–SEM tomography. Angew. Chem. Int. Ed. 50, 1294–1298 (2011). https://doi.org/10.1002/anie.201006031
Lin, K., Pescarmona, P., Vandepitte, H., Liang, D., Vantendeloo, G., Jacobs, P.: Synthesis and catalytic activity of Ti-MCM-41 nanoparticles with highly active titanium sites. J. Catal. 254, 64–70 (2008). https://doi.org/10.1016/j.jcat.2007.11.017
Lin, K., Pescarmona, P.P., Houthoofd, K., Liang, D., Van Tendeloo, G., Jacobs, P.A.: Direct room-temperature synthesis of methyl-functionalized Ti-MCM-41 nanoparticles and their catalytic performance in epoxidation. J. Catal. 263, 75–82 (2009). https://doi.org/10.1016/j.jcat.2009.01.013
Liu, K.-L., Kubarev, A.V., Van Loon, J., Uji-i, H., De Vos, D.E., Hofkens, J., Roeffaers, M.B.J.: Rationalizing inter- and intracrystal heterogeneities in dealuminated acid mordenite zeolites by stimulated Raman scattering microscopy correlated with super-resolution fluorescence microscopy. ACS Nano 8, 12650–12659 (2014). https://doi.org/10.1021/nn505576p
Buurmans, I.L.C., Pidko, E.A., de Groot, J.M., Stavitski, E., van Santen, R.A., Weckhuysen, B.M.: Styrene oligomerization as a molecular probe reaction for zeolite acidity: a UV–Vis spectroscopy and DFT study. Phys. Chem. Chem. Phys. 12, 7032–7040 (2010). https://doi.org/10.1039/C002442B
Van Loon, J., Janssen, K.P.F., Franklin, T., Kubarev, A.V., Steele, J.A., Debroye, E., Breynaert, E., Martens, J.A., Roeffaers, M.B.J.: Rationalizing acid zeolite performance on the nanoscale by correlative fluorescence and electron microscopy. ACS Catal. 7, 5234–5242 (2017). https://doi.org/10.1021/acscatal.7b01148
Kumar, N., Kalirai, S., Wain, A.J., Weckhuysen, B.M.: Nanoscale chemical imaging of a single catalyst particle with tip-enhanced fluorescence microscopy. ChemCatChem 11, 417–423 (2019). https://doi.org/10.1002/cctc.201801023
Liu, Y., Meirer, F., Krest, C.M., Webb, S., Weckhuysen, B.M.: Relating structure and composition with accessibility of a single catalyst particle using correlative 3-dimensional micro-spectroscopy. Nat. Commun. 7, 12634 (2016). https://doi.org/10.1038/ncomms12634
de Winter, D.A.M., Meirer, F., Weckhuysen, B.M.: FIB–SEM tomography probes the mesoscale pore space of an individual catalytic cracking particle. ACS Catal. 6, 3158–3167 (2016). https://doi.org/10.1021/acscatal.6b00302
Hendriks, F.C.: Accessibility, structure and reactivity of individual catalyst particles studied by fluorescence microscopy. PhD Thesis, Utrecht University (2017)
Hauser, M., Wojcik, M., Kim, D., Mahmoudi, M., Li, W., Xu, K.: Correlative super-resolution microscopy: new dimensions and new opportunities. Chem. Rev. 117, 7428–7456 (2017). https://doi.org/10.1021/acs.chemrev.6b00604
Aloi, A., Vilanova, N., Albertazzi, L., Voets, I.K.: iPAINT: a general approach tailored to image the topology of interfaces with nanometer resolution. Nanoscale 8, 8712–8716 (2016). https://doi.org/10.1039/C6NR00445H
Nehme, E., Weiss, L.E., Michaeli, T., Shechtman, Y.: Deep-STORM: super-resolution single-molecule microscopy by deep learning. Optica 5, 458–464 (2018). https://doi.org/10.1364/OPTICA.5.000458
Fu, D., van der Heijden, O., Stanciakova, K., Schmidt, J.E., Weckhuysen, B.M.: Disentangling reaction processes of zeolites within single-oriented channels. Angew. Chem. Int. Ed. (2020). https://doi.org/10.1002/anie.201916596
Falcaro, P., Okada, K., Hara, T., Ikigaki, K., Tokudome, Y., Thornton, A.W., Hill, A.J., Williams, T., Doonan, C., Takahashi, M.: Centimetre-scale micropore alignment in oriented polycrystalline metal–organic framework films via heteroepitaxial growth. Nat. Mater. 16, 342–348 (2017). https://doi.org/10.1038/nmat4815
Cotlet, M., Vosch, T., Habuchi, S., Weil, T., Müllen, K., Hofkens, J., De Schryver, F.: Probing intramolecular Förster resonance energy transfer in a naphthaleneimide–peryleneimide–terrylenediimide-based dendrimer by ensemble and single-molecule fluorescence spectroscopy. J. Am. Chem. Soc. 127, 9760–9768 (2005). https://doi.org/10.1021/ja042656o
Dill, M., Baier, M.C., Mecking, S., Wöll, D.: Enhanced accuracy of single-molecule diffusion measurements with a photocleavable energy-transfer dyad. Angew. Chem. Int. Ed. 52, 12435–12438 (2013). https://doi.org/10.1002/anie.201303658
Acknowledgements
We thank Dr. Z. Ristanović (Leiden University) and Dr. F. T. Rabouw (Utrecht University) for fruitful discussions. This work was supported by the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), an NWO Gravitation Program funded by the Ministry of Education, Culture and Science of the Government of the Netherlands, a European Research Council (ERC) Advanced Grant (No. 321140), and a NWO VIDI Grant to F.M.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maris, J.J.E., Fu, D., Meirer, F. et al. Single-molecule observation of diffusion and catalysis in nanoporous solids. Adsorption 27, 423–452 (2021). https://doi.org/10.1007/s10450-020-00292-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-020-00292-7