
Abstract
Multiple Sorption Model (MSM) is used to simulate sorption isotherms and the effect of the multiplicity of physicochemical parameters is reduced by introducing an invariant procedure using a few sorbates that are small molecules. This study presents the use of water, methanol, carbon dioxide and methane as test molecules to determine the structure and texture of coal and energy parameters. Parallel calculations for a set of sorption systems on the same coal sample recursively yield the most probable estimates. The procedure was tested for 6 coal samples with different carbon content. Effect of simulations made by MSM is evaluated on measurements of sorption isotherms. Result obtained by the analysis shows that smaller submicropores are in better contact with sorbate molecule and bigger one contact is weaker-contacts play vital role in energy contribution to the molecule. Tendency of significant absorption for CO2 and CH3OH and insignificant for H2O and CH4 is confirmed on the basis of thermodynamic dissertation/calculation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
As carbon is a complex copolymer containing a plurality of components, and thus a complicated structure and texture, examining its properties as an adsorbent is a demanding task. The classic adsorption theories assume a large simplification of the structure of this material and for this reason the calculated parameters are only of comparative nature.
Even today, some researchers consider sorption in polymeric materials as dual, despite the obvious fact that there are pores smaller than the penetrant molecules (Camboni and Sokolov 2016). On the other hand, in the Multiple Sorption Model (MSM) developed in the team, a multiplicity of sorption phenomena from absorption/solubility (Tsoulou et al. 2008), through expansion to multi-layer adsorption, is assumed (Jodłowski and Wójcik 2013; Jodłowski et al. 2007). This fact is confirmed by laboratory tests that show that swelling depends both on the volume of the penetrant molecules and on the space between the objects in the polymer (Ulrich et al. 1999).
Such a multitude of phenomena significantly complicates the problem of modeling sorption in a carbon copolymer. Therefore, it was decided to use the procedure of simultaneous/parallel simulation of sorption isotherms on the same carbon sample for several sorbates of various structure. From the previous studies, the picture emerges that steam, methanol vapor, carbon dioxide and methane are good and sufficient probes for determining the structure and texture of coal.
The simultaneous use of polar and apolar sorbates of various shapes and sizes allows to describe the structure and texture of the carbon sorbent in a sufficient way.
2 Modeling of sorption process
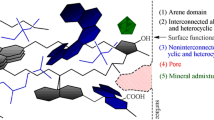
The model is constructed on the base of fundamental physico-chemichal laws, starting from analysis of free energy formula. The analysis of enthalpy of polymer solution and entropy of mixing results in the set of equations describing a behavior of sorbate molecules in the sorption system. Theory of polymer solution [Flory 1953] is enhanced and developed for coal treated as complex copolymer in the way of Flory–Huggins model of polymer. Finally, the classes of object are proposed for representation of coal structure:
-
arene domains—small quasi-crystal forms built with condensed benzene rings plates; the number of plates and number of benzene rings in plates as well as compactness factor of arene domain remains in a loose relation to coalification degree;
-
crosslinked chains—a set of aliphatic, alicyclic and heterocyclic chains which connect each-other or connect arene domains composing limitedly rigid network of macromolecular phase (together with arene domains);
-
non-crosslinked chains—a set of aliphatic, alicyclic and heterocyclic chains which compose limitedly elastic molecular phase, these objects are located in the cells of macromolecular phase.
Arene domains together with crosslinked chains compose macromolecular phase which is limitedly rigid and non-crosslinked chains forms molecular phase which is elastic. The mutual ratio of these phases to some extent has an impact on the occurrence of the absorption phenomenon. Two classes of objects are introduced to model not connected to organic composition:
-
mineral admixtures—small pieces of non-organic rocks playing negligible role in the sorption process;
-
pores—empty spaces in coal mass treated as high-energy contacts to sorbate molecules.
The last class of objects is vital for evaluation of entropy and enthalpy of the sorption system. Objects are divided to elements having size of the network cell. Different sizes of cells could be assumed for computations. The most often used size is so-called “base cell” which volume is equal to the volume of segment –CH2– from aliphatic chain. It gives simple relations: water molecule volume is nearly the same (18 cm3 for water and 16 cm3 for the cell), benzene ring is represented by 2 cells and alicyclic ring by 4 cells. All objects are represented by the set of cells and the rule is that it is not allowed to place a segments of objects in the same network cell except pores and sorbate molecule, which can simultaneously take the same location.
The determination of the actual sorbate molecule volume in a sorption system is important for modeling the sorption process. The space occupied by the objects of the sorption system has an influence on determining the entropy of mixing. Segment volumes of sorbate are determined using a dedicated state equation developed by the team specifically to model sorption. The mixing entropy is calculated according to the theory of Flory's polymer solutions. The enthalpy of the system is determined on the basis of the analysis of the number of contacts of the spherical segments of the components of this system and the value of the unit energy assigned to the contact in accordance with the Berthelot rule (Boda 2008; Ziółkowska 2016). Segments are part of a spatial network with spherical cells in which the network constant (maximum number of contacts) is 12. At the same time, the number of contacts with assigned energy values determines the allocation of a molecule to a selected sorption subsystem. Physically, sorption is divided into three subsystems:
-
adsorption, where at most half of the maximum number of contacts of segment is involved and only adhesion forces are included;
-
expansion, where number of segment contacts is bigger than half and lower than maximum with involvement of adhesion and cohesion forces;
-
absorption, where molecules of sorbate (so called penetrant in this particular case) occupy the space previously occupied by the sorbent segments and only cohesion forces are involved.
The term segment is used interchangeably with the term molecule for sorbates with sizes comparable to the cell of the network. The segment, on the other hand, is a more general term, which may include parts of objects (molecules) larger than the cell of the network. In particular, this applies to the components from which the carbon material is built (see Fig. 1). The problem of varying the shape and size of objects is solved by assuming that they could be considered as agglomerates of cells of equal size. The volume of the object represents the average number of x cells that make up its composition. Default volume of the cell is equal to volume of aliphatic chain segment -CH2- and has volume 16 cm3/mole and it is called base cell. The model gives possibility of use different cell volume so called sorbate cell (e.g. water—18 cm3/mole).

Relation between object and segment in relation to components of sorbent and to sorbates
The set of segments represented by spheres in Fig. 1 forms a model object of the sorption system. One segment corresponds to the assumed cell of the sorption space. Some approximations have necessarily been used in this drawing. Above is the representation of component objects (although benzene and cyclohexane can also be sorbates), while the reference to sorbate molecules is shown below.
The invariant procedure, in addition to the constancy of the coal structure parameters, also assumes the same monolayer volume for selected sorbates. This assumption seems justified due to the better adaptation to the texture of small molecules, and therefore a larger number of them fills the difference in the molar volume of sorbates (see Fig. 2).

Idea of monolayer volume as invariant parameter for texture analysis
The number of molecules filling the pore in the monolayer for smaller sorbet (water) is bigger than number of molecules of larger sorbate (methane or carbon dioxide). Some differences appears between location of methanol and carbon dioxide molecules (shadowed circles)—small discrepancies in the molar volume (Table 1) could cause different accounting of molecules to the monolayer. In presented example CO2 molecule (shadow circle) is accounted to the next layer because number of contacts to other sorbate molecules is bigger than number of contacts to sorbent surface (Fig. 3). In the case of methanol small difference in volume cause accounting the molecule in the similar location to monolayer because the decrease of contacts number to other molecules of sorbate. Certainly, in the model space sorbate molecules are represented by the segments/cells of the network. Moreover, in the range of submicropores and bigger pores the differences in the number of molecules located in monolayer could be more distinctive. In our opinion assumption on the similar volume of monolayer is statistically valid (Table 2).

In addition, detailed enthalpy analysis allows the isolation of the monolayer adsorption isotherm from the adsorption sub-system. This property of the model gives possibility the introduction of the invariant analysis procedure, making the sorption estimates more precise basing on a parallel sorption analysis of several sorbates on the same sorbent. The above properties of the model allow to analyze accurately the structure of the coal matter as a sorbent, and molecules of low molecular weight sorbents such as water, methanol, methane and carbon dioxide can be structural probes.
For numerical reasons, the process is divided into 11 sub-processes, two of which are for adsorption and absorption, and of the intermediate ones are in the range of pure expansion. Expansion subprocess is divided a priori to 9 subsystem in mathematical way as the normal distribution with maximum at RB—dominant radius of submicropores being the factor for simulations and standard deviation σ. This division allows us to analyze the distribution of sorbate molecules between individual subsystems in submicropores. Parameters of a distribution are related to the shape of total sorption isotherm in low range of pressure (lower RB values generate more convex isotherms). Sorption formula is transformed to the set of eleven formulas; ten of them parameterized by radius values (related to radius of sorbate molecule—see Table 1 "criterion") and it and last one for adsorption developed in other way to represent the heterogeneity of coal surface (LBET—polylayer model of adsorption with limited capacity of layers). Simulated eleven isotherms are called poly-sorption (green bars on the upper-left chart from Figs. 4, 5, and 6). It is possible to evaluate the starting volume distribution of submicropores (green bars on the bottom-left chart from Figs. 4, 5, and 6) similar like in (Jin et al. 2017). In the next step, the intermediate 9 isotherms are summed up, while the absorption and adsorption isotherms remain unchanged, in this way the multi-sorption isotherms are obtained together with the total theoretical sorption isotherm obtained after summing all subsystems. The last transformation aims at visualization in order to more easily analyze the global phenomenon and evaluate the total sorption isotherm by comparing with the measurement result during subsequent simulations. It should be borne in mind that the final swelling of coal is the result of two opposing phenomena of expansion and contraction of submicropores, while absorption generates swelling in its entirety. Difference between distribution of submicropores volume and submicropores capacity distribution is the result of this phenomena.

Distributions of submicropores volume Uh and their capacity UAm (left side) as well as distribution of energy correction factor Z and sorption energy Qbc (as balance of cohesive "+" and adhesive "−" share) for sample B82 and CH4 sorption

Distributions of submicropores volume Uh and their capacity UAm (lest side) as well as distribution of energy correction factor Z and sorption energy Qbc (as balance of cohesive "+" and adhesive "−" share) for sample B82 and CO2 sorption

Distributions of submicropores volume Uh and their capacity UAm (lest side) as well as distribution of energy correction factor Z and sorption energy Qbc (as balance of cohesive "+" and adhesive "−" share) for sample M85 and CO2 sorption
This approach allows to analyze several sorption isotherms in parallel, where a set of constant parameters, such as the structure of carbon (i.e. shares of arene domains, cross-linked and non-crosslinked chains, volume and compactness of arene domains) and the volume of a monolayer occurs (Jodlowski 2014, 2016). As a result, for each individual isotherm, the texture parameters, i.e. the volume of pores, including submicropores and micropores, the distribution of molecules between sub-systems and the energy parameters of sorption, can be refined. The normal distribution of pores volume and capacity within expansion sub-systems is assumed (Karney 2016; Das 2019; Luo 2019).
3 Results
The set of sorbates used are polar substances such as water and methanol, non-polar but with a quadrupole moment, such as carbon dioxide and non-polar like methane. Properties of sorbates are presented in Table 1.
A set of isotherms taken from the literature and measured by the author was used for numerical research. The coal samples on which the sorption was measured came from various types of coal, what allows analysis of various coal structures. The parameters of hard coals selected for analysis are presented in Table 2.
Different sorbates are used for isotherms measurements in selected samples. Full set of sorbates for every sample is not acquired yet but for every sample are measured at least sorption isotherms for two sorbate and some other sets contain sorption isotherms for three sorbates. An important feature of the choice of sorption systems is that sorbates of different polarity are included in the set, which allows to differentiate the results in terms of the content of polar groups on the surface of coal in statistical terms. Eventually, one can follow selected sets of sorption systems in the Table 4.
The model describes a complicated coal structure using a number of parameters. Of course, almost all of them have a physical sense and refer to the structure and texture of hard coal or to the properties of the sorption system. The model also contains 2 mathematical parameters related to the description of the distribution curve, which are selected only for the optimal shape of the distribution curve of the molecules in sorption subsystems. However, this distribution is normalized by physicochemical estimates and mathematical parameter cannot be manipulated freely. The number of parameters describing the properties of the sorption system is over 20. Therefore, some of these parameters must be precisely identified for the same sample of coal at several sorbates always with the same value, while others are only evaluated for the selected sorbate. First type of parameters we call invariant, however, they are found by parallel calculations until an optimal structure is found for each sorption system under study (i.e. in each of them it must be possible to reasonably identify the texture with the simulation of the sorption isotherm covering the measurement points). The set of invariant parameters is presented in the Table 3. Parameters Udo, Ue1, Ue2 and Uh are unit less molar partition of component in coal substance. Last parameter Vh is estimated invariant parameter on the basis of parallel calculation with the condition of optimal sorption isotherm simulation and means volume of monolayer in cm3of sorbate per gram of sorbent. It must be remembered that this is not a parameter corresponding to the total sorption capacity.
After obtaining the set values of invariant parameters, the texture parameters, such as the contribution of submicropores and the distribution of their volume vs. radius relative to the size of sorbate molecule, and later the distribution of sorption capacity in submicropories, can be further specified. Selected parameters of sorption system are presented in Table 4.
Further description of sorption system is presented on Figs. 3, 4, and 5 presenting selected distributions of submicropores, sorbate molecules and energy factors vs. relative radius or pores.
Distributions of pores volume (Uh—standardized and unitless) and capacity (UAm—in mmol/g) are presented on the left side of Figs. 4, 5, and 6. In every case maximum of capacity distribution (RS) is shifted down by radius with comparison to submicropores initial volume maximum (RB). It is a result of expansion of pores under process of bulk penetration with taking of small submicropore volume and expanding it to the size of penetrant (sorbate) molecule. That process is similar to absorption however pore exist but it size must be increased. Mutual interaction of molecules—the distribution of cohesion-adhesion energy causes the generation of sorption isotherms in the sub-systems associated with specific radii, which in turn produce the distribution of the pore capacity.
Substantial property of model is usage of idea of contacts (like in sphere model). Each contact has depicted value of energy calculated from Berthelot rule and ratio of contacts number to maximum number of contacts (Z). The function Z presented on left-upper corner of Figs. 4, 5, and 6 shows the course of the parameter with inflection point. Lower part of line is defined by parameter ZA which describe a non-ideality of contacts in smaller pores, while upper part is defined by parameter ZB for bigger pores.
Co-existence of cohesion and adhesion forces causes specific behavior of molecules in subsystems. As the diameter of submicropores increases, the share of adhesion energy in interactions increases. These trends are shown on right-bottom charts at Figs. 4, 5, and 6 as the energy distribution (dark-blue bars).
Selected trends for the parameters presented in Table 4 are better seen in the diagrams which are presented in the Figs. 7, 8, and 9.

Tendency of changes geometric factors ZA and ZB versus carbon content Cdaf

Adsorption Qads and absorption Qabs energy tendencies

Absorption partition in total sorption process
One could observe the changes in ZB parameters for studied systems while ZA parameter is more stable. ZA parameterize the smaller pores and they usually better fit to small molecule of sorbate. In bigger pores situation is more complex especially when the change in the structure of the porous system in sub-bituminous, bituminous and anthracite coals is considered. It cause the bigger changes in value of ZB parameters because the contacts number decrease in slit-shape pores and that phenomenon is viewable on Fig. 7.
Adsorption energy Qads for water decrease with carbon content what is natural consequence of decreasing content of surface group (Chen et al. 2012; Baran et al. 2018) not in the direct action but they forms the active sites which increase the surface energy for water molecules in their neighborhood (Yu et al. 2013). In the case of remain sorbates adsorption energy primarily increase and further decrease what is consequence of the changing pores number and pore system structure (Baran et al. 2014, 2015). Tendency of absorption energy Qabs change is in the close relation to absorption level. It is bigger for methane and water vapor and their absorption is minimal, except W31 sample. This single system shows low energy of absorption and simultanously higher content of non-crosllinked chains (the elastic phase of coal matter) and this two factors cause significant share of absorption (see Fig. 9). While in case of methanol and carbon dioxide absorption energy is lower and share of absorption in total sorption process is bigger.
Absorption partition in total sorption for the simulated isotherms shows that methane and water do not penetrate the molecular phase, except the system water vapor-W31 coal sample. If the enthalpies of the process are usually determined by Berthelot rule, in which the effect arises due to opposing large factors, then a slight change in their correlation may significantly affect the result. In this particular case such situation take place. Interested effect is observed for absorption partition of methanol and carbon dioxide. Absorption partition for methanol is twice bigger than carbon dioxide when absorption energies evaluated for the sets are on similar level. Methanol is polar substance and this property lets molecules of this sorbate penetrate the molecular phase easier.
4 Summary
The invariant procedure, despite having some drawbacks in the form of simplifications and approximations, is a good method of analyzing carbon sorbents in terms of their structure as well as interactions occurring during the sorption process. The adoption of a model of hard coal structure and the method of spatial network modeling gives the opportunity to view the phenomena taking place in the sorption process. The simultaneous use of several sorbates with different properties (polar and apolar, single-segment and multi-segment) allows to specify the evaluation of the structure and improve the estimates of texture and energy, which makes the task of modeling sorption more precise, especially in the point of view of surface heterogeneity (Zarebska 2007; Liu and Wilcox 2012).
It is necessary to supplement the sorption systems so that for each coal sample there are at least three sorption isotherms of different sorbates (the more, the better). Therefore, the modeling of the hard coal structure and the sorption process requires further research.
References
Baran, P., Zarębska, K., Nodzeński, A.: Energy aspects of CO2 sorption in the context of sequestration in coal deposits. J. Earth Sci. 25(4), 719–726 (2014)
Baran, P., Zarębska, K., Bukowska, M.: Expansion of hard coal accompanying the sorption of methane and carbon dioxide in isothermal and non-isothermal processes. Energy Fuels 29(3), 1899–1904 (2015)
Baran, P., Krzyżanowski, A., Wójcik, M., Zarębska, K.: Evaluation of surface chemistry of selected hard coals. Adsorp. Sci. Technol. 36, 7–8 (2018)
Boda, D., Henderson, D.: The effects of deviations from Lorentz-Berthelot rules on the properties of a simple mixture. Mol. Phys. 106(20), 2367–2370 (2008)
Camboni, F., Sokolov, I.M.: Sorption of small molecules in polymeric media. Physica A 464, 54–63 (2016)
Ceglarska-Stefańska G, Współzależność procesów sorpcyjnych i dylatometrycznych zachodzących w układach: węgle kamienne - para wodna, dwutlenek węgla, metan, Zeszyty Naukowe AGH, Chemia, z. 16, Kraków, 1990
Chen, Y., Mastalerz, M., Schimmelmann, A.: Characterization of chemical functional groups in macerals across different coal ranks via micro-FTIR spectroscopy. Int. J. Coal Geol. 104, 22–33 (2012)
Das, S.K.: General dusty gas model for porous media with a specified pore size distribution. Chem. Eng. Sci. 203, 293–301 (2019)
Duda, J., Jagiełło, L., Jagiełło, J., Milewska-Duda, J.: Complementary study of microporous adsorbents with DFT and LBET. Appl. Surf. Sci. 253, 5616–5621 (2007)
Flory JP (1953) Principles of Polymer Chemistry. Cornell University Press, New York
Jin, B., Bi, R., Nasrabadi, H.: Molecular simulation of the pore size distribution effect on phase behavior of methane confined in nanopores. Fluid Phase Equilib. 452, 94–102 (2017)
Jodlowski, G.S., et al.: Sorption of methane and carbon dioxide mixtures in Polish hard coals considered in terms of adsorption-absorption model. Appl. Surf. Sci. 253(13), 5732–5735 (2007)
Jodłowski, G., Wójcik, M.: Comparative analysis of sorption of small molecule hydrocarbons and polar substances in polish hard coals. Adsorption 19(2–4), 813–819 (2013)
Jodłowski, G., Wójcik, M.: Identification of bituminous coal structure by using many sorption data. Przemysl Chem. 93(12), 2038–2041 (2014)
Jodlowski, G.S., Wójcik, M., Orzechowska-Zieba, A.: Identification of hard coal surface structure using polar and apolar small molecule substances. Adsorption 22, 847–854 (2016)
Karney, C.F.F.: Sampling exactly from the normal distribution. ACM Trans. Math. Softw. 42(1), 1–14 (2016)
Liu, Y., Wilcox, J.: Molecular simulation of CO2 adsorption in micro- and mesoporous carbons with surface heterogeneity. Int. J. Coal Geol. 104, 83–95 (2012)
Luo, S., Lutkenhaus, J.L., Nasrabadi, H.: Experimental study of pore size distribution effect on phasetransitions of hydrocarbons in nanoporous media. Fluid Phase Equilib. 487, 8–15 (2019)
Milewska-Duda, J., Duda, J.T., Jodłowski, G., Kwiatkowski, M.: A model for multilayer adsorption of small molecules in microporous materials. Langmuir 16(18), 7294–7303 (2000)
Tsolou G et al (2008) Atomistic simulation of the sorption of small gas molecules in polyisobutylene. Macromolecules 41(16):6228–6238
Ulrich J, Brion H-G, Kirchheim R (1999) The influence of sorption of small molecules on the yield stress of glassy polymers. Polymer 40:1807–1814
Yu, J., Tahmasebi, A., Han, Y., Yin, F., Li, X.: A review on water in low rank coals: the existence, interaction with coal structure and effects on coal utilization. Fuel Process Technol. 106, 9–20 (2013)
Zarębska, K.: Methane and carbon dioxide sorption processes measurements: the effect of heterogeneous structure of coals. Polish J. Environ. Stud. 16(3B), 516–519 (2007)
Ziółkowska, M., Milewska-Duda, J., Duda, T.J.: Effect of adsorbate properties on adsorption mechanisms: computational study. Adsorption 22(4–6), 589–597 (2016)
Acknowledgements
The work is done under financial support of Statutory Research of AGH University of Science and Technology in Cracow (Poland), No. 11.11.210.373
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wójcik, M., Jodłowski, G.S. Small molecule substances as molecular probes of structure and texture of coal under sorption process and modelling. Adsorption 26, 1355–1364 (2020). https://doi.org/10.1007/s10450-020-00244-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-020-00244-1