
Abstract
Porous copolymers of 1-vinyl-2-pyrrolidone (VP) with 1,4-di(methacryloyloxymethyl) naphthalene (DMN) have been synthesized by suspension polymerization in the presence of inert diluent. They have different specific surface area and contain various amount of strongly polar pyrrolidone groups. Their sorptive properties have been studied using solution of p-chlorophenol. It was found that adsorption strongly depends on the copolymer polarity. Polymeric sorbents were characterized using elemental analysis, ATR-FTIR, thermogravimetry, differential scanning calorimetry, inverse gas chromatography and their porous structure was determined by low-temperature nitrogen adsorption–desorption method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Phenol and its derivatives are among one of the most harsh pollutant (Sobiesiak et al. 2016; Sobiesiak 2017). Dissolved in water are highly toxic to diverse organisms even at very low concentrations. Despite the high toxicity they are widely used in many branches of industry such as petrochemicals, pharmaceutical, textile, rubber and pesticide production. Since phenol-contained pollutants can cause protein degradation, tissue erosion and paralysis of the central nervous system it is necessary to remove them from the wastewaters. Among miscellaneous approaches to this task, the adsorption method is considered to be the most facile and effective (Dąbrowski et al. 2005; Nakagawa et al. 2004; An et al. 2012; Lu and Sorial 2009; Derylo-Marczewska et al. 2011; Lorenc-Grabowska 2016; Kuśmierek et al. 2014). The driving force of this process is interactions of the solute molecules with the sorbent surface (Velasco and Ania 2011; Garcia et al. 1999; Yang et al. 2016; Pan et al. 2003; Wang et al. 2005). They can be either weak hydrophobic van der Waals forces or stronger dipole–dipole interactions and hydrogen bonding. These stronger interactions only appear when the surface of adsorbent contains polar groups. Therefore introduction of polar groups into the structure of adsorbents is of paramount importance. This goal can be achieved by two main approaches: the chemical modification of hydrophobic polymer skeleton with a suitable polar moiety (Yamada et al. 2017) or direct synthesis of polar adsorbents from a balanced ratio of a hydrophilic monomer and a proper cross-linker (Trochimczuk et al. 2001). The second route seems to be the more convenient, however it is not easy task. Synthesis of polymers for adsorption processes requires considerable degree of crosslinking. Nevertheless, the majority of polar monomers do not copolymerize well with most of the commercially accessible cross-linkers. For instance, 1-vinyl-2-pyrrolidone (VP) which thanks to its nontoxicity, biocompatibility, hydrophilicity and complexibility is one of the most suitable monomers for sorbent synthesis, has the reactivity ratio close to zero in most of the radical copolymerization reaction. For this reason, searching for appropriate cross-linkers is a problem of great importance.
The aim of the present work is to prepare by suspension copolymerization of 1-vinyl-2-pyrrolidone (VP) with 1,4-di(methacryloyloxymethyl)naphthalene (DMN) porous sorbents with pyrrolidone groups distributed evenly throughout the polymeric network and to investigate their physicochemical features and sorptive properties towards p-chlorophenol.
2 Experimental
2.1 Chemicals
VP (Fluka AG, Buchs, Switzerland) was washed with 5% aqueous sodium hydroxide in order to remove inhibitors. Poly(vinyl alcohol) (PVA) and α,α′-azoisobutyronitrile (AIBN) purchased from Fluka were used without purification. Toluene, acetone, methanol (reagent grade) were purchased from POCh (Gliwice, Poland), whereas heptane, hexane, octane, nonane, decane, benzene, butanol, pentan-2-one used in chromatographic measurements were supplied by Merck (Darmstadt, Germany). DMN was obtained in our laboratory according to the procedure described elsewhere (Gawdzik and Matynia 1987; Gawdzik and Czerwińska-Bil 1991; Matynia and Gawdzik 1987).
2.2 Synthesis of porous copolymers
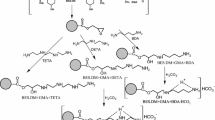
Copolymerization was performed in an aqueous suspension medium. 195 mL of distilled water and 6.5 g of PVA were stirred for 2 h at 80 °C in order to dissolve the suspension stabilizer. Then the solution containing 15 g of monomers (VP and DMN), and 0.2 g of α,α′-azoisobutyronitrile dissolved in 22.5 mL of toluene was prepared and added while stirring to the aqueous medium. Molar ratios of VP to DMN were changed from 1:1 to 4:1. Copolymerization was performed for 20 h at 80 °C. The general scheme of reaction is presented in Fig. 1. Porous beads formed in this process were filtered off, washed with hot water and extracted in a Soxhlet apparatus with acetone, toluene, and methanol. The purified beads were separated into fractions by the sieving.

The general scheme of VP–DMN copolymer synthesis
2.3 Methods of analysis
Textural characterization of the copolymers was carried out by the low-temperature nitrogen adsorption–desorption method. Nitrogen adsorption–desorption isotherms were obtained at the liquid nitrogen temperature using a volumetric adsorption analyzer ASAP 2405 (Micromeritics Inc., USA). The measurements of the porous structure of the copolymers were preceded by outgassing of the samples at 140 °C for 2 h. The specific surface area of the investigated samples were calculated by the Brunauer–Emmet–Teller (BET) method for the adsorption data in the range of a relative pressure p/po 0.05 to 0.3. The total pore volume was estimated from a single point adsorption at a relative pressure of 0.985. The pore size was obtained from the desorption branch of the isotherm using the Barrett–Joyner–Halenda (BJH) procedure.
The microspheres were imaged using a LEO 1430 VP numerical scanning electron microscope (SEM) from Carl Zeiss (Germany) with a countershaft and an energy dispersive X-ray detector.
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were obtained with a Bruker Tensor 27 FTIR spectrometer (Germany). The FTIR spectra were recorded in the spectral range of 600–4000 cm−1 with 16 scans per spectrum with a resolution of 4 cm−1.
The thermal properties of the synthesized copolymers were evaluated on the basis of measurements performed using the STA449, F1 Jupiter analyzer from Netzsch (Günzbung, Germany). The procedure was as follows: about 10 mg of the sample was placed in the pan and heated in helium or in synthetic air atmosphere at a rate of 10 °C/min up to 800 °C. The initial decomposition temperature (IDT), the temperatures of 20% (T20%) and 50% (T50%) mass losses and the final decomposition temperature (FDT) were determined.
The gaseous decomposition products formed under the degradation of copolymers were analyzed by means of a Bruker Tensor 27 FTIR spectrometer (Germany) coupled on-line to a STA instrument by a Teflon transfer line. Each FTIR spectrum was gathered every 10 °C in the spectral range from 600 to 4000 cm−1 with a resolution of 4 cm−1.
Differential scanning calorimetric measurements were carried out with the Netzsch DSC 204 calorimeter (Germany) operating in the dynamic mode. The dynamic scans were performed at the heating rate of 10 °C/min from room temperature to the maximum of 500 °C under argon (30 mL/min) atmosphere. The mass of the sample was about 10 mg. As a reference an empty aluminum crucible was used.
2.4 IGC experiments
Chromatographic measurements were carried out on a Dani GC 1000 gas chromatograph (Dani, Italy) equipped with a packed injector (220 °C), a thermal conductivity detector (TCD, 220 °C), using stainless-steel columns (100 cm × 1.6 mm I.D.), and helium as carrier gas at a flow-rate of 50 mL/min. The samples were manually injected using of a 1 µL syringe (SGE, North Melbourne, Australia).
Chromatographic columns were packed with obtained porous polymers in the form of spherical beads. The diameter of the sorbent beads was always 0.08–0.12 mm. Before measurements all the columns were conditioned in a stream of helium. The columns were conditioned with temperature programming from 60 to 230 °C at 4 °C/min and then overnight isothermally at the final temperature.
2.5 Polarity
The polarity of the studied copolymers was determined according to procedure proposed by Rohrscheider and modified by McReynolds (1970). The method is based on the measurement of the retention times of the standard substances: benzene (x), butanol (y), pentan-2-one (z) and calculation of the retention indices (Ix, Iy, Iz respectively) from the equation proposed by Kovats (Kovats 1958):
where: \({t^{\prime}_{R,x}}\) is the reduced retention time of the substance; \({t^{\prime}_{R,n}}\) is the reduced retention time of the homologous alkane with the nearest shortest retention time; \({t^{\prime}_{R,n+1}}\) is the reduced retention time of the next higher homologue eluted after homologue n; n is the number of carbon atoms in the alkane molecule.
Remaining indices (Iy and Iz) were calculated analogically.
After determination the retention indices on the studied copolymers the McReynolds’ constants (ΔI) were obtained by calculating the difference between the Kovats’ index for benzene, butanol, pentan-2-one on examined stationary phase and graphitized thermally carbon black (GTCB):
McReynolds’ constant for butanol and pentan-2-one were calculated analogically.
The sum of the three calculated McReynolds’ constant was used to define the overall polarity of the phase under study. The measurements were carried out at 140 °C.
2.6 Equilibrium adsorption studies
About 50 mg of the adsorbent were weighed in Erlenmeyer flasks, and next 5 mL of water were added to them and degassed under vacuum. Then, 25 mL initial solutions of p-chlorophenol were added. The flasks were shaken for 14 days in the incubator shaker (New Brunswick Scientific Innova 40R Model). The shaker was set at a temperature of 25 °C and 110 rpm speed. The equilibrium solute concentrations were measured by using the UV–Vis spectrophotometer Cary 100 (Varian Inc., Australia) at λ = 282 and 278 nm. The adsorbed amounts of chlorophenol were calculated from the following mass balance equation:
where aeq is the equilibrium amount adsorbed on the adsorbent (mmol/g), co is the initial concentration of bulk fluid (mmol/L), ceq is the equilibrium concentration of the solution (mmol/L), V is the volume of solution (L), and m is the weight of adsorbent (g).
3 Results and discussion
Porous copolymers of poly(VP–DMN) used in this study were obtained through suspension polymerization. This method is especially suitable for synthesis of porous microspheres with diameter in the micrometric range (Fig. 2). During the synthesis DMN served as a cross-linker and is responsible for the mechanical and thermal properties of the resulting polymeric matrix. VP provides polar groups which are effective for interaction with phenols and its derivatives. The molar ratio of the functional monomer to the cross-linker was increased from 1:1 to 1:4. The amount of the monomers along with copolymers symbols are listed in Table 1.

SEM image of VP–DMN1 copolymer
The course of the reaction was confirmed by the elemental analysis and ATR-FTIR spectroscopy. In the structure of the modified copolymers nitrogen was found. The content of nitrogen increases along with the enlargement of VP in the polymerization mixture, however its amount considerably differs from the theoretical value. On the basis of the elemental analysis the real molar ratio of functional monomer to cross-linker in the polymer matrix was calculated (Table 2). The presence of pyrrolidone units in the copolymers was also confirmed by ATR-FTIR absorption at 1683 cm−1 parallels to the ester absorption at 1723 cm−1 (Fig. 3).

ATR-FTIR spectrum of VP–DMN copolymer
As the aim of the work was synthesis of sorbents with different diameters capable to adsorb chlorophenol, the developed internal structure was a prerequisite parameter. Generally, the internal structure of the polymers is predominantly determined by the phase separation during polymerization. The process of phase separation strongly depends on the concentration of the cross-linker. In the case of high amount of cross-linker phase separation occurs late at greater conversion of monomer to polymer. As a result, a network of interconnected individual microglobules is formed. The network is characterized by high surface area and pore-size distribution with a maximum in the region of micro- to mesopore. Reduced cross-linker concentration leads to early phase separation at lower conversion. This process provokes not only aggregation of microglobules but also in-filling of small pores to form polymer network with low surface area. Figure 4 presents the nitrogen adsorption/desorption isotherms for the copolymer beads with diameter about 90 µm. Calculated on their basis (using BET method) value of specific surface area decreases from 172 m2/g for the VP–DMN1 (molar ratio of di(methacryloyloxy methyl) naphthalene to 1-vinyl-2-pyrrolidone is equal 1:1) to 141 m2/g for VP–DMN4 (molar ratio of di(methacryloyloxy methyl) naphthalene to 1-vinyl-2-pyrrolidone is equal 1:4) (Table 3). At the same time pore volume declines from 0.40 to 0.29 cm3/g. The data obtained for beads with diameter about 160 µm were practically identical.

Nitrogen adsorption/desorption isotherms of VP–DMN1 (a), VP–DMN2 (b), VP–DMN3 (c) and VP–DMN4 (d) at 77 K
Also the curves of pore size distribution (Fig. 5) for the copolymers under study have very similar course and consequently the pore diameters estimated from their maxima are congruous.

Pore size distribution calculated using the BJH method
To determine thermal stability of the obtained copolymers TG and DSC methods were used. Figure 6 displays TG curves of VP–DMN1 copolymer determined in helium and synthetic air atmospheres. The main parameters determined on the basis of these curves are presented in Table 4. As can be seen from these data, VP–DMN1 copolymer is characterized by high thermal resistance. The IDT exceeds 300 °C and is equal 322 °C for decomposition conducted in helium and 311 °C for the one conducted in air. Final decomposition temperature for both atmospheres is greater than 600 °C. Significant thermal stability of the copolymer under study was also confirmed by DSC measurement (Fig. 7). Since the copolymer possesses highly developed internal structure it adsorbs water from the air. As a result an endothermic peak with maximum at about 90 °C is present on the DSC curve. It indicates moisture desorption and is not connected with the thermal degradation of the copolymer. The first step of the degradation process is represented by the second endothermic peak with the maximum at 357 °C. At this temperature the ester bonds decompose. Both α-hydrogen bond scission (formation and emission of aldehydes) and β-hydrogen bond scission (formation and emission of carboxylic and vinyl compounds) are possible. The following exothermic peak can be connected with reaction of the molecules coming from the first step of degradation The last endothermic peak appears at 420 °C. At this temperature decompose the crosslinked parts of the sample and in a consequence, considerable amount of carbon mono- and dioxide is produced. The suggested process of degradation was confirmed by FTIR analysis of gases evolved during thermal decomposition. At the first stage bands coming from water, aldehydes, aliphatic esters, carboxylic spices, compounds containing nitrogen and carbon mono- and dioxide are present. At the second stage significant evolution of carbon dioxide and carbon monoxide connected with decarboxylation processes was observed. Figure 8 shows as example 3D FTIR spectrum of VP–DMN1 copolymer.

TG curves of VP–DMN1 copolymer determined in helium and synthetic air atmospheres

DSC curve of VP–DMN1 copolymer

3D diagram of FTIR spectrum of gases evolved during thermal decomposition of VP–DMN1 copolymer in synthetic air atmosphere
One of the most important parameter in the case of polymeric adsorbents is their polarity. It can be determined by the means of inverse gas chromatography. Microbeads for gas chromatography should be rigid, thermally stable and porous to offer a large interacting surface area. On the basis of conducted analysis it can be stated that all the copolymers under study entirely fulfil these requirements and can be used in chromatographic measurements. Table 5 presents Kovats’ retention indices for the McReynolds’ test substances and overall polarity (ΣΔI) for the porous copolymers determined at 140 °C. As can be seen VP–DMN copolymers indicate polar character. The difference between overall polarity of hydrophobic Porapak Q (an ethylvinylbenzene and divinylbenzene copolymer) and the DMN-VP copolymers is over 400. However, the polarity variation among copolymers with different amount of VP is not considerable. The highest value of overall polarity indicates VP–DMN4 that contains greatest quantity of polar monomer (VP).
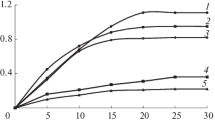
The sorptive properties of the derived copolymers were investigated using p-chlorophenol. Figure 9 present p-chlorophenol isotherms on the investigated copolymers. In the first part of experiment (Fig. 9a) beads with diameter about 90 µm were used. As can be seen, isotherms obtained on VP–DMN1 and VP–DMN2 copolymers have almost identical course. This phenomenon can be explained on the basis of polarity of these materials. The difference in overall polarity VP–DMN1 and VP–DMN2 copolymers is very slight. Similar situation is observed in the case VP–DMN3 and VP–DMN4 copolymers. Nearly the same value of polarity is reflected in the similar course of the isotherms. What is important, the highest value of p-chlorophenol adsorption (2.68 mmol/g) is observed for the most polar copolymer (VP–DMN4), that possesses the lowest value of specific surface area and pore volume. The second part of experiment was performed on copolymeric beads with diameter about 160 µm. Generally, adsorption on larger beads is slightly weaker, but the differences between studied copolymers are more noticeable (Fig. 9b).

Adsorption isotherms (a, b) and normalized adsorption isotherms (c, d) of p-chlorophenol on VP–DMN beads of different diameters
In order to separate the influence of the value of specific surface area and the polarity on the p-chlorophenol adsorption the normalized adsorption isotherms were introduced. They refer to the amount of adsorbate deposited on 1 m2 of the adsorbent. Figure 9c, d display the normalized adsorption isotherms of p-chlorophenol on 90 and 160 micrometric beads. In both cases the normalized sorption is noticeably higher for VP–DMN4 copolymer and much lower for VP–DMN1 one. These are the most polar and the most nonpolar copolymers, respectively, as revealed by inverse gas chromatography (see Table 5). For VP–DMN1 copolymer the value of normalized adsorption is equal 12.6 × 10−3 mmol/m2. Under the same condition the adsorption on VP–DMN4 is about 50% higher and is equal 19 × 10−3 mmol/m2.
4 Conclusion
Porous VP–DMN sorbents were synthesized via suspension polymerization. Application of aromatic cross-linker (DMN) allows to obtain materials with good thermal stability. At the same time VP provides pyrrolidone moiety in the polymer structure. Along with the increasing of the amount of functional monomer in the polymerization mixture increasing of the nitrogen content in the polymer network and decreasing of the value of porous surface area and pore volume were observed. All of the obtained copolymers possess adsorption ability for p-chlorophenol. The difference in their adsorption uptakes was found to be a results of the variance in their polarity.
References
An, F., Du, R., Wang, X., Wan, M., Dai, X., Gao, J.: Adsorption of phenolic compounds from aqueous solution using salicylic acid type adsorbent. J. Hazard. Mater. 201–202, 74–81 (2012). https://doi.org/10.1016/j.jhazmat.2011.11.037
Dąbrowski, A., Podkościelny, P., Hubicki, Z., Barczak, M.: Adsorption of phenolic compounds by activated carbon—a critical review. Chemosphere 58, 1049–1070 (2005). https://doi.org/10.1016/j.chemosphere.2004.09.067
Derylo-Marczewska, A., Buczek, B., Świątkowski, A.: Effect of oxygen surface groups on adsorption of benzene derivatives from aqueous solutions onto active carbon samples. Appl. Surf. Sci. 257, 9466–9472 (2011). https://doi.org/10.1016/j.apsusc.2011.06.036
Garcia, A., Ferreira, L., Leitão, A., et al.: Binary adsorption of phenol and m-cresol mixtures onto a polymeric adsorbent. Adsorption 5, 359–368 (1999). https://doi.org/10.1023/A:1008904631147
Gawdzik, B., Czerwińska-Bil, U.: Porous copolymer-based cation exchanger for the off-line preconcentration of aromatic amines from water. Chromatographia 32, 167–170 (1991). https://doi.org/10.1007/BF02325021
Gawdzik, B., Matynia, T.: Influence of synthesis conditions of porous copolymers of 1,4-di(methacryloyloxymethyl) naphthalene with divinylbenzene on their structure. II. Influence of diluent composition. Angew. Makromol. Chem. 125, 33–35 (1987). https://doi.org/10.1002/apmc.1987.051520104
Kovats, E.: Gaschromatographische Charakterisierung organischer Verbindungen. Teil 1: Retentionsindices aliphatischer Halogenide, Alkohole, Aldehyde und Ketone. Helv. Chim. Acta. 41, 1915–1932 (1958). https://doi.org/10.1002/hlca.19580410703
Kuśmierek, K., Sankowska, M., Świątkowski, A.: Kinetic and equilibrium studies of simultaneous adsorption of monochlorophenols and chlorophenoxy herbicides on activated carbon. Desalination Water Treat. 52, 178–183 (2014). https://doi.org/10.1080/19443994.2013.780984
Lorenc-Grabowska, E.: Effect of micropore size distribution on phenol adsorption on steam activated carbons. Adsorption 22, 599–607 (2016). https://doi.org/10.1007/s10450-015-9737-x
Lu, Q., Sorial, G.A.: A comparative study of multicomponent adsorption of phenolic compounds on GAC and ACFs. J. Hazard. Mater. 167, 89–96 (2009). https://doi.org/10.1016/j.jhazmat.2008.12.086
Matynia, T., Gawdzik, B.: Influence of synthesis conditions of porous copolymers of 1,4-di(methacryloyloxymethyl) naphthalene with divinylbenzene on their structure. I. Influence of mol composition of monomers. Angew. Makromol. Chem. 147, 123–132 (1987). https://doi.org/10.1002/apmc.1987.051470112
McReynolds, W.O.: Characterization of some liquid phases. J. Chromatogr. Sci. 8, 685–691 (1970). https://doi.org/10.1093/chromsci/8.12.685
Nakagawa, K., Namba, A., Mukai, S.R., Tamon, H., Ariyadejwanich, P., Tanthapanichakoon, W.: Adsorption of phenol and reactive dye from aqueous solution on activated carbons derived from solid wastes. Water Res. 38, 1791–1798 (2004). https://doi.org/10.1016/j.watres.2004.01.002
Pan, B.C., Xiong, Y., Su, Q., Li, A.M., Chen, J.L., Zhang, Q.X.: The adsorption process based on the distribution of the organic compounds present initially in the aqueous phase between this phase and the adsorbent. Chemosphere 51, 953–962 (2003). https://doi.org/10.1016/S0045-6535(03)00038-9
Sobiesiak, M.: Chemical structure of phenols and its consequence for sorption processes. In: Soto-Hernández, M. (ed) Phenolic Compounds pp. 1–28. Intech Open, London (2017) ISBN: 978-953-51-2958-5. https://doi.org/10.5772/66537
Sobiesiak, M., Podkościelna, B., Podkościelny, P.: New functionalised polymeric microspheres for multicomponent solid phase extraction of phenolic compounds. Adsorption 22, 653–662 (2016). https://doi.org/10.1007/s10450-015-9749-6
Trochimczuk, A.W., Streat, M., Kolarz, N.: Highly polar polymeric sorbents: characterization and sorptive properties towards phenol and its derivatives. React. Funct. Polym. 46, 259–271 (2001). https://doi.org/10.1016/S1381-5148(00)00056-0
Velasco, L.F., Ania, C.O.: Understanding phenol adsorption mechanisms on activated carbons. Adsorption 17, 247–254 (2011). https://doi.org/10.1007/s10450-011-9322-x
Wang, L.K., Chang, C.C., Shammas, N.K.: Polymeric adsorption and regenerant distillation. In: Wang, L.K., Hung, Y.T., Shammas N.K. (eds.) Physicochemical Treatment Processes. Handbook of Environmental Engineering, vol 3. Humana Press, New York (2005)
Yamada, S., Nagai, S., Soraku, K., et al.: Synthesis and radical polymerization of styrene bearing 2-oxazolidone moiety derived from α-amino acid and investigation of its phenol adsorption behavior. Polym. Bull. 74, 2671–2683 (2017). https://doi.org/10.1007/s00289-016-1858-1
Yang, M., Huang, Y., Cao, H., et al.: A novel polymeric adsorbent by a self-doped manner: synthesis, characterization, and adsorption performance to phenol from aqueous solution. Polym. Bull. 73, 2321–2341 (2016). https://doi.org/10.1007/s00289-016-1610-x
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Maciejewska, M., Rogulska, M. Synthesis and characterization of VP–DMN polymeric sorbents. Adsorption 25, 419–427 (2019). https://doi.org/10.1007/s10450-019-00031-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-019-00031-7