
Abstract
This paper presents results of research on modeling of a coal structure and sorption system properties. Investigations combine a viewpoint on surface heterogeneity, coming from approach aimed at adsorption mechanism identification, when applied to real coal structure in multiple sorption model (MSM) simulations. For imaging of surface heterogeneity in real coal structure two hypothetical models are considered, corresponding to a structure of graphite and active carbon. In particular, adsorbate cohesion energy and volume in adsorption system are estimated modeling selected surface heterogeneity, and employed for determination of corresponding MSM parameters. The newly elaborated MSM simulation procedure is shown and discussed, and the results of its application for analysis of real hard coals properties are shown.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Molecular dynamics based models along with Monte Carlo method are efficient tools used for modeling of different technological processes. One of the possible applications is evaluation of physical and chemical properties of processes and its components, like modeling of char combustion (Jovanović et al. 2015). Moreover, they are also useful in clarification of fundamentals of processes, like: coalification, pyrolysis and char formation, liquefaction and solvent swelling, carbon dioxide sequestration and coalbed methane or spontaneous combustion (Mathews et al. 2011; Mathews and Chaffee 2012). Recently, GCMC simulations were found to give satisfactory results in characterization of a carbonaceous materials structure, like activated carbon (Palmer et al. 2009), and also in adsorption studies, where Lennard-Jones potentials are applied for modeling of adsorbent walls (Di Biase and Sarkisov 2013). Moreover, molecular dynamics and GCMC may be applied for analysis of permeability of CO2 through a coal barrier (Zhang et al. 2016) and gives opportunity to evaluate a competitive adsorption of CO2–CH4 mixture (Zhao et al. 2016a). The distribution of ultramicropores (exchangeable nomenclature of submicropores) used by (Zhao et al. 2016a) and obtained by (Castro-Marcano et al. 2012), in HRTEM microscopy imaging of coal lattice, give the structure nearly identical to that used in our paper.
Chemical and structural complexity of hard coals is continuously attracting an interest of a great deal of researchers, due to wide spectrum of present and possible applications (Baran et al. 2014). Recently, a decreasing demand for high-grade coking coal or production of generator gas was observed. On the other hand, there is a growing demand for hard coal applicability in active carbon production or flue gas purification in coal-fired power stations. Furthermore, recently a significant interest was aimed at development of underground storage in coal seams, to reduce and mitigate greenhouse gases (Corum et al. 2013; Zhao et al. 2016b). Nevertheless, the practical use of hard coals has to be supported by material and sorption system properties characterization, for the most effective and reliable applications. Therefore, a significant interest has been concentrated on elaboration of the most reliable numerical tools for hard coals properties characterization, considering absorption-adsorption phenomena commonly occurring in a coal matter (Milewska-Duda and Duda 1993; Santarosa et al. 2013).For these reasons, the author’s team is currently working on continuous development of multiple sorption model (MSM) aimed at hard coal structure identification and sorption system properties characterization (Jodłowski et al. 2016). The applicability of MSM to identify of various sorption systems parameters is examined, as well as its use for estimation of coal deposits capacity in carbon dioxide sequestration. MSM makes it possible to draw a picture of hard coal structure, as well to estimate energetic parameters of sorption phenomena, including possible simultaneous subprocesses: adsorption, absorption and expansion. Despite, MSM is originated from physicochemical considerations, a calculation procedure itself requires preliminary assumptions on energetic parameters and basic knowledge about an investigated coal structure. Simulations performed applying MSM model requires initial assumptions on thermodynamic parameters values of sorption system. Some of the values are taken from the literature and the remaining part comes from our earlier works (Hombach 1980; Van Krevelen and Te Nijenhuis 2009; Milewska-Duda and Duda 1997). An application of new methods for more precise setting of initial values of these parameters facilitates a simulation procedure and enables to obtain more reliable simulation results.
Simultaneously, team’s works are concentrated on development and applicability of a LBET class adsorption model for microporous heterogeneous structures examination (also implemented in MSM). The basis for the model elaboration is to consider localized adsorption phenomenon for materials of random heterogeneous structure (Duda et al. 2013). The latter, comes from the intuitive understanding of energetical heterogeneity coming from geometrical heterogeneity for real porous materials, such as active carbons. Hence, recently the Author’s team works are focused on the interpretation: in what extent surface diversity effects are correlated with adsorption phenomenon mechanism (Ziółkowska et al. 2016a, b). Such a qualitative interpretation of the adsorption mechanism, gives a cognitive basis for selection of the most appropriate mathematical model for reliable examination of material porous structure.
Having regard the coal structure complexity and heterogeneity, a surface structure picture driven by adsorption mechanisms analyses is considered to be applicable in improvement of MSM parameters determination. In particular, adsorbate cohesion energy and volume are implemented in MSM, making it possible to reduce the uncertainty of estimates and more reliable coal structure identification and sorption system characterization.
2 Experimental isotherms
Sorption isotherms for coal M85 and W42 are chosen from the set of experimental isotherms from our earlier works (Wójcik 1999) and Ceglarska-Stefańska works (Ceglarska-Stefańska 1990). M85 and W42 hard coals are structurally rigid samples due to high content of macromolecular phase, and consecutively high content of arene domains (anthracite of Cdaf≈92.4). This property is required for making possible to determine adsorbate cohesion energy and volume, applying qualitative approach to adsorption mechanism identification (more detailed see Sect. 3.3). M85 and W42 samples parameters are gathered in Table 1.
Measurements of carbon dioxide sorption isotherms on M85 were performed up to 3.5 MPa at 298 K, after degassing of the sample to 0.001 Pa, on a manostate apparatus. Experiment was carried out on coal cube samples of size 15 × 15 × 5 mm. In turn, measurements of methanol sorption on W42 were led at an atmospheric pressure and temperature 298 K, after degassing of the sample to 0.001 Pa, on the microburette apparatus Table 2.
Empirical isotherms are presented in Sect. 4 and compared to theoretical isotherms obtained in MSM simulations.
3 Numerical analysis
3.1 Multiple sorption model
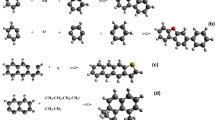
Let us consider sorption phenomena in coals as coexisting subprocesses of sorbate particles absorption, frequently accompanied by the bulk coal expansion in the elastic part, and adsorption in a rigid part of the coal structure. Therefore, to compromise a wide spectrum of assumed simultaneous subprocesses, rigid and elastic parts of the coal structure may be seen as copolymer components (see Fig. 1). Subsequently, the rigid part may be represented by semi-crystalline arene domains (1), occurring as condensed benzene rings, that forms limited inflexible network together with interconnected aliphatic, alicyclic and heterocyclic chains (2). Inter-network spaces are filled with the elastic molecular phase of non-interconnected aliphatic, alicyclic and heterocyclic chains (3). Simultaneously, incorporated pores (4) and mineral admixtures (5) are independently treated, as a part of the coal structure, as well as sorbate particles (6) able to occupy any point of the network space, besides arene domains volume (1). Hence, absorption-adsorption phenomena may be seen as mixing of sorbate particles (6) with individual components of the carbonaceous material (1–5). In order to examine such sorption system, a consistent approach so called MSM, was elaborated in Author’s team (Milewska-Duda et al. 2000; Milewska-Duda and Duda 1993).

Schematic representation of a copolymeric coal structure consisting of individual components: arene domain (1), interconnected aliphatic, alicyclic and heterocyclic chains (2)—black color, noninterconnected aliphatic, alicyclic and heterocyclic chains (3)—blue color, pore (4) red color and mineral admixture (5)—green. (Color figure online)
In particular, coming from general equilibrium thermodynamics, sorption system in MSM is considered comparing to a standard state of pure liquid-like sorbate and dry sorbent. Thus, the energy change due to sorption is considered as a hypothetical three-stage process (see Fig. 2), separately for each indicated subprocess of the adsorptive-absorptive nature and adsorptive or absorptive nature, only. Hence, let us consider energy of sorbate molecule deposition, expressed by the dimensionless energetic parameter χ ac , as a separation of the sorbate molecule from its liquid-like reference state (i.e., evaporation) at the first stage, creation of a hole (i.e., room) for this molecule within the sorbent matter (expansion) and placing the molecule in the created hole (condensation) in second and third stage, respectively (Milewska-Duda et al. 2000; Milewska-Duda and Duda 1997, 1993).

Description of a hypothetical three-stage process of pure liquid-like CO2 sorption on the coal surface. First stage corresponds to the evaporation, viewed as the sorbate molecule separation from its liquid-like reference state and transition to the hypothetical vacuum. Second stage concerns possible creation of the space for this molecule within the sorbent matter i.e., pore expansion. The last stage refers to the molecules transition from the hypothetical vacuum, its condensation and placement of molecules in the designed space. Schematic representation of a copolymeric coal structure consists of individual components: arene domain, interconnected aliphatic, alicyclic and heterocyclic chains—black color, noninterconnected aliphatic, alicyclic and heterocyclic chains—blue color, pore red color and mineral admixture—green. (Color figure online)
A separation of the sorbate molecule from its liquid-like reference state and its transition to a vacuum, at the first stage (Fig. 2 Evaporation), has purely cohesive nature. Simply, it is defined as sorbate molar evaporation energy and may be expressed by a dimensionless energetic parameter χ ph :
where δ p is sorbate solubility parameter, V s is sorbate molar volume in sorption system, R is a gas constant and T is temperature.
At the second stage, the nature of each subprocess may be expressed parameterizing an effective radius of a created hole R ha (Fig. 2 Expansion). In particular, effective radius describes in what extent this hole needs to be expanded to place sorbate molecule of effective radius R p . Quantitatively, it may be represented by a surface expansion ratio ω a , expressed by the following formulae:
An expansion of the hole is energetically diversified due to possibility of its surrounding by different individual components of the carbonaceous matter (Fig. 1 1–5). Therefore, it is dependent on the surface expansion ratio ω a and may be related to the energy required to overcome cohesive forces of the sorbent matter. Hence, following theory of solutions (Milewska-Duda and Duda 1997), average energy of the hole, in sorbent matter, ready to place sorbate molecule may be defined by a dimensionless energetic parameter χ ch :
where δ c is sorbent solubility parameter that may be determined experimentally and theoretically, depending on a various carbon contents Cdaf (%) in coal (Van Krevelen and Te Nijenhuis 2009).
In fact, the parameter expressed in Eq. (3) is a reduced form for pure absorption, where radius R ha = 0 and ω a = 1. Nevertheless, non-isotropic expansion has to be taken into account in case of smaller and bigger holes, using a correction factor f(R ha ) (i.e., if R ha = 0 the correction factor f(R ha ) = 1).
Now let us recall the third-stage, of sorbate molecule placement in the created hole (Fig. 2 Condensation). For nonpolar liquid, energy evolved due to mixing of sorbate molecule with hole placed at the coal matter may be evaluated combining, adsorbate and adsorbent cohesion energies via Berthelot rule (i.e., simply by virtue of Eqs. (2) and (3), taking into account dimensionless parameters). Therefore, sorbate and sorbent molecules are viewed as being in full and ideal contact, and hence possible nonideality coming from losses in contact due to irregularities of the sorbent walls should be taken into account. To this aim, the correction factor ζ(R ha ,R p ) ≤ 1 may be defined, expressing the effective contact fraction of sorbate and sorbent molecules. In particular, the correction factor describes both geometric and energetic properties of the surface and bulk sorbent matter, distinguishing four regions denoted with (I)–(IV):
where R B is a characteristic pore size of the particular coal sorbent sample; Z A and Z B stand for the energy correction in smaller (R ha < R B ) and bigger (R ha > R B ) pores due to geometrical constraints, respectively. (I) is a pure absorption for R ha = 0; (II) is an expansion of submicropores smaller than the size of R B and effective radius of a created hole is 0 < R ha < R B ; (III) is an expansion of submicropores larger than the size of R B and effective radius of a created hole is R B < R ha < R p ; (IV) is a pure adsorption for R ha ≥ R p . Finally, correction factors accounting for an interactions polarity are C pa and C pA . More detailed see (Jodłowski et al. 2016).
Concerning the proposed description of each of the three-stage processes, the overall thermodynamic description of the energy change due to sorption may be expressed by unit-less term (Jodłowski et al. 2016; Milewska-Duda and Duda 1997):
where \(\phi _{h}^{0}\)° is the initial holes fraction in the coal matter and parameter χ ac is a generalization of a Flory–Huggins parameter for porous sorbents (i.e., assumption \(\phi _{h}^{0}=0\) leads to the description of purely absorptive system).
A description of purely adsorptive subprocesses in fourth region (IV) in Eq. (4) was recently strongly developed in Author’s team, both as a completion of MSM, as well as individual numerical tool aimed at identification of materials porous structure, referred as LBET class adsorption models. The LBET approach is far generalization of classical BET formulae enabling as well to consider, multilayer adsorption in bigger pores and also a non-BET stacks formation (Duda et al. 2013; Milewska-Duda et al. 2000).
Reliable and accurate modeling applying MSM with LBET extension for multilayer heterogeneous adsorption has to be completed interpreting liquid-like reference state. To this aim suitable equation of state has to be applied, thus enabling for evaluation of sorbate molar volume and cohesion energy. For a convenience, in order to get these quantities simultaneously, Author’s team elaborated and implemented in MSM, an original PVT relationship for compressed fluids properties characterization (Milewska-Duda and Duda 2009).
MSM computation procedure requires seven fitting parameters, inter alia i.e., R B , Z A , Z B -determined based on empirical data. The simulation is preformed until satisfactory agreement is reached between the experimental and simulated isotherms; as well identified parameters of a sorption system are in a good agreement with general information on the coal structure coming from our database of optimization experiments (Jodłowski et al. 2013).
3.2 Qualitative approach to adsorption mechanism identification
Now, let us consider an individual particle of a volatile phase (adsorptive molecule) freely moving in space V fi of purely adsorptive system, i.e., near geometrically heterogeneous rigid adsorbent. Moreover, let us assume equally probable finding of its mass center at any variables v of the phase-space such that v ≤ V fi , excluding possible collisions with other adsorptive particles. Hence, the space of free movement V fi is bounded by an equipotential surface U bdry (V fi ), at which kinetic energy of two degrees of freedom k B τ i is equilibrated by a decrease in an average solid–fluid potential U avge (V fi ). In such a view, kinematic equilibrium of the particle free movement may be expressed by the following formulae (Ziółkowska et al. 2016a)
where kinetic energy term is represented by a Boltzmann constant k B and average particle “temperature” τ i :
To evaluate average U avge (V fi ) and boundary U bdry (V fi ) potentials, at the given variables v of the phase-space, geometrically heterogeneous adsorbent is represented by a set of carbon atoms forming a predefined nanostructure. Therefore, U bdry (V fi ) may be taken arbitrary enabling to evaluate U avge (V fi ), following:
where u C−fk is Lennard-Jones potential (Lennard-Jones 1931) for single interaction of molecule mass center, at a given k-th position, with carbon atom of the adsorbent surface.
So far, the movement of single particle is taken under considerations it may be assumed that there are no particles in volatile phase, i.e., p = 0. In this case, having the values for the potentials in Eq. (8), the average temperature τ i corresponding to a given V fi may be calculated. For a convenience, let us further use previously defined quantities recalculated to a particle volume, \({{V}_{fi}}\to {{V}_{pi}},\) and next relate them to 1 mol of particles multiplying by Avogadro constant N A . Hence, kinematic equilibrium temperature τ i may be expressed (Ziółkowska et al. 2016a, b):
where R is a gas constant.
An inverse relationship V mpi (τ i ) from Eq. (9) constitutes criteria for adsorption mechanism identification (see Fig. 3), considering volume V mpi (τ i ) < V ind as corresponding to the localized mechanism and V mpi (τ i ) < V max to the possible mobile mechanism. This interpretation follows a reasonable assumption that it is not likely that the space V mpi (τ i ) may be shared with any other molecules when is less or equal to average volume of particle movement at a critical point, i.e. V ind = V c . Otherwise, the space V mpi (τ i ) may be occupied by individual molecules or shared with other j molecules, i.e. j = V mpi (τ i )/V ind assuming that volume V ind corresponds to the maximum separated volume of 1 mol of individual particles. To get more familiar with the considerations underlying a determination of criteria for adsorption mechanism identification, see (Ziółkowska et al. 2016a).

Adsorption mechanism identification scheme based on the relationship V mpi (τ i ), calculated applying Eq. (9). For V mpi (τ i ) < V ind localized monolayer adsorption takes place and V ind is a maximum volume of 1 mol of individual particles that occupies localized sites, at the temperature τ ind . V ind < V mpi (τ i ) < V max may be seen as purely mobile or closer to the clustering based or to locally mobile adsorption mechanisms and V max is a maximum volume of 1 mol of individual particles that occupies or share localized sites, at the temperature τ max
When considering isothermal adsorption, an excess energy evolved due to the adsorbate particle interaction with adsorbent surface may be seen as totally withdrawn to keep the constant temperature T. Simultaneously, having regard to accepted assumptions, the volume V mpi and the quantities U madh (V mpi ), U m (V mpi ) may be seen as an additive (in the range τ ind < τ i ≤ τ max the quantities has to be proportionally shared in terms of the number of j particles, to keep the consistency of the description). Hence, it makes it possible for averaging of the quantities in Eq. (6) with Maxwell–Boltzmann distribution function f(τ i ,T) at a given temperature T, more detailed see (Ziółkowska et al. 2016a). Assuming that τ i temperature distribution is the Maxwell–Boltzmann distribution, for each i-th particle being in the kinematic equilibrium (i.e. at the intervals between collisions with other particles of volatile phase), averaging is performed in analogy to the formulae:
where ̅. refers to quantity averaged over a number of particles N ind that corresponds to the particular temperature τ i , such that τ i ≤ τ ind .
Volume \({{\overline{V}}_{{{m}_{p}}}}\), evaluated with Eq. (10), is considered to be adsorbate molar volume in real adsorption system of localized mechanism. Moreover, it may be employed for evaluation of surface texture parameter ζ, constituted in our previous papers as suitable for the approach validation (Ziółkowska et al. 2016a, b) The definition of ζ comes from the elaborated in author’s team approach aimed at the localized mechanism modeling, exploited in MSM and LBET modeling (Duda et al. 2013; Jodłowski et al. 2016.) Thus, once more let us consider adsorption phenomenon as a condensation of adsorbate molecules being in the reference liquidlike state and mixing of these molecules with empty cells placed at the adsorbent surface (see, sec. 3.1, FFig. 2. In such a view, ζ may be seen as an adsorbate-adsorbent effective contact ratio, defined taking the values for real effective adhesion energy U̅ madh (V̅ mp ) and ideal contact adhesion energy E̅ adh (V̅ mp ):
where adhesion energy of ideal adsorbate-adsorbent contact is evaluated with Berthelot rule:
and E vap is the adsorbate cohesion (evaporation) energy.
The values for V̅ mp and E vap refer to the adsorbate reference state, i.e. liquid state in case of vaporous substances, and the latter is evaluated with PVT relationship for liquid bulk phase (Milewska-Duda and Duda 2009).
3.3 Determination of multiple sorption model parameters
A number of parameters employed in the computation procedure applying MSM may cause an uncertainty of estimates. Thus, in order to get valuable information on particular sorption system it is necessary to complete the procedure by reliable assessment of the resultant model’s parameters. So far, information gained on the coal structure and sorption system was evaluated verifying general agreement of the results with the database of optimization experiments, coming from our earlier works (Jodłowski et al. 2013). Therefore, one may expect that team’s works should be focused on enhancement of the evaluations reliability, to reduce uncertainty and improve the quality of the estimates. To this aim, a qualitative interpretation of adsorption mechanisms is considered as a promising tool.
Let us consider again, the coal structure as copolymer consisting of rigid and elastic parts schematically shown in Fig. 1. In particular, the rigid part of the structure represented by semi-crystalline arene domains may be easily interpreted in view of the qualitative approach to adsorption mechanism identification. Thus, let us assume that condensed benzene rings of arene domains may be represented by carbon atoms spaced like in a graphene, forming a graphite-like structure or active-carbon structure of a predefined shape. Moreover, let us accept that the coal structure may be represented as system of such structures (Fig. 4), enabling to employ in MSM modeling surface structure parameters determined from qualitative approach to adsorption mechanism identification.

A graphite sample (left) and active carbon (right) models (Ziółkowska et al. 2016b), composed of carbon atoms at fixed positions (i.e., chemical heterogeneity is neglected)
In particular, the results presented in our earlier works were taken under considerations (Ziółkowska et al. 2016b) for more precise setting of the adsorbate volume V̅ mp , calculated with Eq. (10), and cohesion energy E vap from Eq. (12). To this aim graphite sample (GS) and active carbon (AC) models were selected as the closest representation of arene domain structure (see, Fig. 4). Analyses for CH3OH and CO2 interaction with GS and AC models were performed on MATLAB® platform, employing software elaborated in author’s team with a Monte-Carlo calculation technique, more detailed see (Ziółkowska et al. 2016b).
What concerns determination of the cohesion energy E vap with PVT relationship for liquid bulk phase (Milewska-Duda and Duda 2009), in this approach the adsorbate volume V̅ mp is treated as a fitting parameter. Thus, in low range of temperatures this may result in inadequate description, since PVT formulas are dedicated for liquid bulk phase only. A calculation results seems to confirm this expected feature, especially when analyzing the relationship for CH3OH, see Fig. 5. However, at the given temperature of experimental isotherms measurements (see sect. 2), the discrepancy in E vap (V̅ mp ,T) is no longer observed.

Cohesion energies E vap (V̅ mp ,T) for CH3OH interacting with carbon atoms of GS model (dashed black line) and AC model (dashed blue line) and CO2 interacting with carbon atoms of GS model (dotted black line) and AC model (dotted blue line) evaluated with PVT relationship for liquid bulk phase (Milewska-Duda and Duda 2009) applying fitting parameter V̅ mp determined with Eq. (10). (Color figure online)
4 Results and discussion
In our earlier papers (Ziółkowska et al. 2016a, b) it was shown that rigorous examination of geometrical heterogeneity effects, when adsorption mechanism identification, makes it possible to describe conditions for localized and possible mobile mechanisms. Moreover, when considering more complex, individual geometrical heterogeneity, this approach is able to detect more than one active adsorption site, and due to assumption of the adsorbate volume V̅ mp additivity is well determinable. However, when employing total volume V̅ mp for determination of E vap (V̅ mp ,T), the description inconsistency is recognized due to discrepancy in evaluation of the volume corresponding to the particular site. Nevertheless, an ambiguity area in an interpretation of these results is not heavily enlarged when the above-mentioned quantities are considered to be total for the particular heterogeneity.
MSM simulations were performed for M85-CO2 and W42–CH3OH sorption systems (see, Sect. 2). To this aim and simultaneously, in order to show in what extent adsorption mechanism analyses are applicable for determination of MSM parameters, the cohesion energy evaluated for GS (1) and AC (2) models is applied as an initial parameter. Furthermore, consecutive simulations are performed correcting remaining parameters of the model to obtain the most satisfactory fitting quality (see, Table 3—variants 1 and 2, respectively). Finally, the obtained results are compared to the best fitted simulated total sorption isotherm (3) and then to the original simulation with optimal parameters for the experimental data fitting with preliminary assumptions (4). On the basis of preliminary analysis of the resultant parameters in Table 3, one may observe a significant reduction in the absorption level, when compared (1), (2) and (3) to (4) variant. It is related to the representation of the arene domains component by much more rigid structures, corresponding to the increase of the sorbent’s cohesion energy density \(\delta _{c}^{2}\). Moreover, such a significant change in \(\delta _{c}^{2}\) value enforces further modifications in parameters describing surface geometrical structure, due to transfer of the molecules from the absorption system to the other subsystems.
For M85—CO2 sorption system the density of the cohesion energy, used in this study, appears to be too high in relation to the data presented by Van Krevelen and Hombach (Van Krevelen 1965, Hombach 1980) on the basis of a coal solvent extraction. It results in significant decrease of absorption contribution in sorption process \({}^{abs\left( 0.8 \right)}\!\!\diagup\!\!{}_{sorp\left( 0.8 \right)}\;\) from 15% to less than 1.5% (see Table 3; Fig. 6), due to increase of the molecular phase cohesion forces and appearing of obstacles for creation of new holes in the coal matter (namely, caused by increase of absorption energy parameter, see Q abs Table 3). Moreover, a decrease of amount of the sorbate molecules in the absorption subsystem is related with its migration to the expansion subsystems, excluding possible migration to the adsorption one (see Fig. 7). Therefore, characteristic radius of distribution is shifted up to the value bigger than the molecule size and may be observed in Fig. 8 on distribution charts for an A parameter (namely, the expansion is due to energetical constraints rather than geometrical). Furthermore, one may notice, a low decrease in adsorption subsystem resulting from a decrease of adsorption energy parameter χ ac . This is not observed in case of MSM parameters applied from AC model, due to completely rigid structure assumption. Hence, the value of geometric fitting of molecules to the pores ζ A (Fig. 9) turned out to be unreliably high for hard coals, even when anthracite is taken under considerations. Simultaneously, Q bc energy distributions vs. submicropores radius R ha /R p is slightly shifted on both cohesion (positive value) and adhesion (negative value) energy sides, see Fig. 9.

Multisorption isotherms(sorption in physical subsystems) of CO2 on M85 coal sample for MSM parameters determined applying GS (1) and AC (2) models, the best fitted simulated total sorption isotherm (3) and original simulation with optimal parameters for the preliminary assumptions (4)Circles—experimental sorption, black solid line—simulated total sorption, red solid line—hypothetical adsorption (obtained by subtracting the absorption of the total sorption, like in the dual sorption model), black dotted line—simulated adsorption, magenta dotted line—simulated monolayer adsorption, black dashed-dotted line—total expansion with absorption (pure expansion is calculated by subtracting the absorption from total expansion), green solid line—absorption. (Color figure online)

Sorption isotherms of CO2 on M85 coal sample in virtual subsystems (isotherms distribution) for different classes of pores for MSM parameters determined applying GS (1) and AC (2) models, the best fitted simulated total sorption isotherm (3) and original simulation with optimal parameters for the preliminary assumptions (4)

Sorption capacity distribution A and function of geometrical correction factors ζ(R ha ,R p ) of CO2 sorption on M85 for MSM parameters determined applying GS (1) and AC (2) models, the best fitted simulated total sorption isotherm (3) and original simulation with optimal parameters for the preliminary assumptions (4)

Submicropores volume distribution U h (red line—total volume, green bars—normalized distribution) and sorption energy parameters Q bc of CO2 sorption on M85 for MSM parameters determined applying GS (1) and AC (2) models, the best fitted simulated total sorption isotherm (3) and original simulation with optimal parameters for the preliminary assumptions (4). (Color figure online)
For W42—CH3OH sorption system MSM simulations were performed applying calculation scheme analogous as for M85—CO2 system. Interestingly it turned out that the best fitted simulated total sorption isotherm, denoted as variant number (3) in the calculation scheme, is the isotherm evaluated for MSM parameters determined applying GS model—variant (1), see Fig. 10. A resultant simulation estimates are observed to be substantially amended, although the density of the cohesion energy is less distinctive comparing to M85—CO2 system. In turn, in Fig. 11 a significant modification in expansion subsystems may be observed for AC model—variant (2), due to change of the cohesion energy density contributing in evaluation of molecule contacts energy via Berthelot rule. However, the isotherms distribution for sorption systems with parameters determined from GS model (1) and original cohesion energy variant (4), are of substantial similarity. It is related to the assignment of the same value describing characteristic pore size parameter R B in both cases. On the other hand, the isotherms distribution for variant with parameters determined from AC model (1) may be seen as completely inappropriate, due to unreliably high values of molecule contacts Z B (see Table 3) as well as characteristic submicropore radius R B , making it impossible to fit theoretical isotherms to experimental data with no effect on simulation assumptions. Moreover, estimated energy parameters are not coherent in this variant, namely if energy coefficient of pores χ ch (meaning the energy of adsorption centers), and energy of molecule-pore contacts χ ac are low, one should expect low values for Q abs absorption energy. Despite for AC model (1) the absorption energy Q abs is relatively high, the absorption capacity turns out to be inappropriately big (i.e., one should expect the decrease in the absorption capacity for increasing absorption energy Q abs ).

Multisorption isotherms (sorption in physical subsystems) of CH3OH on W42 coal sample for MSM parameters determined applying GS (1) and AC (2) models, original simulation with optimal parameters for the preliminary assumptions (4). Circles—experimental sorption, black solid line—simulated total sorption, red solid line—hypothetical adsorption (obtained by subtracting the absorption of the total sorption, like in the dual sorption model), black dotted line—simulated adsorption, magenta dotted line—simulated monolayer adsorption, black dashed-dotted line—total expansion with absorption (pure expansion is calculated by subtracting the absorption from total expansion), green solid line—absorption. (Color figure online)

Sorption isotherms of CH3OH on W42 coal sample in virtual subsystems (isotherms distribution) for different classes of pores for MSM parameters determined applying GS (1) and AC (2) models, original simulation with optimal parameters for the preliminary assumptions (4)
In order to obtain good fitting of the simulated isotherm to the experimental data with AC model (1), parameters describing geometrical properties of the molecule in the sorption system, Z A and Z B , (see Table 3) have to be changed when submicropores distribution in coal matter R B is shifted. As a result of these changes a different picture of submicropores capacity distribution in expansion subsystems may be observed (Figs. 11, 12, see variant (2)), as well in estimated starting pore volume distribution U h and energy profile Q bc in submicropores (Fig. 13).

Sorption capacity distribution A and function of geometrical correction factors ζ(R ha ,R p ) of CO2 sorption on M85 for MSM parameters determined applying GS (1) and AC (2) models, original simulation with optimal parameters for the preliminary assumptions (4)

Submicropores volume distribution U h (red line—total volume, green bars—normalized distribution) and sorption energy parameters Q bc of CH3OH sorption on W42 for MSM parameters determined applying GS (1) and AC (2) models, original simulation with optimal parameters for the preliminary assumptions (4). (Color figure online)
Summarizing, the results of application of new initial parameters for estimation of the cohesive energy are more significantly affected for CO2 sorption than for CH3OH. A substantial decrease in absorption was observed for new estimates of CO2, while for CH3OH sorption only minor modification of the initial parameters turned out to be sufficient to obtain optimal fitting of simulated isotherm. Moreover, it should be noted that cohesive energy for CO2 sorption significantly increases in all variants, i.e. from 22.65 to 28.00 [MPa 1/2], whereas for CH3OH only slight modification is observed from 22.86 to 21.66 [MPa 1/2] for AC model—variant (2) and 23,307 [MPa 1/2] for GS model in variant (1). For CO2 sorption, it became two-fold increase in the absorption energy, and so one should consider reducing the values for the cohesive energy density in accordance to Van Krevelen and Hombach publications (Hombach 1980; Van Krevelen and Te Nijenhuis 2009).
5 Conclusions
The results of studies on adsorption energy, presented in (Di Biase and Sarkisov 2013), are similar to results obtained in our investigations (χ ac *RT = 12.4 kJ/mole at temperature T = 298 K and pressure p = 2.87 MPa) and this is due to an agreement of assumed structure of macromolecular phase. Nevertheless, it has to be taken into account that the presence of surface functional groups is also included in this approach, linking the coal structure picture to real hard coals. Density Functional Theory may be also used for modeling of adsorption on heterogeneous surface of coal (with Monte Carlo method), and the resultant adsorption energy for CO2 is about 16.4 kJ/mole (Liu et al. 2016). On the other hand, the adsorption energy reported in (Xiang et al. 2014), is found to be much higher − 37 kJ/mole. This might be due to allowance of interactions with surface functional groups, and hence adsorption energy has analogous value to the water adsorption case (Xiang et al. 2014).
The development of the numerical tools presented in this paper is a significant progress in works lead in the Author’s team, aimed at mathematical modeling of sorption phenomena. In particular, two independent approaches are combined implementing the viewpoint on the surface heterogeneity from adsorption mechanism identification procedure to describe hard-coals sorption system. Thus, it enables for evaluation of the surface heterogeneity impact on the energetic parameters of adsorption subsystems and to analyze hard coal structure as a complex bulk system with arbitrary taken energy distribution parameters The fitting of theoretical isotherms to experimental data with new assumptions gives promising results, but further works has to be focused on improvement of introducing parameters in model. This is due to serious underestimation of absorption level and overestimation of expansion subsystem content, when the originally derived energetic parameters are applied. In particular, the results for active carbon and graphite sample models were applied from adsorption mechanism identification procedure. It turned out, that optimally fitted theoretical description for M85 and W42 hard-coals samples is closely related to the graphite sample model but in case of M85 sample is not equivalent. Although graphite sample model assumption gives good results in simulations for both M85 and W42 samples, the result with very small participation of carbon dioxide absorption phenomenon in M85—CO2 sorption system is not in an agreement with commonly known properties. This property of simulated sorption system needs further examination.
References
Baran, P., Jodłowski, G.S., Krzyżanowski, A., Zarębska, K.: Experimental testing of methanol sorption on selected coal samples from Upper Silesian Basin. Geol. Geophys. Environ. 40, 261–269 (2014)
Castro-Marcano F., Winans R.E., Chupas P., Chapman K., Calo J.M., Watson J.P., Mathews J.P.: Fine structure evaluation of the pair distribution function with molecular models of the Argonne premium coals. Energy Fuels 26, 4336–4345 (2012)
Ceglarska-Stefańska G.: Współzależność procesów sorpcyjnych i dylatometrycznych zachodzących w układach: węgle kamienne—para wodna, dwutlenek węgla, metan. Zesz. Nauk. AGH (1990)
Corum, M.D., Jones, K.B., Warwick, P.D.: CO2 sequestration potential of unmineable coal-state of knowledge. Energy Procedia 37, 5134–5140 (2013)
Di Biase, E., Sarkisov, L.: Systematic development of predictive molecular models of high surface area activated carbons for adsorption applications. Carbon. 64, 262–280 (2013)
Duda, J.T., Milewska-Duda, J., Kwiatkowski, M., Ziółkowska, M.: A geometrical model of random porous structures to adsorption calculations. Adsorption 19, 545–555 (2013)
Hombach, H.-P.: General aspects of coal solubility. Fuel. 59, 465–470 (1980)
Jodłowski, G.S., Baran, P., Wójcik, M., Nodzeński, A., Porada, S., Milewska-Duda, J.: Sorption of methane and carbon dioxide mixtures in Polish hard coals considered in terms of adsorption-absorption model. Langmuir. 13, 5732–5735 (2013)
Jodłowski, G.S., Wójcik, M., Orzechowska-Zięba, A.: Identification of hard coal surface structure using polar and apolar small molecule substances. Adsorption 22, 847–854 (2016)
Jovanović, R., Marek, E., Maletić, S., Cvetinović, D., Marković, Z.: Lattice Monte Carlo simulation of single coal char particle combustion under oxy–fuel conditions. Fuel. 151, 172–181 (2015)
Lennard-Jones, J.E.: Cohesion. Proc. Phys. Soc. 43, 461–482 (1931)
Liu, X.-Q., Heb, X., Qiu, N.-X., Yang, X., Tian, Z.-Y., Li, M.-J., Xue, Y.: Molecular simulation of CH4, CO2, H2O and N2 molecules adsorptionon heterogeneous surface models of coal. Appl. Surf. Sci. 389, 894–905 (2016)
Mathews, J.P., Chaffee, A.L.: The molecular representations of coal—a review. Fuel. 96, 1–14 (2012)
Mathews, J.P., van Duin A.C.T., Chaffee, A.L.: The utility of coal molecular models. Fuel Process. Technol. 92, 718–728 (2011)
Milewska-Duda, J., Duda, J.: Mathematical modeling of the sorption process in porous elastic materials. Langmuir. 9, 3558–3566 (1993)
Milewska-Duda, J., Duda, J.: Hard coal surface heterogeneity in the sorption process. Langmuir. 13, 1286–1296 (1997)
Milewska-Duda, J., Duda, J.T.: High-accuracy PVT relationships for compressed fluids and their application to BET-like modelling of CO2 and CH4 adsorption. Adsorpt. Sci. Technol. 25, 543–559 (2009)
Milewska-Duda, J., Duda, J.T., Jodłowski, G.S., Kwiatkowski, M.: Model for multilayer adsorption of small molecules in microporous materials. Langmuir. 16, 7294–7303 (2000)
Palmer, J.C., Brennan, J.K., Hurley, M.M., Balboa, A., Gubbins, K.E.: Detailed structural models for activated carbons from molecular simulation. Carbon. 47, 2904–2913 (2009)
Reid, R.C., Prausnitz, J.M., Poling, B.E.: The properties of gases and liquids. McGraw-Hill, New York (1987)
Santarosa, C.S., Crandall, D., Haljasmaa, I. V., Hur, T.B., Fazio, J.J., Warzinski, R.P., Heemann, R., Ketzer, J.M.M., Romanov, V.N.: CO2 sequestration potential of Charqueadas coal field in Brazil. Int. J. Coal Geol. 106, 25–34 (2013)
Van Krevelen, D.W.: Chemical structure and properties of coal. XXVIII-Coal Constitution and Solvent Extraction. Fuel 44, 229–242 (1965)
Van Krevelen, D.W., Te Nijenhuis, K: Properties of Polymers. Elsevier, Amsterdam (2009)
Wójcik, M.: Application of sorption models for analysis of surface properties of hard coals, PhD thesis, AGH Univeristy of Science and Technology, Kraków, Poland (1999)
Xiang, J.H., Zeng, F.G., Liang, H.Z., et al.: Molecular simulation of the CH4/CO2/H2O adsorption onto the molecular structure of coal. Sci. China. Earth Sci. 57, 1749–1759 (2014)
Zhang, J., Clennell, M.B., Liu, K., Dewhurst, D.N., Pervukhina, M., Sherwood, N.: Molecular dynamics study of CO2 sorption and transport properties in coal. Fuel. 177, 53–62 (2016)
Zhao, Y., Feng, Y., Zhang, X.: Selective Adsorption and selective transport diffusion of CO2–CH4 binary mixture in coal ultramicropores. Environ. Sci. Technol. 50, 9380–9389 (2016a)
Zhao, Y., Feng, Y., Zhang, X.: Molecular simulation of CO2/CH4 self- and transport diffusion coefficients in coal. Fuel. 165, 19–27 (2016b)
Ziółkowska, M., Milewska-Duda, J., Duda, J.T.: A qualitative approach to adsorption mechanism identification on microporous carbonaceous surfaces. Adsorption 22, 233–246 (2016a).
Ziółkowska, M., Milewska-Duda, J., Duda, J.T.: Effect of adsorbate properties on adsorption mechanisms: computational study. Adsorption 22, 589–597 (2016b).
Acknowledgements
The research is led within the AGH University of Science and Technology Grant No. 11.11.210.213.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial or non-financial interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jodłowski, G.S., Ziółkowska, M. Characterization of hard coal properties applying multiple sorption model with parameters determined from qualitative approach to adsorption mechanism identification. Adsorption 23, 381–393 (2017). https://doi.org/10.1007/s10450-017-9869-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-017-9869-2