
Abstract
Experimental data of nitrogen adsorption (T = 77.3 K) from gaseous phase measured on commercial closed carbon nanotubes are presented. Additionally, we show the results of N2 adsorption on compressed (using hydraulic press) CNTs. In order to explain the experimental observations the results of GCMC simulations of N2 adsorption on isolated or bundled multi-walled closed nanotubes (four models of bundles) are discussed. We show that the changes of the experimental adsorption isotherms are related to the compression of the investigated adsorbents. They are qualitatively similar to the theoretical observations. Taking into account all results it is concluded that in the “architecture” of nanotubes very important role has been played by isolated nanotubes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Carbon nanotubes (CNTs) are materials of specific structure and properties, making them promising media for highly selective and efficient adsorption and/or transport processes (Harris 2009; Wesołowski et al. 2010). It should be pointed out that the experimental (e.g. high-resolution transmission electron microscopy (HRTEM), adsorption porosimetry, and microcalorimetry measurements) and theoretical investigations (quantum calculations and computer simulations) indicate that the structure of these materials is, unfortunately, very complex. It is well-known that many factors should be considered, i.e. mutual position of nanotubes in the bundle, the outer and inner diameter of tubes, the number of walls, defects, an unavoidable by-products of a typical nanotube growth process i.e. impurities, such as carbon-coated catalyst particles, graphitic and amorphous carbons, and other carbon nanomaterials (Charlier 2002; Harris 2009; Krungleviciute et al. 2004).
It is well-known that one of the simplest methods to investigate of the porous structure of adsorbents is the measurement of adsorption isotherms of various adsorbates (Bansal and Goyal 2005; Lowell et al. 2004; Marsh and Rodriguez-Reinoso 2006; Rouquerol et al. 1999). Typically, the low temperature nitrogen adsorption isotherms are measured and studied because much experimental evidence have been obtained in the past for nitrogen and therefore, the comparison of results is easy. A further reason is low operating cost, simplicity, and quite low time-consumption. Thus N2 is used in most commercial surface area and porosity analyzers. On the other hand, the very important problem is the analysis/interpretation of adsorption isotherm and the calculation of the parameters related to the pore structure on the basis of the measured data (Bansal and Goyal 2005; Lowell et al. 2004; Marsh and Rodriguez-Reinoso 2006; Rouquerol et al. 1999). It is widely accepted that computer simulations can be treated as a useful tool for this purpose (Terzyk et al. 2012a). The process of N2 adsorption in carbon nanotubes has been modeled since the middle of the 1990s (Maddox and Gubbins 1995). It should be noted that more and more complicated models reproducing the structure of real carbon nanotubes (i.e. from ideal cylinders (Agnihotri et al. 2005, 2006, 2007; Arora et al. 2004; Arora and Sandler 2005; Furmaniak et al. 2010; Gauden et al. 2009; Harris 2009; Khan and Ayappa 1998; Kotsalis et al. 2004; Kowalczyk et al. 2005; Maddox and Gubbins 1995; Müller 2008; Ohba and Kaneko 2002; Terzyk et al. 2008; Zhang et al. 2008) to bundles (Alain et al. 2000; Arab et al. 2004; Furmaniak et al. 2008, 2009a; Harris 2009; Jiang and Sandler 2003, 2004; Paredes et al. 2003; Wongkoblap et al. 2009; Terzyk et al. 2012a, Yin et al. 1999) and tubes with defects (Agnihotri et al. 2005, 2006, 2007; Arora and Sandler 2007a, b; Furmaniak et al. 2009a; Gauden et al. 2009; Gauden 2010; Harris 2009; Terzyk et al. 2008; Zhang et al. 2008) have been applied.
In the current study we try to check the influence of the compression (using hydraulic press) of CNTs on the decrease of the available pore space. Nitrogen is treated as the probe molecule. Measuring adsorption from gaseous phase we prove that the “architecture” is observed only at the places of tube contacts, and the isolated tubes play the most important role in adsorption. In order to verify this hypothesis, computer simulation of nitrogen adsorption of various models of CNT bundles will be investigated.
2 Experimental
The commercial, high purity, closed multi-walled carbon nanotubes, labeled as A-1 (Nanostructured & Amorphous Materials, Nanoamor, Houston, TX, USA) were investigated. From the analysis of data provided by producer (Furmaniak et al. 2010; Pacholczyk 2011, Terzyk et al. 2012b) it is seen that outer diameter (D) is lower than 8 nm. However, in the current theoretical studies we consider D = 6 nm, i.e. maximum value for the distribution of diameters obtained from the analysis of HRTEM images. Number of walls (n) is equal to 4–7 (data provided by producer (Furmaniak et al. 2010; Pacholczyk et al. 2011, Terzyk et al. 2012b) and again estimated from the microscopy analysis). The apparent BET surface area (S BET ) is equal to 446 m2/g. This value was calculated on the basis of low temperature nitrogen adsorption data on raw A-1 sample (Furmaniak et al. 2010; Pacholczyk et al. 2011, Terzyk et al. 2012b)—Fig. 1. To check the influence of compression on the “architecture” of studied nanotubes we prepared the samples of A-1 nanotubes using hydraulic press at exerting by 15 min the pressure 200, 500, and 600 atm (A-1-200, A-1-500, A-1-600, respectively). It should be noted that the assumed time of exertion guarantees the stable samples for measurements of nitrogen adsorption isotherms. In order to prepare the series of pressurized adsorbents we take only as-received CNT without drying. The pallets of nanotubes (13 mm diameter) were prepared by weighting of ca. 20 mg of material and placed in 13 mm die (Specac, Britain). All nitrogen adsorption (Fig. 1) isotherms were measured at T = 77.3 K using ASAP 2010 (Micromeritics, Norcross, GA, USA) sorption apparatus. Before measurement CNT samples were outgassed in vacuum at T = 453 K.

Top panel: HRTEM images of studied nanotube (without compression). Middle panel: nitrogen (T = 77.3 K) adsorption–desorption isotherms plotted in normal scale for nanotubes A-1 under and/or without compression for 15 min. Bottom panel: all nitrogen adsorption–desorption isotherms plotted in logarithmic scale and the relationship between the hysteresis loop area (calculated on the basis of adsorption/desorption data) and hydraulic pressure
3 Molecular simulation in grand canonical ensemble
We used the realistic three centre TraPPE type N2 molecule model proposed by Potoff and Siepmann (2001). This model considers not only diatomic character of N2 molecule but also its quadrupole moment. Two of three centres corresponding with the position of nitrogen atoms are the Lennard-Jones (LJ) centres and possess point charges, while the third centre (located at a half-distance between the mentioned two) is only a point charge. Table 1 collects the values of applied interaction parameters. As was shown by Potoff and Siepmann (2001), these values allow to satisfactorily model the properties of bulk nitrogen. The same model was also used successfully for modelling of N2 adsorption on carbon nanotubes (Furmaniak et al. 2010; Wiśniewski et al. 2013), activated carbons (Furmaniak et al. 2009b, Nguyen et al. 2008) and non-porous carbonaceous materials (Herrera and Do 2009). Energy of nitrogen–nitrogen and nitrogen-CNT interactions was calculated as previously (Furmaniak et al. 2010). For LJ interactions between pairs of centres we used simple truncation for distances greater than 5 × σ ij (where σ ij is the collision diameter for interaction of the pair) (Frenkel and Smit 1996). For electrostatic interaction between N2 molecules we also used truncation but for whole molecules (Furmaniak et al. 2009b, 2010). The energy of electrostatic interactions for pair of N2 molecules was neglected for the distance between their centres of mass greater than 1.5 nm (at this distance the energy value is negligible).
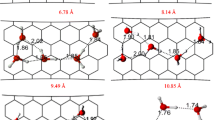
We performed simulations for different systems of tubes (triangle, rectangular, and hexagonal packing arrangements) located at the middle of the simulation box: (i) infinitely long in one direction (series A and series B) and (iii) infinitely long in three directions (series C and series D)—Fig. 2. Thus, four theoretical models of closed bundles related to material A-1 are compared in this figure. We considered nanotubes separated at the various effective distances: from 3.06 nm (A_3.06 (series A) or B_3.06 (series B)—see Figs. 2, 3, 5) to the adjoining tubes, 0.00 nm (A_0.00 or B_0.00—see Figs. 2, 3, 5) and from 3.50 nm (C_3.50 (series C) or B_3.50 (series D)—see Figs. 2, 4, 6) to the adjoining tubes, 0.00 nm (C_0.00 or D_0.00—see Figs. 2, 4, 6). It should be noted that the changes in the available porosity with the progressive rise of separation spacing are limited to the extreme cases in Fig. 2. In order to characterize the systems the following parameters are defined: ∆l tube is the effective distance between tubes and l box is the size of the simulation box (Furmaniak et al. 2010). Bundles were generated basing on triple-walled tube obtained from structures (72,0), (64,0) and (56,0). We limited the number of investigated walls to three due to the simplification of the theoretical calculations. On the other hand, it is well-known that the consideration of higher number of walls is unnecessary from theoretical point of view and this simplification significantly reduces the time of simulations (Furmaniak et al. 2010).

The scheme of four theoretical models of closed bundles related to material A-1 showing the changes in the available porosity in the box with the progressive rise in the separations between tubes (∆l tube is the effective distance between two opposite tubes and l box is the size of the simulation box). Bundles were generated basing on triple-walled tube obtained from structures (72,0), (64,0) and (56,0). It should be noted that this figure and Figs. 5 and 6 were created using the VMD program (Humphrey et al. 1996)

The changes in shapes of nitrogen adsorption isotherms (77.3 K) simulated for bundles of carbon nanotubes {for A [(a) and (b)] and B [(c) and (d)] series} with the progressive decrease in the separations between tubes. Additionally, the data related to isolated tubes are presented—this measurement is treated as the reference one. In (b) and (d) the adsorption isotherms calculated for separated nanotubes are only shown (it should be noted that the behavior of adsorption data is the same as ones from Fig. 1)

The same as Fig. 3 but the results for C [(a) and (b)] and D [(c) and (d)] series are shown
Nitrogen adsorption isotherms (77.3 K) were simulated using the classical Grand Canonical Monte Carlo method. The cubicoid simulation boxes with a tube placed in a box centre (along z axis) having dimensions l box × l box × 4.23 nm were applied. For series A and Series B l box is equal to 40 nm for all systems in contrary to series C and Series D, where l box is the function of ∆l tube . Periodic boundary conditions were used in all three directions, and the internal space of a tube was assumed as inaccessible for adsorbed molecules. For each adsorption point 25·106 iterations were performed during the equilibration, and next 25·106 equilibrium ones, applied for the calculation of the averages (one iteration = an attempt to change the state of the system by creation, annihilation, rotation or displacement). The probability of attempts of changing a system state by creation, annihilation, and rotation and displacement (the latter one is connected with the change in angular orientation) were equal to: 1/3, 1/3, 1/6 and 1/6.
4 Results
In order to evaluate the change of the pore space between nanotubes forming bundles and the mutual orientation of CNTs the investigated adsorbent was compressed using hydraulic press. Figure 1 shows the results of nitrogen adsorption isotherms measured for A-1 series without or under compression. From the analysis of this figure one can easily observe the progressive decrease in maximum adsorption (for p/p s ≈ 1), i.e. the decrease in total pore volume under compression, but at the same time, no remarkable changes in the adsorption for monolayer (i.e. at the B-point) are observed. Consequently, the values of apparent surface area were change insignificantly (i.e. 446 m2/g (A-1), 448 m2/g (A-1-200), 443 m2/g (A-1-500), 450 m2/g (A-1-600)) confirming the mentioned above observations. Taking into account adsorption/desorption data the values of hysteresis loop area were calculated via numerical integration. Final results are collected in Fig. 1. From the analysis of the relationship between the mentioned above area and hydraulic pressure it is seen that the area decreases with increasing compacting pressure. Summing up, the compression of CNTs leads the change of mesoporosity and does not influence on the monolayer region of adsorption isotherm.
The behavior of adsorption isotherms under compression can be explained basing on the GCMC simulation results (Figs. 3, 4). For the case of osculating nanotubes (A_0.00, B_0.00, C_0.00, and D_0.00,—dashed lines in Figs. 3, 4) we observe the decrease in the B—point value on simulated isotherm since the places of the contacts of nanotubes are inaccessible for molecules (lower adsorption). In the case of more separated tubes, i.e. A_0.34, B_0.34, C_0.40, and D_0.40 (in comparison with A_0.00, B_0.00, C_0.00, and D_0.00, respectively) one can observe the significantly different values of adsorption for fixed values of relative pressure in the range of low p/p s (dashed lines in Figs. 3, 4)—this behavior is different in comparison with experimental results (Fig. 1). On the other hand, from the analysis of adsorption isotherms related to A_0.68, B_0.68, C_0.70, and D_0.70 (dashed lines in Figs. 3, 4) it is seen that various values of B-point are also observed comparing to the isolated tubes (treated as reference system), in spite of the similarity of the respective curves for low values of relative pressures. The B-point remains the same and we only observe the decrease in the volume of pores and the vanishing of the phase change for large values of ∆l tube (i.e. for A_1.36 and B_1.36 (Fig. 3) or C_1.50 and D_1.50 (Fig. 4) and for more separated tubes—solid lined in both figures). Moreover, the values of adsorption (the range of adsorption isotherm before pore filling) on isolated CNT are the same as on the mentioned above bundles. Summing up, the progressive approaching of CNTs from the distance equal to 3.06 nm (or 3.50 nm) down to 1.36 nm (or 1.50 nm) leads to the same effect as observed on experimental isotherms for A-1 adsorbents (Figs. 1, 3b, d, 4b, d).
From the analysis of experimental and theoretical isotherms at the low coverages it is seen the significant differences between both techniques. Measured densities at low pressure-coverage range, (inset of Fig. 1) exhibit a layering step associated with the monolayer formation. This is a crystallinity evidence of the nanotube sample. However, the same monolayer transition is clearly steeper in simulations (insets of Figs. 3, 4—low pressure range and adequate interstitial distances) showing that the investigated models are much more homogeneous than the real solid. This mismatch, which is expected, demonstrates the heterogeneity (structural or energetic) of the nanotube sample. These features cannot be captured by simplified models (the CNTs have smooth surfaces and are perfectly parallel and aligned inside the bundles, leaving equally sized interstices) and therefore at low capacities, where solid—fluid interactions prevail, simulated isotherms fail to describe the experimental adsorption behavior. On the other hand, the comparison of experimental data at low pressure-coverage range (Fig. 1) shows that the similarity of the curves. The same results can be observed for isotherms calculated on the basis of computer simulations (Figs. 3, 4). Summing up, one can concluded that increasing the compacting pressure on the commercial sample don not change the crystallinity of the studied samples. It should be pointed out that we are interested in adsorption data for high relative pressures (the mesopore region) where the influence of the mentioned above heterogeneity is negligible small. The next problem is the comparison of simulated adsorption isotherms with experimental ones. As it was expected these values are not expected to coincide (as it strongly depends on how many CNT layers one is using, the diameters (number of walls) of CNTs are dispersed, our model is very simple), however, the qualitative similarity between experimental isotherms and theoretical calculations is observed.
In order to visualize the mechanism of nitrogen adsorption the monolayer was separated from adsorption isotherms, assuming that nitrogen atoms is in the monolayer if the distance between carbon and N2 atoms is smaller than the critical value, σ crit,mono . As mentioned by Birkett and Do (2007) and Cascarini de Torre et al. (1996) proper choosing of this value is not a simple task, especially, for the disordered adsorbents and for molecules composed of two or more atoms. In this study the critical values were chosen based on the observation of special animations of adsorption with marked atoms in the monolayer. It should be noted that Figs. 2, 5 and 6 (showing the snapshots) were created using the VMD program (Humphrey et al. 1996). The value of the critical σ crit,mono for N2-C systems was found empirically as equal to 0.45 nm.

Snapshots showing the configurations of adsorbed nitrogen molecules for selected relative pressures on the triple-walled carbon nanotubes—series B (we only show central part of a simulation box, molecules adsorbed in monolayer are marked in blue and in higher layers as red, note that nanotubes are drawn in the simulation boxes as line to guide the eye)

The same as Fig. 5 but the results for D series are shown
It is well-known that in the case of osculating CNTs three stages for nitrogen adsorption can be observed for closed nanotubes: adsorption in corners between tubes, creation of monolayer on the pore walls and finally condensation (Figs. 5, 6). The increase in the distance between nanotubes makes it possible to create full layers on outer surface of nanotubes (and the absence of adsorption in “corners”). For larger distance between tubes (A_0.68, B_0.68, C_0.70, and D_0.70) two layers of N2 are created at the narrowest fragment between tubes. Here the creation of monolayer on the walls of tubes is accompanied by a partial condensation (A_1.02, B_1.02, C_1.00, D_1.00, A_1.36, B_1.36, C_1.50, and D_1.50). Finally, the complete filling of the main channels between tubes is detected. Further rise in the distance between nanotubes leads, in the case of closed tubes, to the appearance of condensation for higher values of relative pressure. Thus, the existence of monolayer on all nanotubes guarantees the similarity of the mechanism of adsorption for the region of adsorption isotherm related to B-point. Summing up, obtained results (shown in Figs 5, 6) demonstrate that isolated nanotubes under compression approach one to the other, and confirm our idea about the importance of isolated nanotubes in “architecture” of the bundle.
5 Conclusions
Low temperature nitrogen adsorption isotherm data measured at typical temperature (77.3 K) were used for analysis of porosity of nanotubes. The essential role of isolated nanotubes was confirmed by the results of N2 adsorption and the progressive changes in the shapes of adsorption—desorption isotherms measured for the tubes under compression. In order to verify this hypothesis, GCMC simulations of N2 adsorption on isolated or bundled multi-walled closed nanotubes (four models of bundles) were discussed. Using simple geometric considerations (i.e. the decrease of the distance between two opposite tubes) we showed that the similar behaviour of theoretical and experimental adsorption isotherm (the monolayer region of these curves) is observed for the assumption of the isolation of nanotubes. In the other words the separated CNTs play more important role in bundle architecture as it has been assumed till now.
It should be pointed out that this type of study will be continued in future for nanotubes characterizing by lower and higher values of outer diameters and for other carbonaceous materials.
References
Agnihotri, S., Mota, J.P.B., Rostam-Abadi, M., Rood, M.J.: Structural characterization of single-walled carbon nanotube bundles by experiment and molecular simulation. Langmuir 21, 896–904 (2005)
Agnihotri, S., Mota, J.P.B., Rostam-Abadi, M., Rood, M.J.: Adsorption site analysis of impurity embedded single-walled carbon nanotube bundles. Carbon 44, 2376–2383 (2006)
Agnihotri, S., Zheng, Y., Mota, J.P.B., Ivanov, I., Kim, P.: Practical modeling of heterogeneous bundles of single-walled carbon nanotubes for adsorption applications: Estimating the fraction of open-ended nanotubes in samples. J. Phys. Chem. C 111, 13747–13755 (2007)
Alain, E., Yin, Y.F., Mays, T.J., McEnaney, B.: Molecular simulation and measurement of adsorption in porous carbon nanotubes. In: Unger, K.K., Kreysa, G., Baselt, J.P. (eds.) COPS V, Studies in Surface Science and Catalysis, vol. 128, pp. 313–322. Elsevier, Amsterdam (2000)
Arab, M., Picaud, F., Devel, M., Ramseyer, C., Girardem, C.: Molecular selectivity due to adsorption properties in nanotubes. Phys. Rev. B 69, 165401 (2004)
Arora, G., Wagner, N.J., Sandler, S.I.: Adsorption and diffusion of molecular nitrogen in single wall carbon nanotubes. Langmuir 20, 6268–6277 (2004)
Arora, G., Sandler, S.I.: Air separation by single wall carbon nanotubes: Thermodynamics and adsorptive selectivity. J. Chem. Phys. 123, 044705 (2005)
Arora, G., Sandler, S.I.: Nanoporous carbon membranes for separation of nitrogen and oxygen: Insight from molecular simulations. Fluid Phase Equil. 259, 3–8 (2007a)
Arora, G., Sandler, S.I.: Molecular sieving using single wall carbon nanotubes. Nano Lett. 7, 565–569 (2007b)
Bansal, R.C.H., Goyal, M.: Activated Carbon Adsorption. CRC, Boca Raton (2005)
Birkett, G.R., Do, D.D.: On the physical adsorption of gases on carbon materials from molecular simulation. Adsorption 13, 407–424 (2007)
Cascarini de Torre, L.E., Bottani, L.E., Steele, W.A.: Amorphous carbons: Surface structure and adsorptive properties. Langmuir 12, 5399–5406 (1996)
Charlier, J.-C.: Defects in carbon nanotubes. Acc. Chem. Res. 35, 1063–1069 (2002)
Frenkel, D., Smit, B.: Understanding Molecular Simulation. Academic Press, San Diego (1996)
Furmaniak, S., Terzyk, A.P., Gauden, P.A., Lota, K., Frąckowiak, E., Béguin, F., Kowalczyk, P.: Determination of the space between closed multiwalled carbon nanotubes by GCMC simulation of nitrogen adsorption. J. Colloid Interface Sci. 317, 442–448 (2008)
Furmaniak, S., Terzyk, A.P., Gauden, P.A., Wesołowski, R.P., Kowalczyk, P.: Ar, CCl4, and C6H6 adsorption outside and inside of the bundles of multi-walled carbon nanotubes: Simulation study. Phys. Chem. Chem. Phys. 11, 4982–4995 (2009a)
Furmaniak, S., Terzyk, A.P., Gauden, P.A., Harris, P.J.F., Kowalczyk, P.: Can carbon surface oxidation shift the pore size distribution curve calculated from Ar, N2 and CO2 adsorption isotherms? Simulation results for a realistic carbon model. J. Phys. Cond. Matt. 21, 315005–315010 (2009b)
Furmaniak, S., Terzyk, A.P., Gauden, P.A., Harris, P.J.F., Wiśniewski, M., Kowalczyk, P.: Simple model of adsorption on external surface of carbon nanotubes - a new analytical approach basing on molecular simulation data. Adsorption 16, 197–213 (2010)
Gauden, P.A., Terzyk, A.P., Furmaniak, S., Kowalczyk, P.: Adsorption potential distributions for carbons having defined pore structure: GCMC simulations of the effect of heterogeneity. Adsorption 15, 99–113 (2009)
Gauden, P.A.: Monolayer aspects of high-resolution αs-plots. Appl. Surf. Sci. 256, 5285–5291 (2010)
Harris, P.J.F.: Carbon Nanotube Science. Synthesis, Properties and Applications. Cambridge University Press, Cambridge (2009)
Herrera, L.F., Do, D.D.: Effects of surface structure on the molecular projection area. Adsorption of argon and nitrogen onto defective surfaces. Adsorption 15, 240–246 (2009)
Humphrey, W., Dalke, A., Schulten, K.: VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–38 (1996)
Jiang, J., Sandler, S.I.: Nitrogen adsorption on carbon nanotube bundles: Role of the external surface. Phys. Rev. B 68, 245412 (2003)
Jiang, J., Sandler, S.I.: Nitrogen and oxygen mixture adsorption on carbon nanotube bundles from molecular simulation. Langmuir 20, 10910–10918 (2004)
Khan, I.A., Ayappa, K.G.: Density distributions of diatoms in carbon nanotubes: A grand canonical Monte Carlo study. J. Chem. Phys. 109, 4576–4586 (1998)
Kotsalis, E.M., Walther, J.H., Koumoutsakos, P.: Multiphase water flow inside carbon nanotubes. Int. J. Multiph Flow 30, 995–1010 (2004)
Kowalczyk, P., Hołyst, R., Tanaka, H., Kaneko, K.: Distribution of carbon nanotube sizes from adsorption measurements and computer simulation. J. Phys. Chem. B 109, 14659–14666 (2005)
Krungleviciute, V., Heroux, L., Talapatra, S., Migone, A.D.: Gas adsorption on HiPco nanotubes: Surface area determinations, and neon second layer data. Nano Lett. 4, 1133–1137 (2004)
Lowell, S., Shields, J.E., Thomas, M.A., Thommes, M.: Characterization of Porous Solids and Powders: Surface Area Pore Size and Density. Springer, Dordrecht (2004)
Maddox, M.W., Gubbins, K.E.: Molecular simulation of fluid adsorption in buckytubes. Langmuir 11, 3988–3996 (1995)
Marsh, T.H., Rodriguez-Reinoso, F.: Activated Carbon. Elsevier, Amsterdam (2006)
Müller, E.A.: Staggered alignment of quadrupolar molecules inside carbon nanotubes. J. Phys. Chem. B 112, 8999–9005 (2008)
Nguyen, T.X., Cohaut, N., Bae, J.-S., Bhatia, S.K.: New method for atomistic modeling of the microstructure of activated carbons using hybrid reverse Monte Carlo simulation. Langmuir 24, 7912–7922 (2008)
Ohba, T., Kaneko, K.: Internal surface area evaluation of carbon nanotube with GCMC simulation: Assisted N2 adsorption. J. Phys. Chem. B 106, 7171–7176 (2002)
Pacholczyk, A., Terzyk, A.P., Wiśniewski, M., Gauden, P.A., Wesołowski, R.P., Furmaniak, S., Szcześ, A., Chibowski, E., Kruszka, B.: Phenol adsorption on closed carbon nanotubes. J. Colloid Interface Sci. 361, 288–292 (2011)
Paredes, J.I., Suárez-García, F., Villar-Rodil, S., Martínez-Alonso, A., Tascón, J.M.D., Bottani, E.J.: N2 physisorption on carbon nanotubes: Computer simulation and experimental results. J. Phys. Chem. B 107, 8905–8916 (2003)
Potoff, J.J., Siepmann, L.I.: Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 47, 1676–1682 (2001)
Rouquerol, F., Rouquerol, J., Sing, K.: Adsorption by Powders and Porous Solids. Academic, London (1999)
Steele, W.A.: The Interaction of Gases with Solid Surfaces. Pergamon, Oxford (1974)
Terzyk, A.P., Gauden, P.A., Furmaniak, S., Kowalczyk, P.: Heterogeneity on high-resolution alpha(s) plots for carbon nanotubes: GCMC study. Phys. Chem. Chem. Phys. 10, 4551–4554 (2008)
Terzyk, A.P., Furmaniak, S., Gauden, P.A., Harris, P.J.F., Kowalczyk, P.: Virtual porous carbons. In: Tascon, J. (ed.) Novel Carbon Adsorbents, pp. 61–106. Elsevier, London (2012a)
Terzyk, A.P., Pacholczyk, A., Wiśniewski, M., Gauden, P.A.: Enhanced adsorption of paracetamol on closed carbon nanotubes by formation of nanoaggregates. Carbon nanotubes as potential materials in hot-melt drug deposition: Experiment and simulation. J. Colloid Interface Sci. 376, 209–216 (2012b)
Wesołowski, R.P., Terzyk, A.P., Furmaniak, S., Gauden, P.A.: Carbon nanotube membranes. In: Saha, B.B., Ng, K.Ch. (eds.) Advances in Adsorption Technology, pp. 211–240. Nova Science, Huntington (2010)
Wiśniewski, M., Gauden, P.A., Terzyk, A.P., Kowalczyk, P., Pacholczyk, A., Furmaniak, S.: Detecting adsorption space in carbon nanotubes by benzene uptake. J. Colloid Interface Sci. 391, 74–85 (2013)
Wongkoblap, A., Do, D.D., Wang, K.: Adsorption of polar and non-polar fluids in carbon nanotube bundles: Computer simulation and experimental studies. J. Colloid Interface Sci. 331, 65–76 (2009)
Yin, Y.F., Mays, T., McEnaney, B.: Adsorption of nitrogen in carbon nanotube arrays. Langmuir 15, 8714–8718 (1999)
Zhang, Z., Zhang, H., Zheng, Y., Wang, L., Wang, J.: Gas separation by kinked single-walled carbon nanotubes: Molecular dynamics simulations. Phys. Rev. B 78, 035439 (2008)
Acknowledgments
M.W., P.A.G., and A.P.T. gratefully acknowledge the financial support from NSC Grant DEC-2011/01/B/ST5/01192. The authors acknowledge the use of the computer cluster at Poznań Supercomputing and Networking Center and the Information and Communication Technology Center of the Nicolaus Copernicus University (Toruń, Poland).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Gauden, P.A., Terzyk, A.P., Furmaniak, S. et al. Porosity of closed carbon nanotubes compressed using hydraulic pressure. Adsorption 19, 785–793 (2013). https://doi.org/10.1007/s10450-013-9518-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-013-9518-3