
Abstract
Circulating tumor cells (CTCs) photoacoustic detection systems can aid clinical decision-making in the treatment of cancer. Interaction of melanin within melanoma cells with nanosecond laser pulses generates photoacoustic waves that make its detection possible. This study aims at: (1) determining melanoma cell survival after laser pulses of 6 ns at λ = 355 and 532 nm; (2) comparing the potential enhancement in the photoacoustic signal using λ = 355 nm in contrast with λ = 532 nm; (3) determining the critical laser fluence at which melanin begins to leak out from melanoma cells; and (4) developing a time-resolved imaging (TRI) system to study the intracellular interactions and their effect on the plasma membrane integrity. Monolayers of melanoma cells were grown on tissue culture-treated clusters and irradiated with up to 1.0 J/cm2. Surviving cells were stained with trypan blue and counted using a hemacytometer. The phosphate buffered saline absorbance was measured with a nanodrop spectrophotometer to detect melanin leakage from the melanoma cells post-laser irradiation. Photoacoustic signal magnitude was studied at both wavelengths using piezoelectric sensors. TRI with 6 ns resolution was used to image plasma membrane damage. Cell survival decreased proportionally with increasing laser fluence for both wavelengths, although the decrease is more pronounced for 355 nm radiation than for 532 nm. It was found that melanin leaks from cells equally for both wavelengths. No significant difference in photoacoustic signal was found between wavelengths. TRI showed clear damage to plasma membrane due to laser-induced bubble formation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Detection of circulating tumor cells (CTCs) in human blood and lymph systems has the potential to aid clinical decision-making in the treatment of cancer.2,4,8,20 The presence of CTCs may signify the onset of metastasis, indicate relapse, or may be used to monitor disease progression. Initial CTC detection systems were based on histopathologic techniques and have been proven to be time-consuming and subject to reviewers interpretation. Non-optical means exist for CTC detection, including reverse transcriptase polymerase chain reaction (RT-PCR).19,27 Even though PCR led to increased sensitivity and specificity of detection, and removed the subjective influence inherent in earlier CTC detection methods, its clinical implementation is not feasible due to their complexity and concerns regarding the specificity of PCR in detecting CTC due to inconsistency of results and amplification of false products.19 The use of immunohistochemistry for CTC detection relies on antibody recognition of a cancer-specific marker. Immunohistochemistry has shown conflicting opinions about its reliability and specificity.19 CTC detection is still an uncertain research area and optimal detection has yet to be achieved.
Design and construction of a circulating melanoma cell (CMC) detection system is based on the fact that melanoma cells contain a great amount of melanin and less than 5% of melanomas are amelanotic.29,32 Since melanin is a strong optical absorber for the whole visible spectrum,9 laser irradiation has been used to induce acoustic responses in melanin in vivo.11 Based on this principle, a previous study proposed the use of photoacoustic waves for rapid and accurate in vitro detection of CMCs obtained from routine blood draws from metastatic melanoma patients33; the authors demonstrated that at least ten phantom melanoma cells are necessary to maintain a strong photoacoustic signal. A more recent study attempted detection of circulating cells, nanoparticles, and contrast agents in vivo 34; the authors reported threshold sensitivity as low as a single cancer cell in the background of 107 normal blood cells.
While in vivo detection becomes feasible, implementation of this concept in vitro has several advantages: it is fast, inexpensive, and minimally invasive. In vitro detection entails obtaining the mononuclear cell layer (MNCL) derived from lysing and spinning a blood sample from a melanoma patient in a centrifuge. The resulting MNCL is mixed with 20 mL of normal saline and introduced into a flow system consisting of a pump, a fluid receiver, a transparent flow chamber with an integrated acoustic sensor, and a pulsed laser (λ = 450 nm; 5 ns pulse duration) system which creates the conditions for acoustic wave generation.33 Unfortunately, while larger fluences inevitably result in stronger photoacoustic signals, thus increasing the signal-to-noise ratio (SNR), excessively high optical absorption inside the CMC produces localized laser-heat generation that may lead to bubble formation. Bubble formation inside cells may lead to plasma membrane damage, thereby allowing melanin to leak from the cell, and thus preventing continuous photoacoustic detection. The ideal operation condition for a system of this type in a clinical application is to have a continuous detection. For this, it is necessary to ensure that the plasma membrane remains undamaged after laser irradiation, so the melanin does not leak from the cell and diffuse into the circulating solution, reducing the SNR of the photoacoustic signal.
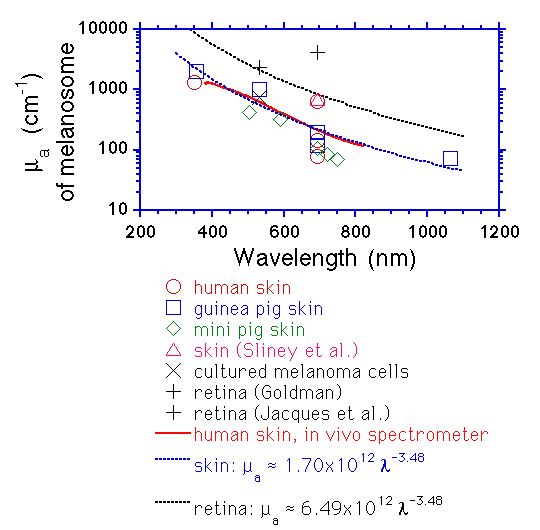
Survival of pigmented melanoma cells after irradiation with laser pulses of 40 ns and 300 μs at λ = 694 nm has been studied before.1 The authors compared the effects of pulse duration using melanoma cells of a mouse-pigmented melanoma cell line (B-16) and a human non-pigmented melanoma cell line (FaDu). It was found that pigmented melanoma cells died only when irradiated with 40 ns pulses, whereas the non-pigmented cells remained viable for both pulse durations. In addition, they demonstrated acoustic wave transients when 8.5 ns laser pulses at λ = 532 and 625 nm were used; these transients are from 2.5 to 3-fold higher for λ = 532 nm wavelength as compared to 625 nm; however, melanosome’s optical absorption coefficient at λ = 532 nm is only 1.75 times higher as compared to λ = 625 nm according to the experimental expression μ a = 1.70×1012λ−3.48 presented in Ref. 9 where μ a is the absorption coefficient and λ is the wavelength expressed in nanometers. Unfortunately, they did not show data for cell survival after irradiation with 8.5 ns.
There are experimental,21,23 numerical,6 and theoretical26 studies that focus on bubble formation around microabsorbers, such as melanosomes and absorbing microbeads in water after laser irradiation with nano- and micro-second pulse durations. Experimental studies3,16,22,28 have revealed that (a) the threshold fluence for bubble formation increases with pulse duration as heat transfer increases; and (b) there is a transition from bubble-driven (mechanical) to protein denaturation-driven (thermal) cell death as the pulse duration is longer. These studies, however, focused on retinal pigment epithelium (RPE) melanosomes. Thus, the motivation for the work presented in this article is the lack of equivalent information melanoma cells from cutaneous origin.
It has been proven that CTCs are photoacoustically detectable using laser pulses of 5 ns duration, 450 nm wavelength, and 0.450 J/cm2 fluence.33 However, this wavelength is difficult to obtain at that pulse duration, as it requires a frequency-tripled Q-switched laser system to pump an optical parametric oscillator (OPO). An OPO is a complex non-linear optical system that increases by a factor of 2—the cost of the laser system used for photoacoustic excitation-detection of CTCs, and it requires maintenance from experienced technicians.
The purpose of this study is to better understand the laser–melanoma cell interactions to assist in the design of an in vitro photoacoustic CTC system developed specifically for pigmented melanoma cell detection. The specific objectives of this study on human melanoma cells are to:
-
(a)
Determine melanoma cell survival after laser pulses of 6 ns33 at wavelengths that are feasible to obtain (e.g., second and third harmonics of a Nd:YAG, Q-switched laser: λ = 532 and 355 nm), and where melanin is still a high linear optical absorber (the 1 ns difference between the pulse duration in some of the experiments in this study and those in Weight et al. 33 should not be of consideration).
-
(b)
Study a potential enhancement of the photoacoustic signals using λ = 355 nm in comparison to λ = 532 nm, since melanin linear absorption at 355 nm is higher than at 532 nm by a factor of 4.
-
(c)
Determine whether melanin leaks from melanoma cells and the critical laser fluence at which this happens, which would adversely affect the SNR of continuous photoacoustic detection.
-
(d)
Develop a time-resolved imaging system with nanosecond resolution to study the intracellular interactions (e.g., laser-induced bubble formation) within the melanoma cells, and their effect on the plasma membrane integrity.
Materials and Methods
Cell Culturing and Sample Preparation
HS936.T (C1) cells were grown as a monolayer in Dulbecco’s Modified Eagle Medium (American Type Culture Collection ATCC, Manassas, VA, USA) with 10% horse serum and 1% of Penicillin–Streptomycin on 6-well cell culture clusters until the culture was confluent. Media were changed every other day. Incubator atmosphere was at 37 °C, 5% CO2 and 90% humidity. Cell monolayers were rinsed with phosphate buffered saline (PBS), and harvested from tissue culturing wells adding Cellstripper (Mediatech, Inc., Herndon, VA, USA) and incubating for 5 min at 37 °C. The cells were resuspended in PBS at a concentration of 2.5 × 106 cells/mL, and sedimented by low-speed centrifugation. Cells were washed and resuspended in PBS twice, and placed on 24-well tissue culture-treated clusters at a cell density of 500 cells/mm2.
Laser Irradiation
Figure 1 shows a schematic of the optical system used to irradiate the samples. Laser irradiation was performed using a Q-switched, Nd:YAG laser and it also has three high-power output ports for the fundamental, second and third harmonics; 1064, 532, and 355 nm wavelengths, respectively.

Experimental setup for laser irradiation of melanoma cells with nanosecond laser pulses with λ = 355 and 532 nm. Second or third harmonic was delivered to the sample inserting or removing the removable mirror (RM). HeNe laser serves as aiming beam. Target’s holder was displaced in the X–Y direction to provide four laser pulses to each sample without overlapping of laser exposure on the same cells
The second harmonic laser beam delivered to the cell cultures was collimated and resized to e −2 spot diameter of 1 mm, using a combination of convergent LC (f = 125 mm) and divergent LD (f = −50 mm) lenses. The energy per pulse was varied using an attenuator made of a half-wave plate HWP1 and a polarizer P1, and it was monitored with a previously cross-calibrated energy meter EM (Ophir, Logan, UT, USA) to a second energy meter placed at the target’s position. The sample was mounted on a 3D translation stage that allowed easy repositioning of the sample. By means of a pellicle beam splitter BS, in line with the Nd:YAG beam, a continuous wave, 0.5 mW, 543 nm wavelength He–Ne laser (Thorlabs, Newton, NJ, USA) served as aiming beam. A total of four pulses were delivered to each sample. Each pulse was delivered at a fresh site; therefore, a single cell was never irradiated more than once. Cells were not irradiated using a single pulse with a large spot size because with such a large spot, the laser fluence that can be achieved with our laser is not high enough to induce damage to the cells. Laser irradiation with the third harmonic (λ = 355 nm) was possible by removing the mirror RM and it was carried out in the same way as it was done with the second harmonic. The distance between lenses LC and LD was adjusted to give the same spot size (as in the second harmonic case) at the sample and the delivered energy was adjusted by using various neutral density (ND) filters.
Cell Damage Determination
Immediately following laser irradiation, a 0.08% solution of Trypan blue, used as a viability assessment dye, was added to the whole sample, including irradiated and non-irradiated cells. Cells were collected from the well cluster and counted using a hemacytometer. The reactivity of Trypan blue is based on the fact that the chromophore is negatively charged and does not interact with the cell unless the membrane is damaged. Therefore, all the cells which exclude the dye are undamaged. Control samples were kept without laser irradiation during the experiment. Long-term effects were not considered as they are irrelevant for continuous photoacoustic CTCs detection.
Melanin Leakage Determination
Immediately following laser irradiation, samples of cells and PBS were collected in Eppendorf tubes. Cells and PBS were then separated by centrifugation at 8000×g for 3 min. Absorbance of the resulting mixture of PBS and melanin from the control and laser-irradiated samples was measured in a nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA) at 220 nm. This spectrophotometer was chosen because it handles sample volume about 3 μL. Differences in PBS absorbance as a function of laser fluence with respect to control samples indicate that there is melanin diluted in PBS that came from the laser-damaged cells.
Comparison of Photoacoustic Signals for λ = 355 and 532 nm
To compare the amplitude of the photoacoustic signal produced by both wavelengths, the experimental setup shown in Fig. 2 was built. Either the second or the third harmonic from a Surelite OPO PLUS (Continuum, Santa Clara, CA, USA), Nd:YAG, Q-switched laser system that emits 5 ns laser pulses were used. The laser beam was focused into a 1-mm spot diameter through a focusing lens and reflected through a right angle prism onto an acrylic well-holder positioned on top of a laboratory-made, unfocused type, polyvinylidene fluoride (PVDF) ultrasonic transducer with sensitivity 0.71 mV/bar. The well-holder contained 50 μL of a 1% intralipid acrylamide disk that was used to protect the transducer. 25 μL of sample suspension was pipetted into the well for testing and a newly made scattering disk was used for each individual test. The signal from the PVDF transducer was sent to a signal amplifier and then into an oscilloscope.

Experimental setup for comparison of photoacoustic pressure at λ = 355 nm and 532 nm. The laser pulse is brought to the melanoma cells (sample) through a focusing lens and a prism. The cells are contained in an acrylic well and diluted in Tryple E to avoid cell clumping. The PVDF ultrasonic transducer is protected from laser damage with a 1% intralipid acrylamide disk
The concentration used for each test was 1 × 106 cells/mL of suspension. The total irradiated volume was 1 μL so the total number of cells irradiated was 1000. In addition, to lessen the effects of cell clumping, the cells were irradiated while suspended in Tryple E.
The photoacoustic signal was quantified as the integrated pressure, which is the result of integrating the oscilloscope trace from 1.6 to 2.5 μs, the calculated acoustic depth of the irradiation well. Integrated pressure units are [volts · seconds]. The thickness of the acrylimide scattering disk was 2.5 mm, although there are some curving effects due to fluid adhesion to the walls of the cuvette. A typical absolute waveform taken at 1 J/cm2 at 32 dB amplification is shown in Fig. 3.

Absolute value of a typical waveform obtained with oscilloscope for comparison of photoacoustic signal at λ = 355 and 532 nm. Signal was integrated from 1.6 × 10−6 to 2.5 × 10−6 s to include the calculated acoustic depth of the irradiation well. Laser fluence was 1 J/cm2, λ = 532 nm and data acquisition was at 32 dB amplification. Inset shows the raw signal as it was acquired with the oscilloscope
TRI of Laser Irradiated Cells
The experimental setup for TRI of laser–cell interaction is shown in Fig. 4. It consists of 2 ns laser systems electronically synchronized. The first, used as the pump, was the EKSPLA laser system previously described (Nd:YAG 1). The second, used as probe, was a Brilliant (Quantel, Les Ulis Cedex, France), Q-switched, Nd:YAG laser that emits 6 ns laser pulses at the fundamental λ = 1064 nm (Nd:YAG 2). This probe wavelength was chosen because melanin has lower absorption as compared to 532 and 355 nm.

Experimental setup for TRI of melanoma cells. Lasers Nd:YAG 1 as pump (λ = 355 or 532 nm) and Nd:YAG 2 as probe (λ = 1064 nm) are externally triggered and synchronized by DG at 10 Hz repetition rate. Second or third harmonic from Nd:YAG 1 laser is released to the sample inserting or removing RM. MS and CCD camera are triggered by a signal generator (SG); both MS and CCD camera’s shutter are open for 100 ms to ensure that only one pump and one probe pulses irradiate the sample and get to the camera, respectively, when the lasers are triggered at 10 Hz repetition rate
Both beams were brought co-linear onto the sample. The green/UV pump beam was resized by means of lenses LC (f = 125 mm) and LD (f = −50 mm) to increase laser fluence on the sample. The delivered energy was varied and recorded in the same way than for the cell damage experiment. The probe beam was slightly focused onto the sample with lens L1 (f = 250 mm). A magnified image of the sample is formed by an aspheric lens L2 (f = 6 mm) and L3 (f = 400 mm) onto a charge-coupled device (CCD) camera (Hamamatsu, Lake Forest, CA, USA). A long-pass filter with cut-off wavelength at λ = 610 nm (RF) positioned in front of the CCD blocked scattered light from the green pump beam.
The delay generator (DG) provided the electronic pulses required to externally trigger both laser systems at 10 Hz repetition rate with a relative delay. Single laser pulses from the pump beam were selectively released by a mechanical shutter (MS) (Uniblitz, Rochester, NY, USA) that opens its aperture for 100 ms allowing only one laser pulse pass through it. Simultaneously, the signal out of the SG was divided and also sent to trigger the CCD whose exposure time was set to 100 ms. This exposure time and the long-pass filter guaranteed that only light from the probe pulse was captured by the CCD.
The pump laser beams for these experiments have a Gaussian intensity profile with e −2 diameter of 910 μm. The diameter of the pump and probe laser beams overfill the CCD field of view.
Results
Cell Damage Determination
Figure 5 shows the normalized cell survival of melanoma cells as a function of the average fluence of the four laser pulses incident on each sample at 532 and 355 nm wavelengths. Cell survival is calculated according to
where C is cell survival, N undamaged is the number of undamaged cells, and N damaged is the number of damaged cells. The denominator in Eq. (1) is the total number of cells per well, not the total number of irradiated cells. Cell survival of irradiated samples was normalized with respect to the average cell survival of control samples, which were not irradiated at all.

Normalized cell survival of melanoma cells irradiated with 6 ns laser pulses with λ = 355 and 532 nm. Significant difference was found in the curves for both wavelengths according to statistical analysis with p < 0.0001. Cell survival was normalized according to Eq. (1) (see body of the text), where the total number of the cells in the sample, and not only cells reached by laser, contributed to the calculation. Each data point is the average of five measurements with its corresponding error bars
In the range of laser fluences used for photoacoustic detection of melanoma cells in suspension, we found that the amount of undamaged cells decreases by 40 and 50% for the 532 and 355 nm wavelengths, respectively, with increasing fluence. The decay of cell survival at both wavelengths is described by exponential curves. The slopes of both curves were found to have significant difference when compared through non-linear, one phase exponential decay fit with p < 0.0001 using GraphPad Prism v.5.00. Nevertheless, for the highest fluences, cell survival tends to be the same at both wavelengths; this means that for such high fluences, the higher absorption for the shorter wavelength is no longer important.
Melanin Leakage Determination
Figure 6 shows absorbance values of the resulting mixture of PBS and melanin when human-pigmented melanoma cells were irradiated. Data are shown normalized with respect to the average of the resulting mixture absorbance of control samples. It is plausible that the large dispersion of data is due to the variable amount of melanin contained in melanoma cells, just as it is for melanosomes.9 However, it is evident that PBS absorbance increases with increasing laser fluence, suggesting that plasma membrane is being damaged by the laser pulse and thus melanin is leaking from the cell.

Normalized PBS absorbance as a function of laser fluence. Absorbance was measured using a nanodrop spectrophotometer with λ = 220 nm. Laser irradiation was carried out at λ = 355 and 532 nm. Data normalization was done with respect to the average of data points obtained with control samples (fluence = 0)
Comparison of Photoacoustic Signals for λ = 355 and 532 nm
Figure 7 shows the integrated pressure generated by the laser–cell interaction as a function of laser fluence for both wavelengths tested. Each data point is the result of averaging five sets of data acquired; each data set is the average of 16 laser shots. As expected, the integrated pressure increases with fluence for both wavelengths. However, the dependence of the integrated pressure on wavelength is not obvious at first sight; therefore, a two-way ANOVA was performed (with α = 0.05) twice: first by considering the whole data up to 1 J/cm2 and, second, by only considering fluences up to 0.6 J/cm2.

Comparison of photoacoustic signal for λ = 532 nm and 355 nm as a function of fluence. Wavelength was found to be of significant difference for fluences up to 0.6 J/cm2, whereas it was not for fluences higher than 0.6 J/cm2
The resulting p-values from the first two-way ANOVA for the two individual factors in the experiment (wavelength and fluence) are 0.9942 and <0.0001, respectively, whereas the p-value for the interaction between them is 0.0011. Such p-values indicate that while the fluence and the interaction between fluence and wavelength are statistically significant for predicting the integrated pressure, the wavelength by itself is unimportant. In contrast, for the second ANOVA, the p-values were 0.007 and <0.0001 for wavelength and fluence, respectively, whereas for the interaction of both the p-values were 0.2737, meaning that both fluence and wavelength are statistically significant for the integrated pressure prediction, whereas the interaction between them is not.
TRI of Melanoma Cells
TRI allowed imaging melanoma cells a few nanoseconds right after its interaction with the pump laser pulse. Figure 8 shows images taken: (a) before, (b) 50 ns after melanoma cells were irradiated using a 6-ns laser pulse with fluence of 0.3 J/cm2 at λ = 355 nm. Figure 8b shows the melanoma cell when “boiling” bubbles are being formed within the cell.

TRI of melanoma cells (a) before and (b) 50 ns after a single, 6 ns laser pulse, λ = 355 nm, 0.3 J/cm2 was delivered to cells. Arrows in (a) show undamaged cells before laser exposure, whereas arrows in (b) clearly show cells where microbubbles were induced by the laser pulse
Figure 9 shows images (a) before, (b) 50 ns after irradiation using a 6-ns laser pulse with fluence of 0.65 J/cm2 at λ = 532 nm. Arrows in Fig. 8a show the melanoma cells without damage prior to laser exposure. Arrows in Fig. 8b show the same cells post-laser exposure when the plasma membrane has been obviously damaged. These results show solid evidence of plasma membrane damage after irradiation with 6 ns laser pulses for both laser wavelengths.

TRI of melanoma cells (a) before and (b) 50 ns after a single, 6 ns laser pulse, λ = 532 nm, 0.65 J/cm2 was delivered to cells. Arrows in (a) show undamaged cells by laser exposure, whereas arrows in (b) clearly show cells that where the plasma membrane was totally destroyed by microbubbles induced by the laser pulse
Discussion
Our results show that the amount of undamaged cells after laser irradiation decreases from 40 to 50% in an exponential decay as a function of delivered fluence. At λ = 355 nm, the amount of cells that survive laser irradiation did not decrease further for fluences higher than 0.35 J/cm2, whereas at 532 nm the amount of undamaged cells does not decrease for fluences higher than 1 J/cm2. It is worth noting that for the higher fluences cell survival tends to the same value. For such fluences, the error bars for both wavelengths in Fig. 5 overlap. The reason for this is because four pulses were delivered to each sample, but the samples were displaced after each pulse, so any given region within the sample received only one pulse and adjacent regions may have not been reached directly by the laser beam. Therefore, the fact that the amount of undamaged cells did not change after certain fluence indicates that all the cells reached by pulses at or above such fluence were killed. It should be mentioned here that all the cells in the sample contributed to the cell survival percentage. A very similar curve was obtained irradiating mouse melanoma cells (B-16) with 40 ns laser pulses at 694 nm in a previous study, although their methodology for laser irradiation and assessing cell survival was different.1 In this study, we use shorter laser pulses and wavelengths. Therefore, it is reasonable to expect stronger laser–melanosomes thermomechanical interactions.
In a related study where single blast-transformed lymphocytes were irradiated with a similar laser pulse duration than in our experiments, the authors tested three different methods to detect damaged cells.15 They found differences in the amount of cells damaged by pulses of the same energy, suggesting that some cells of the same kind are more susceptible to laser damage than others. This may be a plausible explanation for our experiments too, since we observed as much as 20% variation in damage for the same laser fluence and wavelength.
Measurements of absorbance of the mixture of melanin and PBS (Fig. 5) showed that its absorbance increased as laser fluence increased. We observed up to a four-fold increase in the PBS absorbance for the higher fluences tested with respect to the control samples. Data obtained showed large dispersion due to the inherent variability of melanin contained in melanoma cells, just as it is for melanosomes.9 It was not possible to establish a dependence of melanin and PBS mixture absorbance with the wavelength used to irradiate the cells, meaning that both wavelengths damage the cells and melanin, consequently, leaks from them. None of the previous studies about interactions of pigmented melanoma cells or RPE with Q-switched nanosecond laser pulses reported melanin concentration in the cell culturing/PBS medium post-laser irradiation.
Data of integrated pressure as a function of fluence up to 0.6 J/cm2 in Fig. 7 indicate that the 355 nm wavelength offers an advantage to maximize the SNR compared with the 532 nm, as expected due to the four-fold increment in the melanin absorption coefficient at that wavelength. This is in agreement with the information provided in Fig. 5, where cell survival up to that wavelength is clearly higher for 532 nm as compared to the 355 nm.
The cross over of the integrated pressure in Fig. 7 for fluences higher than 0.6 J/cm2 can be attributed to the fact that the total number of cells reached by the laser pulse are damaged at 355 nm, whereas there are still undamaged cells reached by laser at 532 nm, as shown in Fig. 5. The integrated pressure measured in these experiments has two components: the photoacoustic pressure resulting from thermoelastic expansion of the melanosomes within melanoma cells and the pressure originated by the so-called “giant photoacoustic effect” resulting from bubble formation around laser-heated particles. This effect has been reported before in Egerev5 and Lin and Kelly17 and references therein. The pressure resulting from the giant photoacoustic effect has been proven to be higher than that from thermoelastic expansion of heated particles.5
There could be various mechanisms that damage cells due to laser irradiation with a few nanosecond-long laser pulses. Temperature increment within CMCs due to linear absorption of laser light by melanosomes can be calculated under thermal confinement conditions, when the optical absorption coefficient is well known.14
where ΔT is temperature increment, μ a is the optical absorption coefficient of melanosomes at the irradiation laser wavelength, F is fluence or radiant exposure, and ρ and C p are density and specific heat at constant pressure of CMC, respectively. As the different absorbers within a cell are of submicron scale (melanosomes containing melanin, in our case), thermal confinement conditions require the pulse duration to be shorter than the thermal relaxation time, defined as7,15
where τ is thermal relaxation time, d is characteristic length, and α is thermal diffusivity. For a melanosome 1 μm in diameter6 and thermal diffusivity of 1.37 × 10−7 m2/s,30 relaxation time is 7.3 μs, which is three orders of magnitude longer than our 6 ns laser pulses; thus, thermal confinement conditions are fulfilled in our experiments. Although some authors provide other definitions of thermal relaxation time,3,10 with the melanosome size and thermal diffusivity presented here, the thermal relaxation time is two orders of magnitude longer than the laser pulses used for these experiments. Even though more complex theoretical heat transfer models have been developed to study radial and temporal temperature distributions surrounding laser-irradiated absorbing microspheres,3,14,16,21,26 the approximation provided by Eq. (2) is valid for the purposes of this study.
Thermal denaturation of biomolecules in the cell may also be another mechanism of cell damage. Protein denaturation kinetics has been studied within a time resolution of several hundred of microseconds.10 For thermal denaturation, an extrapolation of the Arrhenius equation leads to denaturation temperatures between 370 and 470 K for time range from pico- to nanoseconds. This temperature range is well above the threshold for bubble formation around melanosomes; in other words, like in the case studied here, it would be very difficult to kill cells thermally in the nanosecond time regime.
The most likely damage mechanism for the case studied herein is expansion of laser-induced bubbles inside cells around melanosomes. Laser-induced bubbles without optical breakdown formation have two possible formation mechanisms: boiling and/or cavitation.21,23–25 As melanoma cells contain a large amount of melanosomes (containing melanin), these serve as nucleation centers for bubble formation. When bubbles around single melanosomes expand they merge forming larger bubbles that disrupt the cell membrane as they expand. Boiling nucleation on melanosomes and absorbing beads has been studied previously.21,23 In these studies, experimental results show that the threshold fluence for bubble formation decreases linearly with increasing ambient temperature for different laser pulse durations. The authors extrapolated their data and showed that a bubble is formed around the melanosomes for 12 ns pulse duration at 532 nm wavelength when a temperature of 136 ± 23 °C is reached. Table 1 shows calculations of the temperature increment calculated with Eq. (2) for the lowest and highest fluences used in our experiments, considering the thermal properties in Gerstman et al. 7 and the absorption coefficients of skin melanosomes in Ref. 9 and not retina melanosomes. Our calculations show that the temperature increments are well above the reported nucleation temperatures for the highest fluence, whereas with the combination of lowest fluence and absorption coefficient, temperature increments are near and below nucleation temperatures.
Another study reports detection of laser-induced bubbles in hemoglobin solution and in individual red blood cells at temperatures as low as 30 °C.14 Other authors have experimentally studied bubble formation at temperatures below 100 °C in absorbing solutions, gels, and soft tissues.24,25 Under stress confinement conditions, tensile stresses are responsible for a change of phase below 100 °C, thereby reducing the threshold fluence for bubble formation. Tensile stresses in those experiments are the result of the material optical properties and experiment geometry, in which a compression wave experiments a reflection with opposite sign becoming a tensile wave because of the acoustic mismatch at the boundary. For a melanosome 1 μm in diameter and with acoustic velocity of water, the acoustic confinement time is about 0.7 ns, which is one order of magnitude shorter than the pulse duration in these experiments; however, if two melanoma cells 10 or 20 μm in diameter contained a large amount of melanosomes and were very close to one another, then an acoustic characteristic length in the order of the cell size can be considered, and stress confinement conditions could be fulfilled. Although the complex geometry of the interior of individual cells makes it difficult to affirm that this is the case herein, this mechanism for laser-induced bubble formation inside cells has been proposed previously,14 and some of our data for low fluence combined with the low-absorption coefficient of melanosomes seem to suggest that this mechanism is indeed plausible.
In the study where a similar experiment with melanocytes was carried out,18 the authors reported that the cell was not “blown to pieces”; unfortunately, they do not specify at which fluences this phenomenon occured. In our case, it is evident that the cell membranes were disrupted and, therefore, melanin leaked out and diluted in the PBS solution, which was determined with the nanodrop spectrophotometer.
There is evidence that UV and VIS radiation produce damage to subcellular structures via photosensitization, a process in which a molecule absorbs light and produces reactive species that alter cellular molecules and initiate cellular responses, e.g., apoptosis.12,13 It was proven that when Rose Bengal is used as photosensitizer, 532 nm light induces oxygen singlets, whereas 355 nm radiation induces both oxygen singlets and highly reactive free radicals. Free radicals were found to damage membrane functions more effectively than oxygen singlets.12 Furthermore, for the 355 nm wavelength, melanoma cells are more likely to go through a combination of photochemical decomposition biomolecules due to the UV light31 than the 532 nm wavelength. These biological and chemical mechanisms, along with a stronger photothermal interaction of melanosomes with UV light, may explain the differences in the cell survival rate we observe at both wavelengths.
Long-term effects on cell viability were not considered in this study as these are not significant for the design of the CMC photoacoustic detection system and are beyond the scope of this investigation. Further research with shorter time resolution is required for better understanding of the boiling phenomenon. The required time resolution is possible by synchronizing the pump nanosecond laser pulse with a femtosecond laser as a probe pulse.
Conclusions
Four main conclusion statements can be extracted from this study:
-
(1)
There is statistically significant difference in the amount of cells that survive laser irradiation for both wavelengths. λ = 355 nm kills cells at a higher rate (vs. fluence) than λ = 532 nm.
-
(2)
There is statistically significant difference in the integrated pressure resulting from irradiation of melanoma cells with both wavelengths up to 0.6 J/cm2, as expected due to the four-fold increment in the melanin absorption coefficient at λ = 355 nm with respect to λ = 532 nm.9 For fluences higher than 0.6 J/cm2, the higher amount of damaged cells at 355 nm may be due to the superposition of pressure induced by the thermoelastic expansion of melanosomes and that resulting from the giant optoacoustic effect.
-
(3)
The post-laser irradiation absorbance of the mixture of melanin and PBS increases equally for the two wavelengths, which suggests that although the λ = 355 nm irradiation is killing cells at a greater rate, melanin is leaking from them just as much as it does for λ = 532 nm irradiation.
-
(4)
The main plasma membrane damage mechanism is microbubble formation within the cell, although it is not linearly correlated to laser energy absorption only; thus, mechanisms other than laser light absorption contribute to the plasma membrane damage. From a practical standpoint, λ = 355 nm wavelength does not offer a much larger SNR of the photoacoustic signal with respect to λ = 532 nm.
References
Ara, G., R. R. Anderson, K. G. Mandel, M. Ottesen, and A. R. Oseroff. Irradiation of pigmented melanoma cell with high intensity pulsed radiation generates acoustic waves and kills cells. Lasers Surg. Med. 10:52–59, 1990.
Braun, S., and B. Naume. Circulating and disseminated tumor cells. J. Clin. Oncol. 23:1623–1626, 2005.
Brinkmann, R., G. Huttmann, J. Rogener, J. Roider, R. Birngruber, and C. P. Lin. Origin of retinal pigment epithelium cell damage by pulsed laser irradiance in the nanosecond to microsecond time regimen. Lasers Surg. Med. 27:451–464, 2000.
Cristofanilli, M., G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. M. M. Terstappen, and D. F. Hayes. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 351:781–791, 2004.
Egerev, S., S. Ermilov, O. Ovchinnikov, A. Fokin, D. Guzatov, V. Klimov, A. Kanavin, and A. Oraevsky. Acoustic signals generated by laser-induced metal nanoparticles. Appl. Opt. 48:C38–C45, 2009.
Faraggi, E., B. S. Gertsman, and J. Sun. Biophysical effects of pulsed lasers in the retina and other tissues containing strongly absorbing particles: shockwave and explosive bubble generation. J. Biomed. Opt. 10(6):064029, 2005.
Gerstman, B. S., C. R. Thompson, S. L. Jacques, and M. E. Rogers. Laser induced bubble formation in the retina. Lasers Surg. Med. 18:10–21, 1996.
Ghossein, R. A., S. Bhattacharya, and J. Rosai. Molecular detection of micrometastases and circulating tumor cells in solid tumors. Clin. Cancer Res. 5:1950–1960, 1999.
Huttmann, G., and R. Birngruber. On the possibility of high-precision photothermal microeffects and the measurement of fast thermal denaturation of proteins. IEEE J. Sel. Top. Quantum Electron. 5:954–962, 1999.
Jacques, S. L., et al. The melanosome: threshold temperature for explosive vaporization and internal absorption coefficient during pulsed laser irradiation. Photochem. Photobiol. 53:769–775, 1991.
Kochevar, I. E., C. R. Lambert, M. C. Lynch, and A. C. Tedesco. Comparison of photosensitized plasma membrane damage caused by singlet oxygen and free radicals. Biochimica et Biophysica Acta 1280:223–230, 1996.
Kulms, D., and T. Schwarz. Molecular mechanisms involved in IV-induced apoptotic cell death. Skin Pharmacol. Appl. Skin Physiol. 15:342–347, 2002.
Lapotko, D. O., and E. Lukianova. Laser-induced micro-bubbles in cells. Int. J. Heat Mass Transfer 48:227–234, 2005.
Lapotko, D. O., E. Y. Lukianova, and A. I. Shnip. Photothermal detection of laser-induced damage in single intact cells. Lasers Surg. Med. 33:320–329, 2003.
Lee, H., C. Alt, C. M. Pitsillides, and C. P. Lin. Optical detection of intracellular cavitation during selective laser targeting of the retinal pigment epithelium: dependence of cell death mechanism on pulse duration. J. Biomed. Opt. 12:064014–064034, 2007.
Lin, C. P., and M. W. Kelly. Cavitation and acoustic emission around laser-heated microparticles. Appl. Phys. Lett. 72:2800–2802, 1998.
Lin, C. P., M. W. Kelly, S. A. B. Sibayan, M. A. Latina, and R. R. Anderson. Selective killing by microparticle absorption of pulsed laser radiation. IEEE J. Sel. Top. Quantum Electron. 5:963–968, 1999.
Loberg, R. D., Y. Fridman, B. A. Pienta, E. T. Keller, L. K. McCauley, R. S. Taichman, and K. J. Pienta. Detection and isolation of circulating tumor cells in urologic cancers: a review. Neoplasia 6:302–309, 2004.
Mocellin, S., D. Hoon, A. Ambrosi, D. Nitti, and C. Rossi. The prognostic value of circulating tumor cells in patients with melanoma: a systematic review and meta-analysis. Clin. Cancer Res. 12:4605–4613, 2006.
Neumann, J., and R. Brinkmann. Boiling nucleation on melanosomes and microbeads transiently heated by nanosecond and microsecond laser pulses. J. Biomed. Opt. 10:024001–024012, 2005.
Neumann, J., and R. Brinkmann. Cell disintegration by laser-induced transient microbubbles and its simultaneous monitoring by interferometry. J. Biomed. Opt. 11:041111–041112, 2006.
Neumann, J., and R. Brinkmann. Nucleation dynamics around single microabsorbers in water heated by nanosecond laser irradiation. J. Appl. Phys. 101:114701–114709, 2007.
Oraevsky, A. A., S. L. Jacques, and F. K. Tittel. Mechanism of laser ablation for aqueous media irradiated under confined-stress conditions. J. Appl. Phys. 78:1281–1290, 1995.
Oraevsky, A. A., S. L. Jacques, R. O. Esenaliev, and F. K. Tittel. Pulsed laser ablation of soft tissues, gels, and aqueous solutions at temperatures below 100°C. Lasers Surg. Med. 18:231–240, 1996.
Pustovalov, V. K., and B. Jean. Theoretical investigations of the processes of selective laser interaction with melanin granules in pigmented tissues for laser applications in medicine. Laser Phys. 16:1011–1028, 2006.
Ring, A. E., L. Zabaglo, M. G. Ormerod, I. E. Smith, and M. Dowsett. Detection of circulating epithelial cells in the blood of patients with breast cancer: comparison of three techniques. Br. J. Cancer 92:906–912, 2005.
Roegener, J., R. Brinkmann, and C. P. Lin. Pump-probe detection of laser-induced microbubble formation in retinal pigment epithelium cells. J. Biomed. Opt. 9(2):367–371, 2004.
Swetter, S. Malignant melanoma. www.emedicine.com/DERM/topic257.htm, 2008.
Vitkin, I. A., J. Woolsey, B. C. Wilson, and R. R. Anderson. Optical and thermal characterization of natural (Sepia officinalis) melanin. Photochem. Photobiol. 59:455–462, 1994.
Vogel, A., and V. Venugopalan. Mechanisms of pulsed laser ablation of biological tissues. Chem. Rev. 103:577–644, 2003.
Wain, E., C. Stefanato, and R. Barlow. A clinicopathological surprise: amelanotic malignant melanoma. Clin. Exp. Dermatol. 33:365–366, 2008.
Weight, R. M., J. A. Viator, P. S. Dale, C. W. Caldwell, and A. E. Lisle. Photoacoustic detection of metastatic melanoma cells in the human circulatory system. Opt. Lett. 31:2998–3000, 2006.
Zharov, V. P., E. I. Galanzha, E. V. Shashkov, N. G. Khlebtsov, and V. V. Tuchin. In vivo photoacoustic flow cytometry for monitoring of circulating single cancer cells and contrast agents. Opt. Lett. 31:3623–3625, 2006.
{kind=link}
Acknowledgments
The authors thank Dr. Rosaura Aparicio Fabre for her assistance in cell culturing, as well as Prof. Bahman Anvari for lending the Hamamatsu camera for TRI experiments. Financial support from UC MEXUS-CONACyT scholarship for graduate studies and 2009 UC MEXUS Dissertation Research Grant for FGPG is greatly appreciated. G.G. thanks DGAPA-UNAM support. G.A. acknowledges the support provided by UCR through the 2007–2008 Academic Senate Research Grant. S.C.L. acknowledges support from CONACyT Grant 57309. J.A.V. acknowledges Missouri Life Sciences Research Board Grant #09-1034 and NIH 1R21CA139186.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Lisa X. Xu oversaw the review of this article.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pérez-Gutiérrez, F.G., Camacho-López, S., Evans, R. et al. Plasma Membrane Integrity and Survival of Melanoma Cells After Nanosecond Laser Pulses. Ann Biomed Eng 38, 3521–3531 (2010). https://doi.org/10.1007/s10439-010-0101-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-010-0101-8