
Abstract
Harmonising methodology between countries is crucial in transborder population monitoring. However, immediate application of alleged, established DNA-based methods across the extended area can entail drawbacks and may lead to biases. Therefore, genetic methods need to be tested across the whole area before being deployed. Around 4,500 brown bears (Ursus arctos) live in Norway, Sweden, and Finland and they are divided into the western (Scandinavian) and eastern (Karelian) population. Both populations have recovered and are connected via asymmetric migration. DNA-based population monitoring in Norway and Sweden uses the same set of genetic markers. With Finland aiming to implement monitoring, we tested the available SNP-panel developed to assess brown bears in Norway and Sweden, on tissue samples from a representative set of 93 legally harvested individuals from Finland. The aim was to test for ascertainment bias and evaluate its suitability for DNA-based transnational-monitoring covering all three countries. We compared results to the performance of microsatellite genotypes of the same individuals in Finland and against SNP-genotypes from individuals sampled in Sweden (N = 95) and Norway (N = 27). In Finland, a higher resolution for individual identification was obtained for SNPs (PI = 1.18E-27) compared to microsatellites (PI = 4.2E-11). Compared to Norway and Sweden, probability of identity of the SNP-panel was slightly higher and expected heterozygosity lower in Finland indicating ascertainment bias. Yet, our evaluation show that the available SNP-panel outperforms the microsatellite panel currently applied in Norway and Sweden. The SNP-panel represents a powerful tool that could aid improving transnational DNA-based monitoring of brown bears across these three countries.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Large carnivores are capable of long-distance dispersal and their populations thus often straddle administrative and international borders. Consequently, implementing an adequate monitoring scheme that operates on a sufficient geographical scale while simultaneously being simple to apply and preferably harmonised transnationally presents a challenge (Bartoń et al. 2019; Bischof et al. 2016). Molecular tools have extended our toolbox, particular in the monitoring of wildlife for rare and elusive species such as large carnivores. DNA-based monitoring of noninvasively collected samples as well as samples taken from dead recoveries enable us to assess populations across areas of various scales (see e.g., Schwartz et al. 2007).
The brown bear (Ursus arctos) made a successful comeback in Sweden and Finland (Chapron et al. 2014) after decades of intense persecution that wiped out the species almost completely (Mykrä and Pohja-Mykrä 2015; Swenson et al. 1995). Today, the brown bear population is divided into the population on the Scandinavian peninsula (Norway and Sweden) in the west and the Karelian population (north-eastern Norway and Finland) in the east (Kopatz et al. 2021). Since the start of regular population size estimations in the 1970s, brown bear numbers have gradually increased to the latest estimates of about 127 (95% credible interval: 111–142) individuals in Norway, 2,616 (95% credible interval: 2,499-2,732) in Sweden and about 1,830 (95% credible interval: 1,740-1,925) individuals were estimated in Finland before hunting season (Bischof et al. 2020; Dupont et al. 2023; Heikkinen et al. 2023). Latest research documented the restoration of the once lost connectivity between both populations. However, the documented gene flow appears to be a comparably recent event and thus should be carefully monitored and managed in order to maintain connectivity and counter potential negative effects of isolation (Frankham 1995; Kopatz et al. 2021; Lowe and Allendorf 2010).
For an effective and sustainable population and conservation management, Norway, and Sweden place high emphasis on their monitoring of large carnivores. The governments of both countries have set specific population management goals that require detailed monitoring tools. Because all large carnivore populations in Norway and Sweden represent transborder populations, transnational collaboration on the monitoring is highly aligned and embedded on different levels in management and ministries, further solidified through signed, bilateral Memorandum of Agreements. Additionally, dissemination of the monitoring results and reports to the public is largely harmonised, including the use of one, common database. As brown bears tend to be involved in conflict with humans, predator management frequently requires rapid DNA analyses for individual identification, necessitating quick response times for delivering results. Currently, more than 70,000 brown bear samples are registered in the publicly accessible database Rovbase (www.rovbase.no or www.rovbase.se). Furthermore, management has requested more comprehensive knowledge such as inferring familial relationships among individuals to assess dispersal and migration. In summary, these requirements emphasise the need for a reliable, cost-effective, and flexible molecular tool capable of handling both small and large sample sizes addressing various management needs.
In Norway and Sweden, the brown bear population is monitored using the same twelve short tandem repeats (STRs) or microsatellite markers (Kopatz et al. 2012). Every monitoring season, 1,000 to 5,000 samples are analysed by both countries. The noninvasively collected biological material for monitoring consists mainly of faeces and hairs. But also tissue samples from legally harvested individuals as well as dead recoveries are subject to analysis. While tissue samples ensure unproblematic genetic analyses, faeces and hairs can pose several challenges. Such samples are notorious in displaying low quality and quantity of DNA for analysis due to degradation, which can lead to potential bias in the results (Piggott and Taylor 2003), low amplification rates and genotyping errors (Creel et al. 2003; Dewoody et al. 2006). Previous studies have suggested single-nucleotide polymorphisms (SNPs) as a reliable alternative to the standard analysis of microsatellites using capillary electrophoresis in DNA-based population monitoring (Fitak et al. 2015; von Thaden et al. 2017). Advanced genotyping with microsatellites, such as high-throughput sequencing, can also offer an alternative (De Barba et al. 2017). However, if samples cannot be analysed in a batch and results need to be provided swiftly, as for rapid analyses in conflict cases, then high-throughput sequencing is not feasible.
Overall, the application of genetic methods has become more accessible and significantly less expensive while allowing for automatization of the analysis pipeline (Kumar et al. 2012). SNP-markers amplify shorter DNA-fragments and are therefore more suitable in genotyping samples with potential degraded DNA (Morin et al. 2004; Seddon et al. 2005). Poor sample quality, as with faeces or hair, can result in allelic dropout, false alleles, and genotyping errors with both markers. However, SNPs have shown to be less prone to these challenges than standard microsatellites and reduce the possibility for homoplasy (Kraus et al. 2015; Norman and Spong 2015; Nussberger et al. 2014; von Thaden et al. 2017). Application of both marker systems requires experienced personnel as allele-calling can be challenging in both microsatellites and SNPs (Pompanon et al. 2005; Wang and Morrissey 2017). SNPs further enable potentially less problematic harmonisation among different laboratories using the same markers, without the need for calibration of allele-calling (Vignal et al. 2002). This is already common practice in the DNA-based transborder population monitoring of grey wolf (Canis lupus) and wolverine (Gulo gulo) in Norway and Sweden (see latest monitoring reports by Kleven et al. 2023; Svensson et al. 2023).
In Finland, a DNA-based monitoring scheme for brown bears is currently under development with the aim to harmonise the transnational DNA-based monitoring of the species with Norway and Sweden. Therefore, the aim of our study was to test the usability of the available brown bear SNP-panel for transnational population monitoring of brown bears across Norway, Sweden, and Finland, especially regarding potential ascertainment bias. This bias can occur when applying genetic markers to other populations, outside the population these markers have been ascertained for (Geibel et al. 2021; Lachance and Tishkoff 2013). We used a representative set of male brown bears from one generation in Finland (Fig. 1A). As the dispersing sex it should be more representative of the genetic variation to be found across the Finnish brown bear population compared to philopatric female brown bears (Støen et al. 2006). Additionally, male brown bears should be highly informative regarding the overall genetic variation found due to the strong gene flow with the Russian population (Kopatz et al. 2014; Tammeleht et al. 2010).

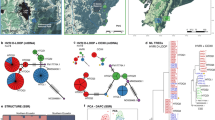
Testing the application of the Scandinavian (Norway and Sweden) SNP-panel (Norman et al. 2013) on brown bears (Ursus arctos) on individuals sampled in Finland. A: Sampling locations of the 93 brown bears from Finland and their assignment to one of the two genetic clusters identified (see Material and Methods). B: Expected heterozygosity (He) of the 82 autosomal SNPs in Finland (sorted from lowest to highest) in comparison to genotypes from Norway and Sweden. C: Mean likelihood of Bayesian clustering for K = 1 to 10 clusters over 10 independent runs of 93 male, Finnish brown bears with the program STRUCTURE enabling admixture model, disabled LOCPRIOR option (Pritchard et al. 2000). D: Estimate of the most likely number of genetic clusters (ΔK) using the approach described by Evanno et al. (2006)
Materials and methods
Sample collection and molecular analyses
We used 93 tissue samples collected 2011–2017 from legally harvested male brown bears in Finland. The legal harvest of brown bears in Finland adheres to an annual quota corresponding to the estimated abundance and distribution of brown bears and the sampling in this study followed this distribution throughout Finland (Fig. 1A). Tissue samples were obtained by our co-authors of the Natural Resources Institute Finland (Luke). No ethical permit was required, as the sample collection did not involve live animals. These individuals were previously genotyped with 12 STR markers used for the DNA-based monitoring of brown bears in Sweden and Norway (Kopatz et al. 2021). Here, we genotyped the same samples and material with the 96 × 96 SNP panel, comprising 85 autosomal, four mtDNA, four y-linked and three x-linked SNP-markers, developed for Swedish brown bears (Norman et al. 2013) including its latest adjustments (Norman and Spong 2015). The SNP-genotyping was performed on a BioMark (Fluidigm Corporation, San Francisco, USA) at the laboratory of NINAGEN Centre for Biodiversity Genetics at the Norwegian Institute for Nature Research in Trondheim following manufacturer’s instructions. Negative and positive controls were included.
Statistical analysis and population genetic structure
After SNP-genotyping, the results were evaluated by comparing with the results based on STRs as described in previous studies (Waits et al. 2001): we calculated the probability of identity (PI), probability of identity among siblings (PIsib), probability of exclusion (PE), observed (Ho) and expected heterozygosity (He) with GenAlEx v6.5 (Peakall and Smouse 2012). For comparison of the performance, we included a representative set of unpublished, previously SNP-genotyped brown bears from Sweden (N = 95) and Norway (N = 27). We used the 93 SNP-genotypes for an analysis in STRUCTURE (Pritchard et al. 2000) employing a burn-in of 100,000 and iterations of 1,000,000. The results were postprocessed with Evanno (Evanno et al. 2005) as well as Clumpak (Jombart et al. 2010) to reveal the most likely number of genetic clusters present.
Results
Genotyping results
Due to a technical error, one of the 85 autosomal SNPs failed to amplify in all samples from Finland. Additionally, two other autosomal SNPs had missing data for more than 10% of the previously genotyped samples from Norway and Sweden. As a result, these three autosomal SNPs were removed prior to analyses. For the remaining 82 autosomal SNPs, the mean call rate for the 93 Finnish samples was 0.995, ranging from 0.939 to 1.000.
Statistical results and population genetic structure
The available SNP-panel provided higher resolution for individual identification in Finland, with probability of identity (PI) being lower for SNPs (PI = 1.18E-27) compared to microsatellites (PI = 4.2E-11). The probability of identity as well as probability of identity among siblings (PIsib) was higher for Finnish brown bears (PIsib=1.33E-14) compared to SNP-genotypes from brown bears sampled in Norway (PI = 2.81E-33 and PIsib=1.05E-17) and Sweden (PI = 7.03E-34 and PIsib=4.75E-18, see Table 1). The probability of exclusion (PE) using SNPs was high in Sweden (PE = 0.99993) and Norway (PE = 0.99990), and only slightly lower in Finland (PE = 0.99896). Expected heterozygosity (He) was lower in Finland, with 19 SNP markers showing He<0.3 (Fig. 1B). Observed (Ho) and expected heterozygosity (He) were lower in Finland (Ho=0.37 and He=0.38) compared to Norway and Sweden, both with Ho=0.45 and He=0.47 and He=0.46 respectively (see Table 1).
Based on their SNP-genotypes, the brown bear population in Finland is subdivided into two, geographically overlapping genetic clusters (Fig. 1A, C and D). One cluster represented the genotypes from individuals sampled in the north, while the second cluster genotypes from individuals sampled in the south (Fig. 1A).
Discussion
The results of our application test demonstrate that the available SNP-panel developed to monitor brown bears in Norway and Sweden (Norman et al. 2013) provides a lower probability of identity (PI) compared to the currently applied STRs or microsatellite markers (Andreassen et al. 2012; Kopatz et al. 2012) and can be used to identify individuals in Finland with much higher statistical power. Probability of exclusion was high and comparable among the results of all three countries. While both marker systems provide sufficient thresholds for individual identification, given the large number of samples to be processed, SNPs allow for more feasible assessment of family relationships among individuals as well as increased efficiency and automatization during analysis and scoring. Additionally, using similar SNP-methodology as already applied in the monitoring of other large carnivores such as grey wolf and wolverine (Kleven et al. 2023; Svensson et al. 2023) allows for comparably cost-effective analysis and application. The spatial population genetic structure of the Finnish brown bear population has been intensively studied (Kopatz et al. 2014, 2017, 2021; Saarma and Kojola 2007; Tammeleht et al. 2010). Our Bayesian analysis for population structure showed that SNP-genotypes group similarly into the two, previously described northern and southern genetic clusters in Finland (Fig. 1A). Earlier studies also showed that the population structure found in Finland, as well as Norway and Sweden, displays isolation-by-distance (Kopatz et al. 2014; Schregel et al. 2018).
Results on heterozygosity however indicate an ascertainment bias (Fig. 1B). The cause of ascertainment bias is the use of a part or non-representative population or population segment when ascertaining the genetic markers. These specific markers may not perform as originally intended when analysing samples from another segment or population. The ascertainment bias is revealed by deviations from expected allele patterns, most commonly by lower heterozygosity (Geibel et al. 2021; Lachance and Tishkoff 2013). The main reason is likely the fact that the used SNP-panel has been developed using individuals sampled from Norway and Sweden (Norman et al. 2013). The genotypes of these individuals are to some extent genetically different from brown bears from Finland, as the populations have been separated for at least 30 to 40 years (Curry-Lindahl 1972; Swenson et al. 1995), which correspond to three to four brown bear generations (Tallmon et al. 2004). Ascertainment bias can therefore affect the assignment of an individual genotype to a population with a different genetic makeup. For instance, genotypes may be less variable at a certain locus of one or more SNP-markers. Due to less informativeness of the markers applied on other geographical and genetically distant populations, a feasible assessment combining individuals from all populations might prove challenging (Giangregorio et al. 2018; Lachance and Tishkoff 2013; Nygaard et al. 2022). Therefore, to enhance its suitability for a comprehensive transnational monitoring and assessment of genetic relationships, one solution could be to replace the 19 SNP-markers showing He<0.3 with more variable loci also found in the Finnish bear population to capture the genetic variation. Another solution could be the development of an all-new SNP-panel based on brown bear genotypes from all three populations, preferably also including individuals from neighbouring countries.
Documenting the status and trajectories of wildlife populations is crucial in a changing world, and large-scale transnational monitoring would be needed for comprehensive assessments of species with continuous distribution (Lamb et al. 2019). For more than a decade, brown bears in Norway and Sweden have been monitored using eight to twelve microsatellites markers for individual identification. Inter-population connectivity of the populations in northern Europe, especially from the Finnish-Russian brown bear populations, has recently become a focus of wildlife management of Norway and Sweden (Kopatz et al. 2021). Maintaining sufficient genetic variation, especially for species capable of long-distance dispersal such as brown bears, is important for a population’s resilience and viability (Driscoll et al. 2014; Heller and Zavaleta 2009). Therefore, migration and gene flow among populations is crucial (Kopatz et al. 2021). Improved DNA-methodologies and technical advancements to survey natural populations will enable new pathways how we monitor our natural world in the future (Abrams et al. 2019; De Barba et al. 2023). High-resolution genetic data will enable effective species and population conservation and management. Therefore, especially refined, and harmonised population monitoring across all three countries (Norway, Sweden, and Finland) will be essential.
Our study provides a practical example of the challenges encountered when expanding the spatial area of DNA-based monitoring or genetic assessment, particularly when incorporating new populations of the target species with potentially different genetic makeup. We demonstrated how an existing genetic tool, in this case SNPs, can be tested beyond its original target population for which it was developed. Overall, the currently available SNP-panel would be sufficient for DNA-based population monitoring enabling individual identification and the determination of the sampled individual’s sex. However, for deeper, and more comprehensive assessments, including genetic diversity and pedigrees to study individual dispersal and gene flow, the utility may be limited (Giangregorio et al. 2023; Hauser et al. 2011). Furthermore, we used tissue samples from Finland for our evaluation. However, when applied to noninvasive sample material, such as faeces and hairs, the issue of genotyping errors needs to be addressed adequately and error rates should be reported as well as the potential risk of one sample collected containing DNA from more than one individual (von Thaden et al. 2017). For a reliable and cost-effective monitoring scheme, transborder harmonisation should be based on recent, accessible and affordable genomic knowledge (Heuertz et al. 2023). In our case, an overhaul of the current SNP-panel, by exchanging the subset of less informative markers among the existing ones, or by designing an additional, second SNP-panel, would ameliorate the ascertainment bias found.
Data availability
The generated and quality checked genotype data from Finland is available at the Dryad depository under DOI: https://doi.org/10.5061/dryad.t76hdr87v.
References
Abrams JF, Hörig LA, Brozovic R, Axtner J, Crampton-Platt A, Mohamed A, Wong ST, Sollmann R, Yu DW, Wilting A, Wheeler H (2019) Shifting up a gear with iDNA: from mammal detection events to standardised surveys. J Appl Ecol 56:1637–1648. https://doi.org/10.1111/1365-2664.13411
Andreassen R, Schregel J, Kopatz A, Tobiassen C, Knappskog PM, Hagen SB, Kleven O, Schneider M, Kojola I, Aspi J, Rykov A, Tirronen KF, Danilov PI, Eiken HG (2012) A forensic DNA profiling system for northern European brown bears (Ursus arctos). Forensic Sci International: Genet 6:798–809. https://doi.org/10.1016/j.fsigen.2012.03.002
Bartoń KA, Zwijacz-Kozica T, Zięba F, Sergiel A, Selva N (2019) Bears without borders: long-distance movement in human-dominated landscapes. Global Ecol Conserv 17. https://doi.org/10.1016/j.gecco.2019.e00541
Bischof R, Brøseth H, Gimenez O (2016) Wildlife in a politically divided World: Insularism inflates estimates of brown bear abundance. Conserv Lett 9:122–130. https://doi.org/10.1111/conl.12183
Bischof R, Milleret C, Dupont P, Chipperfield J, Tourani M, Ordiz A, de Valpine P, Turek D, Royle JA, Gimenez O, Flagstad O, Akesson M, Svensson L, Broseth H, Kindberg J (2020) Estimating and forecasting spatial population dynamics of apex predators using transnational genetic monitoring. Proceedings of the National Academy of Sciences. https://doi.org/10.1073/pnas.2011383117
Chapron G et al (2014) Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346:1517–1519. doi:10.1126/science.1257553%JScience
Creel S, Spong G, Sands JL, Rotella J, Zeigle J, Joe L, Murphy KM, Smith D (2003) Population size estimation in Yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol Ecol 12:2003–2009. https://doi.org/10.1046/j.1365-294x.2003.01868.x
Curry-Lindahl K (1972) The Brown Bear (Ursus arctos) in Europe: Decline, Present Distribution, Biology and Ecology. Paper presented at the Bears: Their biology and management, Calgary
De Barba M, Miquel C, Lobreaux S, Quenette PY, Swenson JE, Taberlet P (2017) High-throughput microsatellite genotyping in ecology: improved accuracy, efficiency, standardization and success with low-quantity and degraded DNA. Mol Ecol Resour 17:492–507. https://doi.org/10.1111/1755-0998.12594
De Barba M, Baur M, Boyer F, Fumagalli L, Konec M, Miquel C, Pazhenkova E, Remollino N, Skrbinsek T, Stoffel C, Taberlet P (2023) Individual genotypes from environmental DNA: fingerprinting snow tracks of three large Carnivore species. Mol Ecol Resour e13915. https://doi.org/10.1111/1755-0998.13915
Dewoody J, Nason JD, Hipkins VD (2006) Mitigating scoring errors in microsatellite data from wild populations. Mol Ecol Notes 6:951–957. https://doi.org/10.1111/j.1471-8286.2006.01449.x
Driscoll DA, Banks SC, Barton PS, Ikin K, Lentini P, Lindenmayer DB, Smith AL, Berry LE, Burns EL, Edworthy A, Evans MJ, Gibson R, Heinsohn R, Howland B, Kay G, Munro N, Scheele BC, Stirnemann I, Stojanovic D, Sweaney N, Villaseñor NR, Westgate MJ (2014) The trajectory of Dispersal Research in Conservation Biology. Syst Rev PLoS One 9:e95053. https://doi.org/10.1371/journal.pone.0095053
Dupont P, Milleret C, Brøseth H, Bischof JK R (2023) Estimates of brown bear density, abundance, andpopulation dynamics in Norway 2012–2022. MINA fagrapport 86. Norwegian University of Life Sciences. Ås. 33 pages
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Fitak RR, Naidu A, Thompson RW, Culver M (2015) A new panel of SNP markers for the Individual Identification of North American Pumas. J Fish Wildl Manage 7:13–27. https://doi.org/10.3996/112014-jfwm-080
Frankham R (1995) Conservation Genetics. Annu Rev Genet 29:305–327. https://doi.org/10.1146/annurev.ge.29.120195.001513
Geibel J, Reimer C, Weigend S, Weigend A, Pook T, Simianer H (2021) How array design creates SNP ascertainment bias. PLoS ONE 16:e0245178. https://doi.org/10.1371/journal.pone.0245178
Giangregorio P, Norman AJ, Davoli F, Spong G (2018) Testing a new SNP-chip on the Alpine and Apennine brown bear (Ursus arctos) populations using non-invasive samples. Conserv Genet Resour 11:355–363. https://doi.org/10.1007/s12686-018-1017-0
Giangregorio P, Mucci N, Norman AJ, Pedrotti L, Filacorda S, Molinari P, Spong G, Davoli F (2023) Performance of SNP markers for parentage analysis in the Italian Alpine brown bear using non-invasive samples. Nat Conserv 53:105–123. https://doi.org/10.3897/natureconservation.53.86739
Hauser L, Baird M, Hilborn R, Seeb LW, Seeb JE (2011) An empirical comparison of SNPs and microsatellites for parentage and kinship assignment in a wild sockeye salmon (Oncorhynchus nerka) population. Mol Ecol Resour 11(Suppl 1):150–161. https://doi.org/10.1111/j.1755-0998.2010.02961.x
Heikkinen S, Kojola I, Mäntyniemi S (2023) Karhukanta Suomessa 2022. Luonnonvara- ja biotalouden tutkimus 23/2023. Natural Resources Institute Finland. Helsinki. 16 pages
Heller NE, Zavaleta ES (2009) Biodiversity management in the face of climate change: a review of 22 years of recommendations. Biol Conserv 142:14–32. https://doi.org/10.1016/j.biocon.2008.10.006
Heuertz M, Carvalho SB, Galindo J, Rinkevich B, Robakowski P, Aavik T, Altinok I, Barth JMI, Cotrim H, Goessen R, González-Martínez SC, Grebenc T, Hoban S, Kopatz A, McMahon BJ, Porth I, Raeymaekers JAM, Träger S, Valdecantos A, Vella A, Vernesi C, Garnier-Géré P (2023) The application gap: Genomics for biodiversity and ecosystem service management. Biol Conserv 278. https://doi.org/10.1016/j.biocon.2022.109883
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94. https://doi.org/10.1186/1471-2156-11-94
Kleven O, Spets MH, Königsson H, Spong G, Milleret C, Dupont P, Bischof R, Brøseth H (2023) DNA-based monitoring of the scandinavian wolverine population 2023. NINA Report 2386. Norwegian Institute for Nature Research. Trondheim, p 33
Kopatz A, Eiken HG, Hagen SB, Ruokonen M, Esparza-Salas R, Schregel J, Kojola I, Smith ME, Wartiainen I, Aspholm PE, Wikan S, Rykov AM, Makarova O, Polikarpova N, Tirronen KF, Danilov PI, Aspi J (2012) Connectivity and population subdivision at the fringe of a large brown bear (Ursus arctos) population in North Western Europe. Conserv Genet 13:681–692. https://doi.org/10.1007/s10592-012-0317-2
Kopatz A, Eiken HG, Aspi J, Kojola I, Tobiassen C, Tirronen KF, Danilov PI, Hagen SB (2014) Admixture and gene flow from Russia in the recovering northern European brown bear (Ursus arctos). PLoS ONE 9:e97558. https://doi.org/10.1371/journal.pone.0097558
Kopatz A, Eiken HG, Schregel J, Aspi J, Kojola I, Hagen SB (2017) Genetic substructure and admixture as important factors in linkage disequilibrium-based estimation of effective number of breeders in recovering wildlife populations. Ecol Evol 7:10721–10732. https://doi.org/10.1002/ece3.3577
Kopatz A, Kleven O, Kojola I, Aspi J, Norman AJ, Spong G, Gyllenstrand N, Dalén L, Fløystad I, Hagen SB, Kindberg J, Flagstad Ø (2021) Restoration of transborder connectivity for fennoscandian brown bears (Ursus arctos). Biol Conserv 253. https://doi.org/10.1016/j.biocon.2020.108936
Kraus RH, vonHoldt B, Cocchiararo B, Harms V, Bayerl H, Kuhn R, Forster DW, Fickel J, Roos C, Nowak C (2015) A single-nucleotide polymorphism-based approach for rapid and cost-effective genetic wolf monitoring in Europe based on noninvasively collected samples. Mol Ecol Resour 15:295–305. https://doi.org/10.1111/1755-0998.12307
Kumar S, Banks TW, Cloutier S (2012) SNP Discovery through Next-Generation sequencing and its applications. Int J Plant Genomics 2012:831460. https://doi.org/10.1155/2012/831460
Lachance J, Tishkoff SA (2013) SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it. BioEssays 35:780–786. https://doi.org/10.1002/bies.201300014
Lamb CT, Ford AT, Proctor MF, Royle JA, Mowat G, Boutin S (2019) Genetic tagging in the Anthropocene: scaling ecology from alleles to ecosystems. Ecol Appl 29:e01876. https://doi.org/10.1002/eap.1876
Lowe WH, Allendorf FW (2010) What can genetics tell us about population connectivity? Mol Ecol 19:3038–3051. https://doi.org/10.1111/j.1365-294X.2010.04688.x
Morin PA, Luikart G, Wayne RK, group. tSw (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216. https://doi.org/10.1016/j.tree.2004.01.009
Mykrä S, Pohja-Mykrä M (2015) Back-calculation of large carnivore populations in Finland in 1865–1915. Ann Zool Fenn 52:285–300. https://doi.org/10.5735/086.052.0504
Norman AJ, Spong G (2015) Single nucleotide polymorphism-based dispersal estimates using noninvasive sampling. Ecol Evol 5:3056–3065. https://doi.org/10.1002/ece3.1588
Norman AJ, Street NR, Spong G (2013) De novo SNP discovery in the scandinavian brown bear (Ursus arctos). PLoS ONE 8:e81012. https://doi.org/10.1371/journal.pone.0081012
Nussberger B, Wandeler P, Camenisch G (2014) A SNP chip to detect introgression in wildcats allows accurate genotyping of single hairs. Eur J Wildl Res 60:405–410. https://doi.org/10.1007/s10344-014-0806-3
Nygaard M, Kopatz A, Speed JMD, Martin MD, Prestø T, Kleven O, Bendiksby M (2022) Spatiotemporal monitoring of the rare northern dragonhead (Dracocephalum Ruyschiana, Lamiaceae) — SNP genotyping and environmental niche modeling herbarium specimens. Ecol Evol 12. https://doi.org/10.1002/ece3.9187
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Piggott MP, Taylor AC (2003) Extensive evaluation of faecal preservation and DNA extraction methods in Australian native and introduced species. Australian J Zool 51:341–355. https://doi.org/10.1071/Zo03012
Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Genotyping errors: causes, consequences and solutions. Nat Rev Genet 6:847–859. https://doi.org/10.1038/nrg1707
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Saarma U, Kojola I (2007) Matrilineal genetic structure of the brown bear population in Finland. Ursus 18:30–37 doi:Doi 10.2192/1537–6176(2007)18[30:Mgsotb]2.0.Co;2
Schregel J, Remm J, Eiken HG, Swenson JE, Saarma U, Hagen SB (2018) Multi-level patterns in population genetics: Variogram series detects a hidden isolation-by-distance-dominated structure of scandinavian brown bears Ursus arctos. Mol Biol Evol 9:1324–1334. https://doi.org/10.1111/2041-210X.12980
Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33. https://doi.org/10.1016/j.tree.2006.08.009
Seddon JM, Parker HG, Ostrander EA, Ellegren H (2005) SNPs in ecological and conservation studies: a test in the scandinavian wolf population. Mol Ecol 14:503–511. https://doi.org/10.1111/j.1365-294X.2005.02435.x
Støen O-G, Zedrosser A, Sæbø S, Swenson JE (2006) Inversely density-dependent natal dispersal in brown bears Ursus arctos. Oecologia 148:356. https://doi.org/10.1007/s00442-006-0384-5
Svensson L, Wabakken P, Maartmann E, Nordli K, Flagstad Ø, Danielsson A, Hensel H, Pöchhacker K, Åkesson M (2023) Bestandsovervåking av ulv vinteren 2022–2023. Beståndsstatus för stora rovdjur i Skandinavien 1-2023. Rovdata and SLU Viltskadesenter. Trondheim and Grimsö. 65 pages
Swenson JE, Wabakken P, Sandegren F, Bjärvall A, Franzén R, Söderberg A (1995) The near extinction and recovery of brown bears in Scandinavia in relation to the bear management policies of Norway and Sweden. Wildl Biology 1:11–25
Tallmon DA, Bellemain EVA, Swenson JE, Taberlet P (2004) Genetic monitoring of Scandinavian Brown Bear Effective Population size and immigration. J Wildl Manag 68:960–965. https://doi.org/10.2193/0022-541X(2004)068[0960:GMOSBB]2.0.CO;2
Tammeleht E, Remm J, Korsten M, Davison J, Tumanov I, Saveljev A, Männil P, Kojola I, Saarma U (2010) Genetic structure in large, continuous mammal populations: the example of brown bears in northwestern Eurasia. Mol Ecol 19:5359–5370. https://doi.org/10.1111/j.1365-294X.2010.04885.x
Vignal A, Milan D, SanCristobal M, Eggen A (2002) A review on SNP and other types of molecular markers and their use in animal genetics. Genet Selection Evol 34:275–305. https://doi.org/10.1186/1297-9686-34-3-275
von Thaden A, Cocchiararo B, Jarausch A, Jungling H, Karamanlidis AA, Tiesmeyer A, Nowak C, Munoz-Fuentes V (2017) Assessing SNP genotyping of noninvasively collected wildlife samples using microfluidic arrays. Sci Rep 7:10768. https://doi.org/10.1038/s41598-017-10647-w
Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10:249–256. https://doi.org/10.1046/j.1365-294X.2001.01185.x
Wang J, Morrissey M (2017) Estimating genotyping errors from genotype and reconstructed pedigree data. Methods Ecol Evol 9:109–120. https://doi.org/10.1111/2041-210x.12859
Acknowledgements
We would like to thank the Finnish Hunters and Finnish Wildlife Agency, the Swedish Environmental Protection Agency, and the Norwegian State Nature Inspectorate for collecting the brown bear samples used in this study. Further, we would like to thank Sanna Kokko-Timonen and Anita Kenttälä from the Natural Resources Institute Finland for their support, Helena Königsson from the Department of Wildlife, Fish, and Environmental Studies at the Swedish University of Agricultural Sciences in Umeå, Sweden genotyping of the samples from Sweden and Line Birkeland Eriksen, Hege Brandsegg and Merethe Hagen Spets from the Norwegian Institute for Nature Research and NINAGEN, Centre for Biodiversity Genetics, for handling and genotyping of the samples from Finland and Norway. Lastly, we would like to thank the two anonymous reviewers for valuable comments and constructive feedback on an earlier version of the manuscript.
Funding
Funding for the analyses of this study was provided by the Norwegian Institute for Nature Research (NINA).
Open access funding provided by Norwegian institute for nature research
Author information
Authors and Affiliations
Contributions
All authors conceived, designed, and contributed to the study. Material preparation, data collection was performed by AK, AN, GS, MV, IK, JA, JK, ØF, OK. Data analysis was performed by AK and OK. The first draft of the manuscript was written by AK and OK, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
No ethic permit was required, as the sample collection did not involve live animals.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kopatz, A., Norman, A.J., Spong, G. et al. Expanding the spatial scale in DNA-based monitoring schemes: ascertainment bias in transnational assessments. Eur J Wildl Res 70, 53 (2024). https://doi.org/10.1007/s10344-024-01808-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10344-024-01808-0