
Abstract
Molecular diversity analysis of deciduous pubescent oaks was conducted for populations from Calabria, Sicily and Sardinia. The aims of this study were twofold. First, to provide data on the genetic diversity of pubescent oaks from an understudied area which currently exhibits one of the highest concentrations of pubescent oak species in Europe. Second, to verify if these groups of oaks are genetically distinct and if their identification is in accordance with the current taxonomic classification. Molecular analyses of leaf material of 480 trees from seventeen populations belonging to putatively different pubescent oak species (Quercus amplifolia, Q. congesta, Q. dalechampii, Q. ichnusae, Q. leptobalanos, Q. virgiliana) were performed. Twelve gene-based Expressed Sequence Tag-Simple Sequence Repeat markers were selected, and genetic diversity and differentiation were calculated. The results showed relatively high values of allelic richness, heterozygosity and number of private alleles for the populations investigated. A weak but positive correlation between geographical and genetic distance was detected. Genetic assignment (STRUCTURE) and principle coordinate analyses exhibited a weak separation into two genetic groups which, however, did not correspond to the taxonomic, chorological and ecological features of the populations investigated. Sardinian populations formed one group which was separated from the Calabrian and Sicilian populations. In light of the results obtained, the taxonomic classification for the pubescent white oaks currently reported in the major Italian floras and checklists for the study area was not confirmed by molecular analyses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The deciduous oak woods represent the most abundant forest vegetation type in southern Europe (Mucina et al. 2016). On the Italian Peninsula they are dominant throughout the whole Apennine range with an increase in the sclerophyllic evergreen oak component (Quercus ilex, Q. suber and Q. coccifera/Q. calliprinos) moving southwards (Blasi and Di Pietro 1998; Blasi et al. 2004; Di Pietro et al. 2010). However, even at the southernmost tip of Italy and Sicily the thermophilous deciduous oak forests cover a wider area than evergreen oak forests (Gianguzzi et al. 2015) whereas the opposite is true for Sardinia where only 15% of the territory is potentially covered by deciduous oaks (Bacchetta et al. 2009). Both taxonomic and phytosociological literature report that the thermophilous broad-leaved forests of southern Italy, Sicily and Sardinia are characterized by different pubescent oak species occurring in sympatry. Pubescent oaks belong to the white oaks (subgenus Quercus; section Quercus) and are characterized by pubescent leaves and twigs that allow them to be distinguished from other white oak species such as Q. petraea and Q. robur. The high concentration of putative pubescent white oak species suggests that southern Italy, Sicily and Sardinia acted as primary refugia for the oak forest vegetation during the Quaternary cold periods (Sadori and Narcisi 2001; Fineschi and Vendramin 2004). It follows the well-established theory according to which several thermophilous tree species survived the glacial periods in the coastal and hilly belts of the Iberian, Italian and Balkan Peninsulas (Huntley and Birks 1983; Watts et al. 1996; Brewer et al. 2002; Tzedakis et al. 2002). Furthermore, the degree of geographic isolation may have played a non-marginal role in the current degree of phenotypic diversification of the pubescent oaks of the study area. Southern Calabria is a narrow mountainous promontory dividing Tyrrhenian and Ionian Seas, while Sicily and Sardinia are the largest Mediterranean islands that experienced different paleogeographic vicissitudes—as testified by their different type of floristic endemic components (Bacchetta et al. 2005; Médail and Quézel 1997; Brullo et al. 2011; Pignatti 2011; Sciandrello et al. 2015)—which may have affected the evolution of the Quercus genetic pools in a different way (see Petit et al. 2002b; Fineschi and Vendramin 2004).
On the basis of the oak classification frameworks reported in the most recently published National floras and in papers on the taxonomy of the Quercus genus (e.g., Pignatti 1982; Brullo et al. 1999; Mossa et al. 1999; Pignatti et al. 2018, 2019) seven pubescent oaks are considered as occurring in southern Italy. These are: Q. amplifolia Guss., Q. congesta C. Presl., Q. dalechampii Ten., Q. ichnusae Mossa, Bacch. and Brullo, Q. leptobalanos Guss., Q. pubescens Willd. and Q. virgiliana Ten. In the recent checklist of the Italian vascular Flora (Bartolucci et al. 2018) only four of these species are considered as valid names (Q. pubescens, Q. dalechampii, Q. congesta and Q. ichnusae) the remaining three being considered as synonyms (Peruzzi et al. 2015, 2019). It is noteworthy that Sicily, Sardinia and southern Calabria are loci classici for five of the aforementioned seven pubescent-oak species and that some of these species (e.g., Q. congesta, Q. dalechampii and Q. virgiliana) are considered “good species” not only in Italy but also in several other European countries. In fact, the debate on the taxonomic value of all these pubescent oak species is very heated throughout Europe. Nonetheless there are very few studies that addressed the problem of oak taxonomy using a multidisciplinary approach where molecular analyses are carried out in support of previous morphological analyses (Franjic et al. 2006; Di Pietro et al. 2016, 2020; Musarella et al. 2018). While it is true that cpDNA markers can be useful to establish the conformity of a given material to the populations of its origin and to trace possible routes of migration at a broad geographic scale, cpDNA provides limited taxonomic information on the systematic status of interfertile, sympatric species (see Curtu et al. 2007a, b; Neophytou and Michiels 2013; Blanc-Jolivet and Liesebach 2015). In contrast, co-dominant markers, such as microsatellites, have successfully been tested to study genetic structures and distinguish oak species at regional or local scale (Degen et al. 1999; Gomory 2000; Gugerli et al. 2007; Guicoux et al. 2011b; Hoeltken et al. 2012). Only recently, population genetic studies based on co-dominant nuclear markers have been carried out in white oak populations from restricted areas of southern Italy (Antonecchia et al. 2015; Fortini et al. 2015; Di Pietro et al. 2016, 2020). These studies were based on sampling protocols, which provided a high number of reference samples per population and a high number of populations per unit area.
The aim of this paper is twofold. First, to provide first insights in the genetic diversity from an area that, although being considered as highly important for the European white oaks diversity, has not been characterized at genetic markers. Second, to verify if groups of oak individuals (or populations) are distinguishable on the basis of their genetic features in order to confirm, or support the assumption of the occurrence of different pubescent oak species.
Materials and methods
Study area
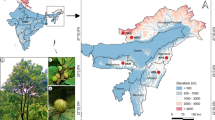
This study was carried out in Southern Italy, in mixed deciduous forest habitats which are located in administrative regions of Calabria (the southernmost end of this region included between the Serre Calabre and the Aspromonte massifs), Sicily and Sardinia (41° 18′ N–7° 23′ E; 36° 19′ N–18° 16′ E) (Fig. 1).

Study sites in Calabria, Sardinia and Sicily
Oak tree material
Leaf material of 17 pubescent-oak populations was collected during autumn of 2017 and 2018. The taxa investigated are: Q. amplifolia Guss., Q. congesta C. Presl., Q. dalechampii Ten., Q. ichnusae Mossa, Bacch. and Brullo, Q. leptobalanos Guss., and Q. virgiliana Ten. As already mentioned in the introduction, some authors consider these taxa as valid species while others consider them as morphotypes included in the natural morphological variability of Q. pubescens Willd. The morphological characters used to distinguish these taxa are for the most part quantitative characters (Di Pietro et al. 2020) with overlapping values between species in the identification keys. For this reason, and to ensure that both the choice of collection sites and the interpretation of the results of the genetic analysis were subject to the lowest possible degree of subjectivity, we have preferred to maintain a neutral position regarding the name of the species, avoiding to provide our a priori identification. In fact, the six putative species were collected in populations in which these oaks were already identified and published as guide species by other authors expert in the taxonomy and phytosociology of oaks (Table 1) and the abundance indexes of each species were already published in phytosociological tables. These tables were originally assigned to the following associations: Erico arboreae-Quercetum virgilianae Brullo et Marcenò 1985, Festuco heterophyllae-Quercetum congestae Brullo et Marceno 1985; Ilici aquifolii-Quercetum leptobalani Maniscalco et Raimondo 2009; Lonicero implexae-Quercetum virgilianae Bacchetta et al. 2004, Oleo sylvestris-Quercetum virgilianae Brullo 1984, Ornithogalo pyrenaici-Quercetum ichnusae Bacchetta et al. 2004, Glechomo-Quercetum congestae Bacchetta et al. 2004, Arabido turritae-Quercetum congestae Brullo et Marcenò 1985, Quercetum leptobalani Brullo 1984 (Brullo 1984; Brullo and Marcenò 1985; Brullo et al. 2001, 2008; Bacchetta et al. 2004, 2009; Maniscalco and Raimondo 2009). Collection sites SIC03, CAL09, SIC11, SAR15 were located in the proximity of the loci classici of Q. congesta, Q. dalechampii, Q. leptobalanos and Q. ichnusae, respectively. The other sites were selected from those for which published taxonomic or phytosociological references were available that attest the occurrence of an oak species among those investigated in this research. A total of 480 oak trees were analyzed. In each stand, leaves were collected from a minimum of 15 and a maximum of 34 individuals. The minimum distance between the collected trees was at least 30 m.
DNA extraction
For all samples from Calabria and Sicily (a total of 393 samples), the DNA was extracted from leaves using the Invisorb® Spin Plant Mini Kit (INVITEK Molecular GmbH, Berlin, Germany) and the work was carried out in the Plant Biology Laboratory of Molise University (Isernia, Italy). For those samples coming from Sardinia (96 samples), DNA was extracted using the Qiagen DNeasy 96 plant kit (Qiagen, Hilden, Germany) and the work was carried out in the Forest Genetics and Forest Tree Breeding Laboratory at the University of Göttingen (Germany).
Twelve gene-based microsatellite markers Expressed Sequence Tag-Simple Sequence Repeats (EST-SSRs) were used: PIE239, PIE227, PIE223, PIE215, PIE020, PIE152, PIE243, PIE242, PIE267, PIE102, PIE258, PIE271 (Durand et al. 2010).
This set of EST-SSR markers (PIE) was chosen according to other recent studies on European white oaks (Lepais et al. 2009; Guichoux et al. 2011a, b; Neophytou et al. 2010; Curtu et al. 2015; Antonecchia et al. 2015; Di Pietro et al. 2020). The primer pairs were combined into three different multiplex reactions, called Mu1, Mu2 and Mu3 (Table 2). The dye type and primers are also shown in Table 2. A PCR Mastermix was obtained blending 1 μL DNA, 1.5 μL 10× reaction buffer B (Solis BioDyne, Tartu, Estonia), 1.5 μL MgCl2 (25 mM), 1 μL dNTPs (2.5 mM each dNTP), and 0.2 μL (5 U/μL) HOT FIREPol Taq DNA polymerase (Solis BioDyne, Tartu, Estonia). To this admixture, we have added EST-SSR primers. The volume, concentration and dye labels of each primer are shown in Table 2.
The PCR reactions were conducted using a touchdown program as follows: denaturation at 95 °C for 15 min, followed by 10 touchdown cycles of 94 °C for 1 min, 60 °C (−1 °C per cycle) for 1 min, and 72 °C for 1 min. The second step consisted of 25 cycles at 94 °C for 1 min, 50 °C for 1 min, and 72 °C for 1 min, followed by a final extension step of 72 °C for 20 min. PCR reactions were performed in a DNA Biometra Thermocycler TOptical Gradient 96 (Biometra, Goettingen, D, EU). The subsequent separation of fragments was performed using GS 500 ROX (Applied Biosystems, Foster City, USA) as size standard in an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, USA). Allele scoring was done with the GeneMapper 4.0 software (Applied Biosystems, Foster City, CA, USA).
Genetic assignment
The main genetic statistics were obtained using GenAlEx software v. 6.5 (Peakall and Smouse 2012). Basic molecular statistics, the mean number of alleles (Na), observed heterozygosity (Ho), expected heterozygosity (He), fixation index (GST) per locus and population, and inbreeding coefficient (FIS) per locus were calculated. In addition, pairwise GST values between populations based on 1000 permutations were calculated. This data set (Pairwise Population Matrix of GST Values) was also used to perform a Principal Coordinates Analysis (PCoA) based on covariance with data standardisation (using the tri distance matrix). Using GenAlEx software v. 6.5, we also tested for significant correlations between pairwise co-dominant genotypic distance and geographical distance by applying simple Mantel tests with 9999 permutations.
The allelic richness (Ar) was calculated with rarefaction to the lowest sample size using the HP-Rare software v. June-6-2006 (Kalinowski 2005).
FSTAT v. 2.9.4 (Goudet 2001) was used for obtaining the inbreeding coefficient (FIS) per population using 1000 permutations to test for significant differences from “zero”. Number of alleles per locus (K), null allele frequencies (Fnull), polymorphic information content (PIC) and deviations from Hardy–Weinberg Equilibrium (HW) were calculated using Cervus 3.0.7 (Marshall et al. 1998).
Analysis of molecular variance (AMOVA) was performed with Arlequin v. 3.5.2.2 (Excoffier and Lischer 2010).
In addition, the software Populations v. 1.2.32 (Langella 1999) was used to calculate phylogenetic trees based on pairwise distances between populations and between individuals using the chord genetic distance of Cavalli-Sforza and Edwards (1967). The phylogenetic tree of populations was obtained with 1000 bootstraps on loci, using MEGA 7.0.26 software (Kumar et al. 2016) to display the trees.
In order to infer molecular clusters and to assign individuals to populations, STRUCTURE v. 2.3.4 based on the Bayesian clustering method was used (Pritchard et al. 2000). We performed genetic analysis with STRUCTURE under the admixture model (Alpha, α) without prior information on the geographical location of populations or their taxonomical classification and applied the correlated allele frequency model (Lambda, λ). Owing to the relatively high number of populations sampled and the unbalanced sampling we have decided to follow Wang (2017) using an Alpha value much smaller than the default. Accordingly, the degree of admixture “Alpha” was set to be inferred for each population and the starting value was set to 1/N (where N = 17). According to Porras-Hurtado et al. (2013), the effect of the parameter of the distribution of allelic frequencies (λ) is expected to be more important with dense genotyping while some studies using SSRs (e.g., Owusu et al. 2015; Thanou et al. 2017) estimate lambda from the data. In this paper, we have followed the suggestion reported in STRUCTURE manual so that λ value has been set to be inferred for each population (starting from λ = 1). To assess the number of clusters that best fit the data, a burn-in period of 50,000 and Markov chain Monte Carlo (MCMC) simulations of 100,000 were used, considering values of K from one to ten, with twenty replications for each value of K. STRUCTURE HARVESTER (Earl and VonHoldt 2012) was used to observe the log-likelihoods over different values of K (Evanno et al. 2005) while CLUMPAK software (Kopelman et al. 2015) was used for obtaining graphic representation and summary of STRUCTURE results.
Results
The analysis exhibited a mean of 7.9 different alleles per locus (Na) for a total of 169 alleles over all populations (Table 3). The locus that exhibited the highest number of alleles (K) was PIE102 with 20 alleles. The mean number of different alleles per locus (Na) over populations ranged from 2.9 (PIE227) to 11.5 (PIE152), the observed heterozygosity (Ho) ranged from 0.227 (PIE227) to 0.834 (PIE271), and the expected heterozygosity (He) ranged from 0.208 (PIE227) to 0.864 (PIE152). PIE227 exhibited the lowest value for Na, Ho, He among all loci. The FIS values were significantly different from zero for four loci and ranged from − 0.005 (PIE020) to 0.398 (PIE239). High and positive FIS values and significant evidence for null alleles were only detected for PIE239 and PIE258.
Mean diversity indices for all populations over all loci are shown in Table 4. The mean number of alleles (Na) per locus ranged from 6.5 in SAR17 (Q. congesta from West Sardinia, only 15 individuals) to 8.8 in both SIC01 and SIC13 (Q. leptobalanos and Q. congesta from West and East Sicily, respectively) (total mean value 7.9). Mean allelic richness (Ar) ranged from 5.8 for SAR16 (Q. virgiliana—NW Sardinia) to 7.2 for SIC01 and SIC02 (Q. leptobalanos and Q. virgiliana from West Sicily) whereas the mean value across populations was 6.7. The observed heterozygosity (Ho) ranged from 0.589 for SIC12 (Q. virgiliana—NE Sicily) to 0.688 for CAL09 (Q. dalechampii—SW Calabria) with a total mean value of 0.649. The expected heterozygosity (He) ranged from 0.619 for SAR16 (Q. virgiliana—NW Sardinia) to 0.727 for SIC13 (Q. congesta—NE Sicily) with a total mean value of 0.683. The mean FIS value was 0.066 while the minimum FIS was − 0.013 for SAR15 (Q. ichnusae—W Sardinia) and the maximum value was 0.137 for SIC12 (NE Sicily). The total number of private alleles Np found was 63. The highest number of private alleles (23) was found in SIC13 (Q. congesta—Sicily) while the second highest number was found in SIC02 (Q. virgiliana—Sicily) with 11 alleles. No private alleles were found in CAL07 and CAL08 (Q. dalechampii and Q. congesta from Calabria) and SAR16 (Q. virgiliana—NW Sardinia). The values of genetic diversity calculated for each administrative region (Calabria, Sicily and Sardinia) showed that the Na values increased from Sardinia (7.1) to Calabria (7.9) and Sicily (8.2). A similar pattern was found for allelic richness (Ar) which was 6.3 for Sardinia, 6.7 for Calabria and 6.8 for Sicily. Mean He was lowest for Sardinian populations (0.637), while it was very similar for Sicilian (0.698) and Calabrian (0.695) populations. The ANOVA (Table 5) showed that differences in He values calculated among administrative regions were statistically significant whereas those in Ho were not. Values for He were significantly lower in Sardinian populations as compared to the Calabrian and Sicilian populations. It is possible that He data could be influenced by the lower total number of individuals and a lower number of individuals per population in two populations (21 individuals in SAR16 and 15 individuals in SAR17). However, also the HT values per region showed the lowest value for Sardinia and the highest for Sicily. The FIS value was slightly negative for Sardinia (− 0.017), while it was positive in Calabria and Sicily where it was found to be 0.069 and 0.102, respectively (Table 4).
The genetic distance (GST) between populations belonging to the same geographical region (Table 4) showed significantly higher values for Sicily (0.084) than for Calabria and Sardinia (0.011 and 0.037, respectively). Pairwise Gst values between geographical regions (Table 6) showed a higher degree of differentiation between Sardinia and Calabria (0.048), and Sardinia and Sicily (0.030) as compared to Sicily and Calabria (0.013).
The highest pairwise GST value (ESM, Table S1) was observed between SAR16 (Q. virgiliana—NW Sardinia) and CAL09 (Q. dalechampii—SW Calabria). Only one non-significant GST value was found in the pairwise matrix, between SIC05 (Q. virgiliana—Sicily) and SIC06 (Q. virgiliana—Sicily) (p value 0.292). These are two oak populations located at similar altitudes at the base of Etna volcano and distant about 4 km from each other as the crow flies.
The PCoA (Fig. 2) showed that all the populations from Sardinia (especially SAR16) segregated in the right part of the diagram, far away from the other populations investigated. Populations from Sicily and Calabria formed a mixed group on the left side of the diagram.

Principal coordinate analysis (PCoA) of the 17 populations sampled in Calabria, Sardinia and Sicily (coordinate 1 and coordinate 2 explain 33.31% and 15.94% of the variation between populations, respectively)
The Mantel test (Fig. 3) showed a positive, although weak, correlation (R2 = 0.085; p = 0.001) between genetic distance (Gen by POP GD) and geographic distance (Geographic POP GGD) of populations.

Isolation-by-distance patterns for individuals, plotting pairwise co-dominant genotypic distance (Gen by POP GD) versus pairwise geographic distances (Geographic POP GGD). The figure includes statistical significance (p = 0.001) obtained by simple Mantel tests in GenAlEx, version 6.5. Each point (diamond) represents a pairwise comparison
According to the global AMOVA (Table 7), most of the genetic variation (92.05% percentage of variation) was found “within individuals” (p value 0.001), followed by that “among individuals within populations” (5.15%) and “among populations” (2.80%).
The Neighbor joining-based tree of populations showed the occurrence of three main clusters (A, B and C) exhibiting a low degree of significance (Fig. 4). Only populations from Sardinia form a distinct cluster with significant bootstrap support (51%). Group A is divided into two main subgroups, one of which (A1) is composed of all the Calabrian populations (CAL07-10) and the other (A2) of two Sicilian populations (SIC05 and SIC06) located very close to each other geographically. Group B comprises two Sicilian populations, one of these (SIC03) referred to a Q. congesta population from the montane belt of Etna volcano and the other (SIC12) to a Q. congesta population of the lower hilly belt of Nebrodi Mountains. Group C is the most numerous and is composed of a well-distinguishable subgroup (C1), which includes the four Sardinian populations and a set of single Sicilian populations that segregate more or less individually, except for SIC01 and SIC02. Genetic distances, which characterize the three main groups and the four further subgroups, are very low except for the Sardinian subgroup (C1). The generally low bootstrap values indicate low phylogenetic signal, suggesting historically high gene flow among populations.

Neighbor-joining tree (NJT) of the 17 populations sampled in Calabria, Sardinia and Sicily based on the chord genetic distance of Cavalli-Sforza and Edwards (1967)
The Neighbor joining-based tree of individuals (ESM, Fig. S1) is less easily interpretable than that based on populations. However, also in this case the individuals belonging to the Sardinian populations (SAR14, SAR15, SAR16 and SAR17) group together in the same cluster while the individuals of the Sicilian and Calabrian populations tend to mix with each other.
The Bayesian analysis revealed K = 2 as the most probable number of genetic clusters obtained with the ad hoc statistic ∆K (ESM, Fig. S2 and Tables S2, S3). A total of 177, out of 480 samples analyzed exhibited Q values > 0.90 of which 74 belonging to cluster 1 and 103 to cluster 2. All the Q > 90 samples coming from the four Sardinia populations belong to cluster 2 whereas almost all the Sicilian and Calabrian populations were found to be composed of samples belonging to both clusters (Fig. 5 and ESM, Fig. S3). The only exceptions were populations SIC04 and CAL08 which presented Q > 90 individuals all belonging to a single cluster (cluster 2 and cluster 1, respectively).

STRUCTURE analysis (K = 2) for all the 17 populations sampled in Calabria Sardinia and Sicily
Plotting the STRUCTURE (Fig. 6) results on the geographic map shows that individuals not clearly assigned to cluster 1 or 2 prevail for most of the investigated populations. Individuals assigned to cluster 1 are absent from Sardinia, while cluster 2 individuals are more frequent and mixed individuals are less frequent in Sardinia than in Calabria and Sicily.

Distribution of individuals with Q > 90 for each cluster in all 17 populations sampled in Calabria Sardinia and Sicily on the geographical base map
Discussion
In the last 25 years we have witnessed an increase in molecular studies in Europe aimed at identifying possible distinctive characteristics within white oaks (Bacilieri et al. 1995; Bruschi et al. 2000, 2003; Csaikl et al. 2002; Curtu et al. 2007a, b; Fortini et al. 2009; Lepais et al. 2009; Lepais and Gerber 2011; Enescu et al. 2013; Yücedag and Gailing 2013; Fortini et al. 2015). However, no references were available about in-detail inter-population genetic studies undertaken for the southernmost part of Italy, Sicily and Sardinia even though all these areas are unanimously considered of great importance for the evolution and phenotypic differentiation of white oaks. In fact, some interesting phylogeographic studies based on cpDNA diversity (Fineschi and Vendramin 2004) had already shown that some of the haplotypes observed in central and northern Europe originated from Italy and in particular from the southern and island regions as result of postglacial recolonization (Fineschi et al. 2002, 2004; Petit et al. 2002a).
The present study fills a gap in the genetic knowledge of the genus Quercus in Italy and provides a better understanding of phenotypic and taxonomic diversity among pubescent white oaks in southern Italy which is considered a center of diversity for pubescent oaks in Europe (see Tutin et al. 1993; Pignatti et al. 2018, 2019). However, there is still a very lively debate, especially among the taxonomists of southern Europe, about the possibility of keeping all these pubescent-oak taxa at the species or subspecies ranks or whether to consider the phenotypic diversity observed as included in the morphological variability pattern of a single widely distributed species (e.g., a pan-European Q. pubescens Willd.). Accordingly, in addition to shedding light on genetic diversity of white oaks in an area still without detailed molecular studies, the aim of this work was to evaluate whether this high phenotypic and taxonomic diversity corresponded to an equally significant level of genetic diversity or biogeographical identity.
The populations investigated show a fair level of genetic polymorphism. Taking into account individual loci, we have found a rather high average number of alleles per locus (14.08) ranging between 8 (PIE227) and 20 (PIE102). The average number of alleles per population and locus was found to range between 2.9 (PIE227) and 11.5 (PIE152). For Ho and He we found values of 0.649 and 0.683, respectively. All these values appear to be quite high when compared with those obtained in a similar study performed in the Apulian Peninsula in the south-easternmost sector of Italy (Di Pietro et al. 2020, Table 2). These results were unexpected, especially considering that Sicily, Sardinia and southern Calabria exhibit a higher geographical isolation when compared to that of the Apulian Peninsula. In fact, the degree of gene polymorphism for the study area was expected to be lower than that from continental areas where, at least in theory, it is conceivable that there may be a greater possibility of gene flow among populations. It is possible that the rugged geomorphology of southern Calabria, Sicily and Sardinia when compared with that of Apulia played a major role in determining such differences in the genetic diversity of these two areas. The Apulian Peninsula is composed of carbonate plateaus (1116 m the highest culmination) separated from the rest of the Italian Peninsula by a vast cultivated plain where the forest stands are scattered in a general matrix of olive groves, vineyards and wheat fields or separated from each other by mosaics of Mediterranean maquis and steppe-like grasslands (Di Pietro and Misano 2009; Biondi et al. 2010; Terzi et al. 2010). On the other hand, Sicily, Sardinia and southern Calabria are all characterized by remarkable mountainous systems whose highest peaks are all ranging between 1600 and 2000 m (see Aspromonte, Nebrodi, Madonie, Gennargentu, Supramonte massifs) with Etna volcano main culminations well above 3000 m. The presence of these mountains has probably made available a greater variety of habitats (and consequently of refuge sites) for the forest vegetation during the climatic oscillations of the Quaternary that allowed a greater territorial contiguity for the surviving oak woods. Three loci (PIE020, PIE239 and PIE242) showed an excess of homozygosity (deviation from Hardy–Weinberg equilibrium p < 0.05 for the first two loci and p < 0.01 for the third). For PIE020, PIE239 null alleles were also detected and therefore possible non-random distribution of genotypes and distorted values of heterozygosity (Brown et al. 2005).
The level of intra-population genetic diversity in the study area shows an average value of alleles per population of 7.9 with an average allelic richness of 6.7. Ho and He values are also quite high (0.64 and 0.68, respectively) and do not show substantial differences if we consider the three study areas (Sicily, Southern Calabria and Sardinia) separately (Table 5). Instead, GST values between populations are very different if the three study areas compared. It is possible, however, that the high values in Sicily when compared to Calabria and Sardinia are influenced by the number of populations analyzed which for Sicily are more than double those of the other two regions. In general, the genetic diversity indices that emerge from this study were found to be significantly higher than those exhibited by the pubescent oak populations of the Apulian Peninsula but lower if compared with those found for a pubescent oak population from the southern/central Apennines (Mount Vairano) which were analyzed at the same markers (ESM, Table S5). It is possible that the higher indices found in the Mount Vairano Q. pubescens forests are due to the co-occurrence and potential hybridization with other white oak species (e.g., Q. frainetto and Q. petraea) and to the spatial contiguity of the Q. robur stands occurring in the foothills.
Genetic assignment, Principal Coordinate Analysis and the Neighbor-Joining tree separated Sardinian populations from Sicilian and Calabrian populations. This separation into two groups can probably be addressed to the greater insularity of Sardinia as compared to Sicily, the latter being separated from southern Calabria by only a narrow stretch of sea (3 km). Despite the close geographical proximity between Calabria and Sicily, however, the interactions between the oak populations of these two territories may have been less than one might expect. In a study on the non-coding regions of chloroplast DNA of Italian populations of deciduous oaks, Fineschi and Vendramin (2004) hypothesized that the missing seed migration from Calabria to Sicily of an eastern haplotype was related to the depth of the Ionian Sea which prevented its freezing even during the phases of maximum glacial extension and prevented the establishment of a land corridor between the two regions. However, not all authors agree on this point. According to some paleontologists during the Quaternary period territorial connections were established between Calabria and Sicily through which several mammalian taxa from continental areas dispersed into Sicily (Bonfiglio et al. 2002).
The correlation between genetic and geographical distance among populations revealed by the Mantel test was found to be positive and statistically significant, however very low. In fact, most of the genetic diversity found is observed within single individuals (92.05%) followed by genetic diversity among individuals within the same populations (5.15%) and among different populations (2.80%).
On the basis of Bayesian cluster analysis (STRUCTURE), the most probable number of clusters considering all individuals from all the populations is two (K = 2).
It is interesting to note that about one third of the individuals (177) had a value of Q > 90 so they could be considered as genetically “pure”. It therefore seems plausible to hypothesize the existence of two distinguishable ancestral populations for the pubescent oaks in the investigated area which for the most part are mixed (in variable proportions) in single individuals. However, genetic clusters do not represent different taxonomic units, but rather differentiation, between Sardinian and Calabrian/Sicilian populations, and this genetic differentiation does not correspond with taxonomic descriptions in the published taxonomic-phytosociological reports. For example, all the Sardinian populations belong to a single genetic cluster (cluster 2) so the records for Q. congesta, Q. amplifolia, Q. ichnusae, Q. virgiliana reported in the taxonomic and phytosociological literature for Sardinia should be summarized to one single taxon (Fig. 6). Furthermore, since pure individuals belonging to cluster 2 were also found in Sicilian and Calabrian populations, the presence of an endemic Sardinian species could not be confirmed (e.g., Q. ichnusae). Also, for Sicilian and Calabrian populations genetic differentiation patterns do not suggest the presence of more than one taxon displaying inconsistencies that are difficult to interpret in a taxonomic key. For example, population CAL08 that was described as Q. congesta population (Nebrodi mountains montane belt) is characterized by unadmixed genetically pure individuals all belonging to cluster 1 whereas population SIC13 that should be another Q. congesta stand is characterized by a large dominance of unadmixed individuals assigned to cluster 2.
According to the phytosociological literature, the four Sardinian populations belong to at least three different species. Overall, the dendrogram does not cluster populations according to putative species, but shows a weak phylogeographic pattern with the Sardinian populations separated and populations collected in neighboring sites grouping together (Fig. 4, see subgroups A1 and C2). Both the first and the second level of clustering, bring together different pubescent-oak (putative) species which, at least based on their original diagnosis and current coenological knowledge (Brullo and Marcenò 1985; Brullo et al. 1999; Mossa et al. 1999; Bacchetta et al. 2009) would have a very different ecology from each other. In the group of Sardinian populations of Q. congesta, Q. virgiliana and Q. ichnusae group together. The Calabrian group includes Q. congesta, and Q. dalechampii. However, the two further subgroups of which the main Calabrian subgroup is composed of (CAL09-CAL10 and CAL07-CAL08) are both composed of a population of Q. congesta and one of Q. dalechampii. In particular, CAL09 was described as a Q. dalechampii population of the Meso-thermo Mediterranean bioclimate of the Gioia Tauro plain less than 100 meters a.s.l. while CAL10 was described as a Q. congesta population of the lower mountain belt of the Aspromonte massif at about 1000 m a.s.l. Only the subgroup SIC05-SIC06 clusters two populations belonging to the same putative species (Q. virgiliana).
Taking into account all the results of the work, the most plausible interpretation of the results is that all the oak populations sampled belong to a single oak taxon which is characterized by a large ecological and morphological amplitude. Although there is still no scientific certainty, the morphological and molecular pattern among pubescent white oaks evidenced in this paper, and those already shown in previous papers for other pubescent-oak populations from the central Mediterranean area (Franjic et al. 2006; Viscosi et al. 2009, 2012; Ballian et al. 2010; Di Pietro et al. 2016, 2020), increasingly reinforce the idea that this “single highly variable pubescent oak taxon” could be the result of repeated events of hybridisation and introgression between an ancient pubescent white oak species (which for simplicity we could here name Q. pubescens) and other European white oak species (e.g., Q. petraea, Q. frainetto, Q. robur). These events would have taken place continuously since the Tertiary and may have even intensified during the Pleistocene following the drastic paleogeographic and paleoclimatic events that characterized this Era. Such a consideration, if translated into a taxonomic key, would exclude a too divisive classification within the collective group of Q. pubescens, and indeed would support the “minimalist” view considering just a single pubescent oak taxon at the rank of species. The Bayesian analysis suggests that there are two main genetic clusters among the pubescent oaks of southern Italy and major islands. Further studies involving other white oak species could provide information on the origin and genetic diversity of the clusters identified. We did not find any correlation between these two genetic groups and the current taxonomic classification of the pubescent oaks in southern Italy. Furthermore, this paper shows a genetic diversification among the Sardinian populations as result of geographical isolation. This result was not completely unexpected if we consider that Fineschi and Vendramin (2004) identified a Sardinian-Corsican endemic haplotype for oaks restricted to these two islands. Actually, population SAR16 (a relic population composed of individuals currently identified as Q. virgiliana from a northwestern Sardinian plain) exhibits comparatively low allelic diversity (see Table 4) as reflected in the complete absence of private alleles and the about lowest values for allelic richness and number of alleles among all populations. The comparatively low allelic diversity could be the result of geographic and topographic isolation. This population appears to be composed of few individuals very similar to each other, surrounded by several very young juvenile trees. This suggests that SAR16 population may have experienced selective removal of trees aimed at favoring particular phenotypes that in addition to changing physiognomy and structure could have also influenced the genetic diversity of the forest ecosystem. A narrow selection of seed-producing trees may in fact lead to a lower variability in forest stands (Dostálek et al. 2011) so that it could be assumed that the centuries-old individuals scattered in the SAR16 population (or at least a part of them) are progeny of few progenitor oaks. The hypothesis that SAR16 stand originates from just a few progenitors is in agreement with what reported in Lawson et al. (2018), as the relatively strong genetic drift experienced by these trees would cause them to appear as a discrete cluster.
Conclusion
As a first study on the genetic diversity of the southern Italy and major islands pubescent-oak populations, this paper displayed relatively high values for all parameters of genetic diversity although more than two thirds of the study area was made up of island territory. We hypothesize that the rugged morphology and wide altitudinal amplitude may have played a role in preserving the spatial contiguity between oak woods in the study area during the Quaternary climatic oscillations and therefore in preserving also high levels of gene flow.
A genetic confirmation for a taxonomical classification providing up to seven pubescent oak species as occurring in the study area did not emerge from this study, despite reported by the most recent floras and checklists and by phytosociological papers as well. Such a result is in accordance with morphological and molecular analyses carried out on pubescent oak populations in south-eastern Italy (Di Pietro et al. 2016, 2020) where it was demonstrated that neither morphological nor molecular results supported the occurrence of more than one pubescent oak species whereas four species were reported by previous phytosociological studies (Biondi et al. 2004, 2010). The oak material analyzed in our study did not show a degree of molecular diversity, within and among populations, sufficient to support this wide taxonomical splitting. However, Bayesian analysis, multivariate statistics and the NJ dendrogram separated Sardinian populations from Calabrian and Sicilian populations mirroring the geographic separation of populations. Our results suggest that all populations investigated belong to a single taxon characterized by a wide range of intra-individual and intraspecific genotypic and phenotypic diversity as result of ecological pressures to which particular groups of oak species are subjected (Kremer and Hipp 2019). The comparatively high genetic diversity highlighted among these S-Italy populations may suggest innumerable events of hybridization and introgression that could have happened between an ancestral pubescent oak (which for simplicity we will call here Q. pubescens s.l.) and other sympatric thermophilous white oaks over the ages. Thanks to the favorable geographical location of southern Italy, in Sicily and Sardinia these events could have occurred without significant interruptions even during the coldest periods of the Quaternary where the different oak species (Q. pubescens s.l., Q. petraea, Q. robur and Q. frainetto) were forced to live in very restricted areas. Possible hints of a process of speciation in progress for the Sardinian populations related to the highlighted (weak) correspondence between genetic and geographic distance and to the geographical isolation of this island are premature and will require further and more detailed studies.
Beyond the phylogenetic or taxonomic relevance, the results have implications for forest economy (timber certification) or nature conservation, if we consider that some of the oak names in issue occur in the list of diagnostic species for European Habitats included in the 92/43/EC Directive (European Commission 2013).
References
Antonecchia G, Fortini P, Lepais O, Gerber S, Legér P, Scippa GS, Viscosi V (2015) Genetic structure of a natural oak community in central Italy: evidence of gene flow between three sympatric white oak species (Quercus, Fagaceae). Ann For Res 58(2):205–216
Bacchetta G, Biondi E, Farris E, Filigheddu R, Mossa L (2004) A phytosociological study of the deciduous oak woods of Sardinia (Italy). Fitosociologia 41(1):53–64
Bacchetta G, Iiriti G, Pontecorvo C (2005) Contributo alla conoscenza della flora vascolare endemica della Sardegna. Inform Bot Ital 37(1):306–307
Bacchetta G, Bagella S, Biondi E, Farris E, Filigheddu R, Mossa L (2009) Vegetazione forestale e serie di vegetazione della Sardegna (con rappresentazione cartografica alla scala 1:350.000). Fitosociologia 46(Suppl 1):3–82
Bacilieri R, Ducousso A, Kremer A (1995) Genetic, morphological and phenological differentiation between Quercus petraea (Matt.) Liebl. and Quercus robur L. in a mixed stand of Northwest of France. Silvae Genet 44:1–10
Ballian D, Ivanković M, Gračan J, Perić S, Marjanović H, Bobinac M, Slade D (2010) Analysis of pubescent oak (Quercus pubescens Willd.) by means of chloroplast DNA (cpDNA) in the western part of the Balkan Peninsula. Acta Soc Bot Pol 79(3):189–195
Bartolucci F, Peruzzi L, Galasso G, Albano A, Alessandrini A, Ardenghi NMG, Astuti G, Bacchetta G, Ballelli S, Banfi E, Barberis G, Bernardo L, Bouvet D, Bovio M, Cecchi L, Di Pietro R, Domina G, Fascetti S, Fenu G, Festi F, Foggi B, Gallo L, Gottschlich G, Gubellini L, Iamonico D, Iberite M, Jiménez-Mejías P, Lattanzi E, Marchetti D, Martinetto E, Masin RR, Medagli P, Passalacqua NG, Peccenini S, Pennesi R, Pierini B, Poldini L, Prosser F, Raimondo FM, Roma-Marzio F, Rosati L, Santangelo A, Scoppola A, Scortegagna S, Selvaggi A, Selvi F, Soldano A, Stinca A, Wagensommer RP, Wilhalm T, Conti F (2018) An updated checklist of the vascular flora native to Italy. Plant Biosyst 152(2):179–303. https://doi.org/10.1080/11263504.2017.1419996
Biondi E, Casavecchia S, GuerraV Medagli P, Beccarisi L, Zuccarello V (2004) A contribution towards the knowledge of semideciduous and evergreen woods of Apulia (South-Eastern Italy). Fitosociologia 41(1):3–28
Biondi E, Casavecchia S, Beccarisi L, Marchiori S, Medagli P, Zuccarello V (2010) Le serie di vegetazione della regione Puglia. In: Blasi C (ed) La Vegezione d’Italia. Palombi Editore, Roma, pp 391–409
Blanc-Jolivet C, Liesebach M (2015) Tracing the origin and species identity of Quercus robur and Quercus petraea in Europe: a review. Silvae Genet 64(4):182–193
Blasi C, Di Pietro R (1998) Two new phytosociological types of Quercus pubescens s.l. woodland communities in southern Latium. Plant Biosyst 132(3):207–223
Blasi C, Di Pietro R, Filesi L (2004) Syntaxonomical revision of Quercetalia pubescenti-petraeae in the Italian peninsula. Fitosociologia 41(1):87–164
Bonfiglio L, Mangano G, Marra AC, Masini F, Pavia M, Petruso D (2002) Pleistocene Calabrian and Sicilian bioprovinces. Geobios 35(Suppl. 1):29–39
Brewer S, Cheddadi R, Beaulieu JL, Reille M (2002) Data contributors the migration of deciduous Quercus throughout Europe since the last glacial period. For Ecol Manag 156:27–48
Brown KM, Baltazar GA, Hamilton MB (2005) Reconciling nuclear microsatellite and mitochondrial marker estimates of population structure: breeding population structure of Chesapeake Bay striped bass (Morone saxatilis). Heredity 94:606–615
Brullo S (1984) Contributo alla conoscenza della vegetazione delle Madonie (Sicilia settentrionale). Boll Acc Gioenia Sci Nat 16(322):351–420
Brullo S, Marcenò C (1985) Contributo alla conoscenza della classe Quercetea ilicis in Sicilia. Not Fitosoc 19(1):183–229
Brullo S, Guarino R, Siracusa G (1999) Revisione tassonomica delle querce caducifoglie della Sicilia. Webbia 54(1):1–72
Brullo S, Scelsi F, Spampinato G (2001) La vegetazione dell’Aspromonte. Laruffa Ed, Reggio Calabria, 368 pp
Brullo S, Gianguzzi L, La Mantia A, Siracusa G (2008) La classe Quercetea ilicis in Sicilia. Boll Accad Gioenia Sci Nat 41(369):1–80
Brullo C, Minissale P, Sciandrello S, Spampinato G (2011) Evaluation of the endemic vascular flora of the Hyblaean territory (SE Sicily-Italy). Acta Bot Gallica 158(4):617–631. https://doi.org/10.1080/12538078.2011.10516299
Bruschi P, Vendramin GG, Bussotti F, Grossoni P (2000) Morphological and molecular differentiation between Quercus petraea (Matt.) Liebl. and Quercus pubescens Willd. (Fagaceae) in Northern and Central Italy. Ann Bot 85:325–333
Bruschi P, Vendramin GG, Bussotti F, Grossoni P (2003) Morphological and molecular diversity among Italian populations of Quercus petraea (Fagaceae). Ann Bot 91:707–716
Cavalli-Sforza LL, Edwards AW (1967) Phylogenetic analysis: models and estimation procedures. Evolution 21(3):550–570
Csaikl UM, Burg K, Fineschi S, Koenig AO, Matyas G, Petit RJ (2002) Chloroplast DNA variation of white oaks in the alpine region. For Ecol Manag 156:131–145
Curtu A, Gailing O, Finkeldey R (2007a) Evidence for hybridization and introgression within a species-rich oak (Quercus spp.). BMC Evol Biol 7:218
Curtu AL, Gailing O, Leinemann L, Finkeldey R (2007b) Genetic variation and differentiation within a natural community of five oak species (Quercus spp.). Plant Biol 9:116–126
Curtu AL, Craciunesc I, Enescu CM, Vidalis A, Sofletea N (2015) Fine-scale spatial genetic structure in a multi-oak-species (Quercus spp.) forest. iForest 8(3):324
Degen B, Streiff R, Ziegenhagen B (1999) Comparative study of genetic variation and differentiation of two pedunculate oak (Quercus robur) stands using microsatellite and allozyme loci. Heredity 83(5):597–603. https://doi.org/10.1038/sj.hdy.6886220
Di Pietro R, Misano G (2009) Analisi fitosociologica e considerazioni sintassonomiche sulla vegetazione forestale delle Gravine occidentali dell’Arco Ionico (Murge pugliesi e lucane, Italia meridionale). Inform Bot Ital 41(2):215–246
Di Pietro R, Azzella MM, Facioni L (2010) The forest vegetation of Tolfa-Ceriti mountains (northern Latium—central Italy). Hacquetia 9(1):5–64
Di Pietro R, Di Marzio P, Medagli P, Misano G, Silletti GN, Wagensommer RP, Fortini P (2016) Evidence from multivariate morphometric study of the Quercus pubescens complex in southeast Italy. Bot Serb 40(1):83–100
Di Pietro R, Di Marzio P, Antonecchia G, Conte AL, Fortini P (2020) Preliminary characterization of the Quercus pubescens complex in southern Italy using molecular markers. Acta Bot Croat 78(2):107–115. https://doi.org/10.2478/botcro-2019-0026
Dostálek J, Frantík T, Lukášová M (2011) Genetic differences within natural and planted stands of Quercus petraea. Cent Eur J Biol 6(4):597–605
Durand J, Bodénès C, Chancerel E, Frigerio JM, Vendramin G, Sebastiani F, Mattioni C (2010) A fast and cost-effective approach to develop and map EST-SSR markers: oak as a case study. BMC Genom 11(1):570
Earl DA, VonHoldt BM (2012) Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Cons Genet Res 4: 359-361. https://doi.org/10.1007/s12686-011-9548-7
Enescu CM, Curtu AL, Șofletea N (2013) Is Quercus virgiliana a distinct morphological and genetic entity among European white oaks? Turk J Agric For 37(5):632–641. https://doi.org/10.3906/tar-1210-28
European Commission (2013) Interpretation manual of European Union Habitats. EUR 28. April 2013. DG Environment, Nature ENV B.3, p 144
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol 10:564–567
Fineschi S, Vendramin GG (2004) Choroplastic diversity in Italian oaks: evidence of a higher genetic richness in southern and insular populations. Forest@ 1:82–87. https://doi.org/10.3832/efor0220-0010082
Fineschi S, Taurchini D, Grossoni P, Petit RJ, Vendramin GG (2002) Chloroplast DNA variation of white oaks in Italy. For Ecol Manag 156(1–3):103–114
Fortini P, Viscosi V, Fineschi S, Vendramin GG (2009) Comparative leaf surface morphology and molecular data of five oaks of subgenus Quercus Oerst. (Fagaceae). Plant Biosyst 143:543–554
Fortini P, Antonecchia G, Di Marzio P, Maiuro L, Viscosi V (2015) Role of micromorphological leaf traits and molecular data in taxonomy of three sympatric white oak species and their hybrids (Quercus L.). Plant Biosyst 149(3):546–558
Franjic J, Liber Z, Skvorc Z, Idzojtic M, Sostaric R, Stancic Z (2006) Morphological and molecular differentiation of the Croatian populations of Quercus pubescens Willd. [Fagaceae]. Acta Soc Bot Pol 75(2):123–130
Gianguzzi L, Papini F, Cusimano D (2015) Phytosociological survey vegetation map of Sicily (Mediterranean region). J Maps 12(5):845–851. https://doi.org/10.1080/17445647.2015.1094969
Gomory D (2000) A gene coding for a non-specific NAD-dependent dehydrogenase shows a strong differentiation between Quercus robur and Quercus petraea. For Genet 7:167–170
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices, version 2.9. 3. http://www2.unil.ch/popgen/softwares/fstat.htm
Gugerli F, Walser JC, Dounavi K, Holderegger R, Finkeldey R (2007) Coincidence of small-scale spatial discontinuities in leaf morphology and nuclear microsatellite variation of Quercus petraea and Q. robur in a mixed forest. Ann Bot 99:713–722
Guichoux E, Lagache L, Wagner S, Chaumeil P, Lèger P, Lepais O, Lepoittevein C et al (2011a) Current trends in microsatellite genotyping. Mol Ecol Resour 11:591–611
Guichoux E, Lagache L, Wagner S, Lèger P, Petit RJ (2011b) Two highly validated multiplexes (12-plex and 8-plex) for species delimitation and parentage analysis in oaks (Quercus spp.). Mol Ecol Resour 11:578–585
Hoeltken AM, Buschbom J, Kaetzel R (2012) Die Artintegrität unserer heimischen Eichen Quercus robur L., Q. petraea (Matt.) Liebl. und Q. pubescens Willd. aus genetischer Sicht. Allg Forst Jagdztg 183:5
Huntley B, Birks HJB (1983) An Atlas of past and present Pollen maps for Europe: 0–13000 years ago. Cambridge University Press, Cambridge
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191
Kremer A, Hipp AL (2019) Oaks: an evolutionary success story. New Phytol. https://doi.org/10.1111/nph.16274
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Langella O (1999) Populations 1.2.32 (02/13/2011): a population genetic software. CNRS UPR9034
Lawson DJ, Van Dorp L, Falush D (2018) A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat Commun 9(1):1–11
Lepais O, Gerber S (2011) Reproductive patterns shape introgression dynamics and species succession within the European white oak species complex. Evolution 65(1):156–170
Lepais O, Petit RJ, Guichoux E, Lavabre JE, Alberto F, Kremer A, Gerber S (2009) Species relative abundance and direction of introgression in oaks. Mol Ecol 18:2228–2242. https://doi.org/10.1111/j.1365-294X.2009.04137.x
Maniscalco M, Raimondo FM (2009) Phytosociological study of the acidophilous deciduous oak woods with Ilex aquifolium of Sicily. Fitosociologia 46(2):67–80
Marshall TC, Slate J, Kruuk LEB, Pemberton JM (1998) Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol 7:639–655. https://doi.org/10.1046/j.1365-294x.1998.00374.x
Médail F, Quézel P (1997) Hot-spots analysis for conservation of plant biodiversity in the mediterranean basin. Mediterr Plant Biodivers 84(1):112–127
Mossa L, Bacchetta G, Brullo S (1999) Quercus ichnusae (Fagaceae), a new species from Sardinia. Isr J Plant Sci 47:199–207. https://doi.org/10.1080/07929978.1999.10676774
Mucina L, Bültmann H, Dierßen K, Theurillat JP, Raus T, Čarni A, Šumberová K, Willner W, Dengler J, García RG, Chytrý M, Hájek M, Di Pietro R, Iakushenko D, Pallas J, Daniëls FJA, Bergmeier E, Guerra AS, Ermakov N, Valachovič M, Schaminée JHJ, Lysenko T, Didukh YP, Pignatti S, Rodwell JS, Capelo J, Weber HE, Solomeshch A, Dimopoulos P, Aguiar C, Hennekens SM, Tichý L (2016) Vegetation of Europe: hierarchical floristic classification system of vascular plant, bryophyte, lichen and algal communities. Appl Veg Sci 19:3–264
Musarella CM, Cano-Ortiz A, Piñar Fuentes JC, Navas-Ureña J, Pinto Gomes CJ, Quinto-Canas R, Cano E, Spampinato G (2018) Similarity analysis between species in the genus Quercus L. (Fagaceae) in southern Italy based on the fractal dimension. PhytoKeys 113:79–95. https://doi.org/10.3897/phytokeys.113.30330
Neophytou C, Michiels H-G (2013) Upper Rhine Valley: A migration crossroads of middle European oaks. Forest Ecol Manag 304:89–98
Neophytou C, Aravanopoulos FA, Fink S, Dounavi A (2010) Detecting interspecific and geographic differentiation patterns in two interfertile oak species (Quercus petraea (Matt.) Liebl. and Q. robur L.) us- ing small sets of microsatellite markers. For Ecol Manag 259:2026–2035. https://doi.org/10.1016/j.foreco.2010.02.013
Owusu SA, Sullivan AR, Weber JA, Hipp AL, Gailing O (2015) Taxonomic relationships and gene flow in four North American Quercus species (Quercus section Lobatae). Syst Bot 40(2):510–521
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Peruzzi L, Domina G, Bartolucci F, Galasso G, Peccenini S, Raimondo FM, Albano A, Alessandrini A, Banfi E, Barberis G, Bernardo L, Bovio M, Brullo S, Brundu G, Brunu A, Camarda I, Conti F, Croce A, Iamonico D, Iberite M, Iriti G, Longo D, Marsili S, Medagli P, Pistarino A, Salmeri C, Santangelo A, Scassellati E, Selvi F, Soldano A, Stinca A, Villani M, Wagensommer RP, Passalacqua N (2015) An inventory of the names of vascular plants endemic to Italy, their loci classici and types. Phytotaxa 196:1–217
Peruzzi L, Peruzzi L, Galasso G, Domina G, Bartolucci F, Santangelo A, Alessandrini A, Astuti G, D’Antraccoli M, Roma-Marzio F, Ardenghi NMG, Barberis G, Conti F, Bernardo L, Peccenini S, Stinca A, Wagensommer RP, Bonari G, Iamonico D, Iberite M, Viciani D, Del Guacchio E, Gusso del Galdo G, Lastrucci L, Villani M, Brunu A, Magrini S, Pistarino A, Brullo S, Salmeri C, Brundu G, Clementi M, Carli E, Vacca G, Marcucci R, Banfi E, Longo D, Di Pietro R, Passalacqua NG (2019) An inventory of the names of native, non-endemic vascular plants described from Italy, their loci classici and types. Phytotaxa 410(1):1–215
Petit RJ, Csaikl UM, Bordács S, Burg K, Coart E, Cottrell J, Kremer A (2002a) Chloroplast DNA variation in European white oaks: phylogeography and patterns of diversity based on data from over 2600 populations. For Ecol Manag 156:5–26
Petit RJ, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E, Kremer A (2002b) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manag 156:49–74
Pignatti S (1982) Flora d’Italia, vol 1. Edagricole, Bologna, pp 113–120
Pignatti S (2011) La flora della Sicilia come chiave di lettura per la fitogeografia mediterranea: una visione autobiografica. Biogeographia 30:72–94
Pignatti S, Guarino R, La Rosa M (2018) Flora d’Italia, vol 2. Edagricole-New Business Media, Milano
Pignatti S, Guarino R, La Rosa M (2019) Flora d’Italia, vol 4. Edagricole-New Business Media, Milano
Porras-Hurtado L, Ruiz Y, Santos C, Phillips C, Carracedo Á, Lareu M (2013) An overview of STRUCTURE: applications, parameter settings, and supporting software. Front Genet 4:98
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Sadori L, Narcisi B (2001) The post-glacial record of environmental history from Lago di Pergusa, Sicily. Holocene 11:655–671
Sciandrello S, Guarino R, Minissale P, Spampinato G (2015) The endemic vascular flora of Peloritani Mountains (NE Sicily): plant functional traits and phytogeographical relationships in the most isolated and fragmentary micro-plate of the Alpine orogeny. Plant Biosyst 149(5):838–854. https://doi.org/10.1080/11263504.2014.908978
Terzi M, Di Pietro R, D’Amico FS (2010) Analisi delle specie indicatrici applicata alle comunità a Stipa austroitalica Martinovsky e relative problematiche sin tassonomiche. Fitosociologia 47(1):3–28
Thanou E, Sponza S, Nelson EJ, Perr A, Wanless S, Daunt F, Cavers S (2017) Genetic structure in the European endemic seabird, Phalacrocorax aristotelis, shaped by a complex interaction of historical and contemporary, physical and nonphysical drivers. Mol Ecol 26(10):2796–2811
Tutin TG, Burges NA, Chater AO, Edmondson JR, Heywood VH, Moore DM, Valentine DH, Walters SM, Webb DA (eds) (1993) Flora Europaea, vol 1, 2nd edn. Cambridge University Press, Cambridge
Tzedakis PC, Lawson IT, Frogley MR, Hewitt GM, Preece RC (2002) Buffered tree population changes in a Quaternary refugium: evolutionary implication. Science 297:2044–2047
Viscosi V, Lepais O, Gerber S, Fortini P (2009) Leaf morphological analyses in four European oak species (Quercus) and their hybrids: a comparison of traditional and geometric morphometric methods. Plant Biosyst 143(3):564–574
Viscosi V, Antonecchia G, Lepais O, Fortini P, Gerber S, Loy A (2012) Leaf shape and size differentiation in white oaks: assessment of allometric relationships among three sympatric species and their hybrids. Int J Plant Sci 173(8):875–884
Wang J (2017) The computer program structure for assigning individuals to populations: easy to use but easier to misuse. Mol Ecol Resour 17(5):981–990. https://doi.org/10.1111/1755-0998.12650
Watts WA, Allen JA, Huntley B, Fritz SC (1996) Vegetation history and climate of the last 15,000 years at Laghi di Monticchio, southern Italy. Quat Sci 15:113–132
Yücedag C, Gailing O (2013) Morphological and genetic variation within and among four Quercus petraea and Q. robur natural populations. Turk J Bot 37:619–629
Acknowledgements
The authors wish to acknowledge two anonymous referees and the Editor for valuable feedbacks on an earlier version of the manuscript and Dr. Orazio Caldarella for his help in collecting oak populations in Sicily. This project was funded by funds from Department of Bioscience and Territory, sect. Nature and Forest (NAF) University of Molise, Italy. Dr. A.L. Conte enjoyed a traineeship grant from European Union Erasmus+ Programme.
Funding
Open access funding provided by Università degli Studi di Roma La Sapienza within the CRUI-CARE Agreement. Funding provided by the project “Impatto dei cambiamenti climatici e antropici sulla natura, sull’ambiente e sul paesaggio” (PROGET_20162019300117LOYDIVISIONENAF).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Christian Ammer.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Di Pietro, R., Conte, A.L., Di Marzio, P. et al. Does the genetic diversity among pubescent white oaks in southern Italy, Sicily and Sardinia islands support the current taxonomic classification?. Eur J Forest Res 140, 355–371 (2021). https://doi.org/10.1007/s10342-020-01334-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-020-01334-z