
Abstract
Pyrethroids, targeting the voltage gated sodium channel (VGSC), are fundamental for the control of arboviral disease circulation. The spread of pyrethroid resistance among vector species represents thus a major public health concern. Culex pipiens is one of the most abundant European mosquito species and main vector of West Nile virus, leading cause of arboviral encephalitis worldwide. Despite this, monitoring of its resistance status and the understanding of underlying mechanisms are widely neglected. Herein, we performed an oligo-hybridization capture approach on 82 Cx. pipiens specimens from Italy and Greece to investigate the whole coding sequence of the vgsc gene for the presence of known and potential knock-down resistance (kdr) mutations associated with target-site resistance to pyrethroids in insects. Among the 26 non-synonymous substitutions revealed by the analysis, the super-kdr haplotype—i.e. the association of kdr-alleles 918T and 1014F, known for conferring a strongly enhanced resistance phenotype in Musca domestica – was revealed for the first time in mosquitoes. Three more potential kdr alleles were detected for the first time in Cx. pipiens and multiple kdr variants were observed for locus 1014, with allele 1014F, reaching frequencies > 80%. Overall, results depict a worrisome situation that could affect the ability to control West Nile virus outbreaks in southern Europe. To avoid this, resistance monitoring needs to be intensified and an enhancement of the diagnostic tool box for the easy detection of different kdr-variants (including in particular the super-kdr haplotype) and for subsequent functional studies on the resistance phenotype of detected variants, is required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Key message
-
Resistance to insecticides is on the rise in mosquitoes capable of transmitting diseases to humans.
-
Little is known on resistance mechanisms in Culex pipiens, one of the most dangerous mosquitoes.
-
We looked for genetic traits linked to insecticide resistance in European Cx. pipiens populations
-
We revealed widespread presence of known and novel genetic features associated with resistance
-
It is crucial to clarify possible risks of those VGSC alleles for disease vector control
Introduction
Insecticide resistance is rapidly spreading across vector populations worldwide and has been acknowledged as one of the main public health challenges to be faced in the near future (WHO 2012). Indeed, without adequate preventive actions, insecticide resistance could have a significant operational impact, limiting our ability to control arboviral disease transmission (WHO 2012). To avoid resistance becoming stable in vector populations, strategies to detect resistance and counteract its spread must be put in place as early as possible. The definition of effective insecticide resistance management strategies requires anyway a comprehensive understanding of the resistance phenotype spreading in a population and its underlying mechanisms. Despite being one of the most relevant European arborvirus vector species, information on insecticide resistance in Culex pipiens is severely lacking.
The northern house mosquito Cx. pipiens is one of the most abundant mosquito species in Europe and Italy (Harbach 2012, Mancini et al. 2017; Severini et al. 2009) where it is present with two biotypes, i.e. form pipiens and form molestus (Becker et al. 2012; Harbach 2012; Di Luca et al. 2016; Brugman et al. 2018; Bisia et al. 2020). The above biotypes exhibit important ecophysiological differences (e.g. biting preference, breeding sites and diapause) which may impact also their relative importance as vectors of arboviral diseases (Farajollahi et al. 2011; Harbach 2012; Brugman et al. 2018; Haba and McBride 2022).
Culex pipiens plays indeed a crucial role in the transmission of at least three viruses circulating at present in Europe: West Nile, Usutu, and Sindbis, with West Nile having the greatest epidemiological impact (Brugman et al. 2018). West Nile virus (WNV, Flaviviridae), the most widespread arbovirus and the leading cause of arboviral encephalitis worldwide (Reiter 2010; Ciota 2017), is transmitted in an enzoonotic transmission cycle involving mosquitoes as vectors and birds as main vertebrate hosts, but occasionally humans and equids can get infected (Brugman et al. 2018). In humans, WNV infections are often asymptomatic or mild but can lead also to a neuro-invasive form with a 10–20% fatality rate (Gyure 2009). During the last few decades, Europe has witnessed a steady increase in WNV cases, with the until now largest outbreak in 2018 with more than 1600 confirmed human cases—most of them in Italy, Greece, Romania and Hungary—and a 9% case fatality ratio, (ECDC, Surveillance atlas of infectious diseases). Large outbreaks in Europe were observed subsequently also in 2022 (> 1100 cases) and 2023 (> 700 cases). At present, no specific treatment nor vaccine against WN disease in humans is available; therefore, limiting human-vector contact and control interventions targeting Cx. pipiens are the only tools available to reduce disease transmission.
Mosquito control interventions, aiming at the reduction of the vector population, are considered to have one of the highest returns on investment in public health (WHO 2015, 2017). In Europe, larval control is reported as the most widely adopted method to reduce the abundance of Cx. pipiens and also Aedes albopictus (i.e. vector of arboviruses such as dengue, chikungunya and Zika) and eventually also the risk of disease outbreaks (ECDC 2012, 2020). Adulticidal treatments are recommended mainly in the case of autochthonous WNV human cases, the detection of viral circulation among mosquitoes or sentinel animals or in situations with exceptionally high risk factors (involving a higher than normal concentration of vectors and hosts) (ECDC 2012, 2020). In such scenarios, adulticidal interventions are strongly needed to reduce fast and effectively the adult mosquito population able to transmit the virus. Indeed, aerial ULV adulticiding is the only method with scientific evidence for its effectiveness in reducing the incidence of WNV human cases, but is not authorized in the EU (ECDC 2020).
In Europe only pyrethroids—which target the voltage gated sodium channel (VGSC) and interfere with normal nervous signal transmission—are allowed for adulticidal treatments (EU Directive 2012; EU Directive 1998). However, the often excessive and erroneous usage of pyrethroids to reduce the considerable nuisance linked to mosquito bites (especially in tourist areas), together with the usage of the same active ingredients for the control of agricultural pests, has led to the selection of pyrethroid resistance (PR) in target species, putting possibly at risk our ability to control fast and effectively ongoing disease transmission (Hemingway and Ranson 2000; Soderlund and Knipple 2003; Rinkevich et al. 2013; Zhu et al. 2016).
Main mechanisms underlying pyrethroid resistance in mosquitoes are overexpression of enzymes involved in detoxification pathways (i.e. metabolic resistance) and substitutions within the VGSC (i.e. target-site resistance). Such substitutions, alter the binding and interaction of the pyrethroid with the VGSC, increasing the dose of insecticide required for knock-down and resulting thus in reduced susceptibility of the mosquito. Therefore, such substitutions are commonly referred to as knock-down resistance (kdr) mutations (Davies et al. 2007; Dong et al. 2014). More than 50 such mutations in the VGSC with varying importance in resistance have been described in different insect species (Davies et al. 2007; Rinkevich et al. 2013; Dong et al. 2014), and abundant literature exists for main vector species such as Anopheles spp. malaria vectors (Clarkson et al. 2021), Aedes mosquitoes (Vontas et al. 2012; Moyes et al. 2017) or Culex quinquefasciatus (Komagata et al. 2009; Li et al. 2012).
Information on the resistance status and its underlying mechanisms are instead scarce for Cx. pipiens and few studies have specifically focused on populations from Europe. Phenotypic PR (assessed by bioassays) was reported in Cx. pipiens populations from countries of the Mediterranean basin including Greece, Italy, Spain and Turkey (Ben Cheikh et al. 1998; Zayed et al. 2006; Vasquez et al. 2009; Peery et al. 2012; Kioulos et al. 2013; Fotakis et al. 2017; Tmimi et al. 2018; Guntay et al. 2018; Paaijmans et al. 2019; Ser and Cetin 2019; Pichler et al. 2022; ECDC 2023), as well as in Belgium (Vereecken et al. 2022). In Italy, reduced susceptibility to permethrin was detected by WHO-tube bioassays in 9 populations out of 10 analysed, with lowest mortality rates (< 20%) observed in coastal tourist sites, suggesting a highly worrisome situation and a possible negative impact of PR on vector control measures (Pichler et al. 2022).
Similarly, information on target-site resistance mechanisms in Cx. pipiens is limited: so far only substitution of the leucine in position 1014 of the VGSC with phenylalanine (L1014F; Martinez-torres et al. 1999) and rarely serine or cysteine have been reported (L1014C/S; Scott et al. 2015). In Europe, substitutions L1014F/C have been detected in Greece, Italy and Turkey, reaching worrisome frequencies in some populations (Kioulos et al. 2013; Taskin et al. 2016; Fotakis et al. 2020, 2022; Guz et al. 2020; Pichler et al. 2022; ECDC 2023), while allele 1014S was detected rarely in Greece and Turkey (< 5%; Fotakis et al. 2022). In Italy, Pichler et al (2022) reported the 1014F allele in all 10 populations examined with highest frequencies in those populations from coastal tourist sites where bioassays had detected phenotypic resistance and a significant association between the presence of the 1014F allele and permethrin resistance was confirmed.
Despite these clear evidence -obtained via bioassays and genetic investigations- indicating the spread of PR across Cx. pipiens populations in Europe and its possible negative impact on the effectiveness of vector control measures, data are very sparse in terms of time and space (ECDC 2023) and few studies were designed for a better understanding of resistance mechanisms and phenotypes in Cx. pipiens.
Here we take advantage of the targeted next-generation sequencing (NGS) approach developed by Itokawa et al. (2019) which allows a high coverage sequencing of the whole vgsc gene. This approach allows to detect novel mutations across the whole gene, a fundamental advantage compared to other approaches limited to small stretches of the vgsc (investigated by partial sequencing of single exons) or to single-nucleotide polymorphisms (SNPs; investigated by PCR approaches). Thanks to this approach, it was possible to gather first information concerning the existence, distribution and frequency of known and novel mutations in the VGSC with possible or confirmed impact on PR in Cx. pipiens specimens from Italy and Greece.
Materials and methods
Mosquito sampling
Sampling of Cx. pipiens specimens was performed from June to October 2022 in 8 sites from 4 provinces from northern to southern Italy as well as one site from Attica region in Greece (Table S1).
In Attica region, adult mosquitoes were captured in the framework of a research project for the entomological surveillance of mosquitoes using BG Sentinel traps equipped with lure and CO2, stored on ice for transportation to the laboratory and then preserved in ethanol until DNA extraction. Samples from Italy were collected by collaborating research groups as egg rafts or larvae from larval breeding sites such as road ditches and sewers; sampled eggs were stored on wet filter paper, and larvae were maintained in plastic containers filled with water and then sent to the Department of Public Health and Infectious Diseases (DPHID) at Sapienza University of Rome where eggs were allowed to hatch and larvae were reared at larval density of 100 larvae/l in the insectary of DPHID at T = 26 °C ± 1 °C, RH = 60 ± 5%, 14:10 h light: dark photoperiod and fed with artificial dry cat-food. Pupae were collected daily and transferred into 40 cm3 cages. Emerged adults were identified as Cx. pipiens using morphological keys (Severini et al., 2009), kept at the same temperature and humidity as larvae and fed with 5% sugar solution at libitum. Adult mosquitoes were subsequently freeze-killed and stored at -80 without any preservant.
Molecular analysis
DNA was extracted from whole mosquito carcasses using the DNeasy Blood and Tissue kit (QIAGEN) and subsequently quantified using a microplate reader and the Quant-iT PicoGreen kit (Thermo Fisher Scientific), according to manufacturer’s instructions. Only specimens with > 4 ng/ul DNA concentration were processed; mean DNA concentration was 13 ng/ul varying from a minimum of 4 to a maximum of 49 ng/ul. Identification of biotypes Cx. pipiens f. pipiens and f. molestus was performed following the protocol described by Bahnck and Fonseca (2006).
Targeted sequencing of the vgsc gene was conducted for 82 Cx. pipiens as described by Itokawa et al. 2019. Given the elevated sequence homology observed for the vgsc gene across dipteran species, it was possible to perform library construction and oligo hybridization capture using the biotinylated oligo probes designed for Ae. albopictus (Davies et al. 2007; Itokawa et al. 2019). We modified the probe set from the previous version described in Itokawa et al. (2019), resulting in Culicinae_VGSC_V2 (Table S2). In this version 35 new probes were added to cover several vgsc exons (Exon2 and 16.5 in Aedes aegypti and Exon2, 8, 14, 24, 29, and 32 for Cx. pipiens) which showed relatively weak capture rate due to low homology and/or short exon length (Itokawa et al. 2019). The probes were synthesized by IDT as xGen Lockdown Probes. NGS library preparation and hybridization capture enrichment were conducted with the protocol described previously (Itokawa et al. 2019). Briefly, 10–20 ng gDNA of each sample was fragmented, end-prepped, and indexed adapters ligated using NEBNext Ultra II FS DNA Library Prep Kit for Illumina (NEB) as indicated in the manufacturer’s protocol. After adapter ligation reaction, ligase was heat inactivated by incubating samples at 65 °C for 20 min, and then all indexed libraries were pooled into a single tube. The pooled library was purified by 0.8 × AMpureXP (Beckman Coulter) and eluted into 20 uL H2O. The purified pooled library was subjected to hybridization capture, washing, and final amplification (12 cycles) using xGen Hybridization and Wash Kit (IDT) and the Culicinae_VGSC_V2 probe set as indicated in the manufacturer’s protocol. Paired-end sequencing (PE150) was performed on an Illumina NextSeq1000 obtaining a range of 122,156–480,752 read pairs per individual. Raw NGS reads were deposited to Sequence Read Archive (DRA) of DNA Data Bank of Japan (DDBJ) under accession number DRR528067–DRR528091 and DRR567865–DRR567921.
Bioinformatic analysis
The automated pipeline MoNaS v2.0 (https://github.com/ ItokawaK/MoNaS) was employed as described by Itokawa et al. 2019 to analyse the obtained NGS read data and to filter, call and annotate functionally variants. MoNaS pipeline relies on BWA (Li and Durbin 2009), SAMTools (Li et al. 2009), BCFtools csq (Danecek and McCarthy 2017), and FreeBayes (Garrison and Marth 2012). Scaffold supercont3.182_3 (Itokawa et al. 2019) (https://github.com/ItokawaK/MoNaS), which was constructed by correcting gaps and possible consensus errors in scaffolds in the Cpip_J2 (Arensburger et al. 2010) assembly, was used as the vgsc gene reference. Read coverages on each exon of each sample were calculated with BEDtools (Quinlan and Hall 2010)) “mulitcov” function. Linkage disequilibrium was computed using the likelihood ratio test available in ARLEQUIN ver. 3.5. (Excoffier and Lischer 2010). Chi-square or Fisher’s exact tests were performed to evaluate significant differences in allele frequencies between populations.
Results
High quality sequences of the vgsc gene were obtained for 82 Cx. pipiens specimens, 21 of which were identified as Cx. pipiens f. molestus (6 from Greece and 15 from Italy) and 61 as Cx. pipiens f. pipiens (4 from Greece and 57 from Italy). Reads were aligned to the vgsc gene from Cx. quinquefasciatus with mean coverage, (computed as number of overlapping reads) per exon per specimen varying from 38 to 5927(Figure S1). In our pipeline, variant and genotype calling were conducted by FreeBayes v1.3.6 (Garrison and Marth 2012). Filtering by quality values in VCF (QUAL > 50), identified 218 variable sites (in comparison to reference supercont3.182_3) within the vgsc coding sequences across all 82 mosquitoes. Total read depth per genotype varied from 14 to 478 with median 153. Of the identified variable sites, 215 were polymorphic among the analysed specimens with a distribution/exonic region as detailed in Table 1. Two hundred sites were biallelic SNPs or MNPs (Multiple Nucleotide Polymorphisms, i.e. highly linked mutations observed within few nucleotide positions). The allelic balance values (i.e. depth of the first allele in genotype/total depth) in all heterozygous genotypes ranged between 0.25 and 0.76 with median 0.51.
Among the polymorphic loci, 26 resulted in non-synonymous mutations of which 24 were biallelic while the remaining 2 loci were multiallelic (Table S3). Impact on pyrethroid susceptibility is highly suspected or confirmed for mutations causing substitutions in 5 amino acid positions of the VGSC (Table 2). Overall allelic frequencies for mutations in these 5 loci varied between 0.6% (allele 253L) and 59% (allele 1014F; Table 3 and Fig. 2).
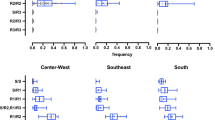
Mutation V253L, situated in exon 7 within the linker connecting helix S4 and S5 of domain I, was detected only once in heterozygosis in one Cx. pipiens f. molestus specimen from Italy (Bari; Fig. 1, Tables 3 and S4).

Results obtained for 82 Culex pipiens specimens from Italy and Greece (split per sampling area) for the genotyping of 5 amino acid positions within the VGSC with potential impact on PR. Number of specimens analysed is shown between parentheses. Panel A) Frequency of wildtype or mutant alleles observed for 5 amino acid positions within the VGSC (253, 918, 1014, 1534, 1879). Panel B) Frequency of different amino acid variants observed for position 1014 of the VGSC. L = wildtype allele, F, C, S = mutant (kdr)alleles
Mutation M918T, situated in exon 19d forming the linker connecting helix S4 and S5 of domain II was detected only in heterozygosis in 8 Italian specimens (4 Cx. pipiens f. molestus and 4 Cx. pipiens f. pipiens) all of which carried also the kdr allele 1014F in homo- or heterozygosis (Fig. 1, Tables 3, S4 and S5).
For locus 1014, situated in Exon 20 forming helix 6 of domain II, 4 different amino acid variants were detected: the wildtype (susceptible) allele 1014L and 3 kdr alleles, 1014F, C, S (Fig. 1, Tables 3 and 4).
The wildtype allele 1014L was observed at an overall frequency of 30%, with frequencies per region ranging from 5% (Attika-region) to 70% (Trentino) and only 12 homozygote specimens (9 Cx. pipiens f. pipiens and 3 Cx. pipiens f. molestus from Italy) across the whole sample (Table 4 and S4). The kdr-allele 1014F, encoded by two different triplets TTT or TTC, represented the most frequent allele with an overall allelic frequency of 59%, varying from 25% (Trentino) to 87% (Ferrara) per province; 41% of specimens overall were identified as homozygotes for the 1014F allele (Table 3). Among the two alternate triplets encoding for allele 1014F the triplet TTT (allele 1014F TTT) was more frequent in both biotypes and was detected at an overall frequency of 46%, varying from 5% (Trentino) to 77% (Ferrara). Allele 1014F TTC was observed at an overall frequency of 13% and, while absent from the Greek sample, in Italy its frequency varied from 9% (Ferrara) to 18% (Bari) (Table S6). Allele 1014C, present at an overall frequency of 10%, was most frequent in Greece (allelic frequency = 50%) where it represented the most common variant in position 1014. In Italy, allele 1014C was detected only in Bari province (in both biotypes) at a frequency of 21%, in heterozygosis with allele 1014F (N = 5) or the wildtype allele 1014L (N = 2). Allele 1014S was observed only in one Cx. pipiens f. pipiens specimen from Trentino region, in heterozygosis with the wildtype allele 1014L (Table 4 and S4).
Mutation F1534L, encoded by triplettes TTA or CTC, situated in Exon 29, forming helix 6 of domain III was detected at an overall frequency of 8% (Figs.1, 2, Table 3). Allele 1534L TTA was detected in all 4 Italian provinces in a total of 10 specimens (4 Cx. pipiens f. molestus and 6 Cx. pipiens f. pipiens), two of which were homozygotes. Allele 1534L CTC was detected only once in heterozygosis in a Cx. pipiens f. pipiens specimen from Bari province (Table S4).

Overall frequencies (bars) of identified kdr mutations along with significant linkage disequilibrium (LD) plot between pairs of mutations; + = significant LD detected; − = no significant LD; * = no LD computed
Mutation P1879S, situated in exon 32 forming the carboxyl terminal domain, was detected only in heterozygosis in 2 Greek Cx. pipiens f. pipiens specimens with both specimens carrying also mutations in position 1014: one was a homozygote for allele 1014F while the other was a heterozygote 1014F/1014C (Fig. 1, Tables 3, S4 and S5).
Differences between biotypes were investigated by performing a Fisher exact test considering only mutations detected at an overall frequency > 5% (i.e. mutations in position M918T, F1534L and L1014F/C) and specimens from Italian provinces where both biotypes were detected. For none of these loci significant differences between biotypes were detected (Tables 5 and S7).
A linkage disequilibrium test performed on all specimens using the likelihood ratio test available in ARLEQUIN ver. 3.5 revealed significant linkage between allele 1534L (encoded by the triplet TTA) and the two triplets encoding 1014F (TTT and TTC), as well as between the mutation M918T and the allele 1014F (encoded by the triplet TTT; see Fig. 2).
Discussion
The novel targeted sequencing approach developed by Itokawa et al. (2019) allowed to: 1) detect for the first time the super-kdr haplotype (918 T + 1014F) in mosquitoes 2) find three substitutions possibly associated with pyrethroid resistance never described before in Cx. pipiens (i.e. variants in position 253, 1534 and 1879) and 3) evaluate the spread of different kdr variants in position 1014 of the VGSC, already known for its importance in conferring resistance in Cx. pipiens.
Mutation M918T, situated in pyrethroid receptor site I (PyR1), is detected herein for the first time in mosquitoes, and in clear linkage disequilibrium with allele 1014F: all specimens carrying the allele 918T (detected only in heterozygosis) were also heterozygotes (N = 4) or homozygotes (N = 4) for allele 1014F (encoded by the triplet TTT). In the latter specimens thus the two alleles 918 T and 1014F are for sure present as a haplotype on the same chromosome; this haplotype, 918T + 1014F, was reported for the first time in Musca domestica and has since been observed in several other insect species (Dong et al. 2014). Electrophysiological studies on insect sodium channels expressed in Xenopus oocytes revealed that the combination of the two alleles is associated with an up to 1000 fold reduction in susceptibility to pyrethroids, compared to a ̴100-fold reduction observed when only allele 1014F is present (Vais et al. 2000), leading thus to define it as super-kdr haplotype (Lee et al. 1999). The increased resistance phenotype of the super-kdr haplotype was confirmed conducting bioassays on M. domestica with resistance ratios being 5 to 10 times higher for strains containing the super-kdr haplotype compared to strains carrying only resistance alleles in position 1014 (Scott et al. 2013; Sun et al. 2016). Given the significant role of this haplotype in enhancing the PR phenotype its first discovery in Cx. pipiens and mosquitoes in general is of highest relevance. It will be crucial to confirm the role of this haplotype on the resistance phenotype also in Cx. pipiens and to monitor its spread across Italy and Europe.
Mutation V253L, found only once in heterozygosis in a specimen also carrying allele 1014F in heterozygosis, is situated in the second pyrethroid receptor site (PyR2), symmetrically to the residue in position 918 in PyR1 (Du et al. 2013; Sun et al. 2022). While nothing is known on the phenotype produced by this exact mutation, recently a substitution in the same position (V253F) was found in Brazilian Ae. aegypti populations where it was detected in linkage with mutation F1534C (situated in PyR1; Itokawa et al. 2021). Electrophysiological studies confirmed the role of allele 253F in reducing the channel sensitivity to both, type I and type II pyrethroids, but also in altering the channel gating, suggesting a potential negative impact on mosquito fitness. Notably, when the V253F mutation was expressed together with allele 1534C, this negative alteration of the channel activity was completely abolished, while the sensitivity to pyrethroids was further reduced compared to the presence of only one of the two mutations (Sun et al. 2022). The impact of this mutation on pyrethroid susceptibility in Cx. pipiens and the synergy between alleles 253L and 1014F should be further addressed in future studies.
Mutation F1534L is situated in PyR1; while absent from the Greek sample, the allele 1534L was detected in all Italian provinces (overall A.F in Italy = 9%) at frequencies ranging from 2 to 20%, representing thus the most frequent allele with possible impact on PR after 1014F in Italy. In the present study we detected two codons producing the 1534L allele, both being one mutational step away from the wildtype allele, suggesting their independent insurgence and thus strong selective pressure on the locus. Different substitutions have been described for this amino acid position in other mosquito species, with mutation F1534L being detected in Ae. albopictus (Chen et al. 2016; Su et al. 2019) and Ae. aegypti (Kushwah et al. 2020). Electrophysiological studies of these substitutions have confirmed their ability to reduce susceptibility to pyrethroids, especially—but not exclusively—of type I, while no significant modifications in channel properties were detected (Zhaonong et al. 2011; Du et al. 2013; Yan et al. 2020; Sun et al. 2022) suggesting low impact on the functionality of the channel and thus mosquito fitness. Substitutions in this position are often found in association with other potential kdr-alleles (e.g. in position 253, see above), but never in linkage disequilibrium with allele 1014F, as instead suggested by the present results (Fig. 2). The phenotype of possible double mutants (1014F + 1534L) needs to be evaluated: being the two mutations situated in the two pyrethroid receptor sites this may produce an increased resistance phenotype similar to other haplotypes combining mutations in both receptor sites (e.g. 253F + 1534C or 918 T + 1014F; Vais et al. 2000; Sun et al. 2022)).
Mutation P1879S is situated in the carboxyl terminal domain known for its role in the fast inactivation and closure of the VGSC (Dong et al. 2014); allele 1879S was detected in the present study only in two Greek specimens in heterozygosis, in linkage with mutations in position 1014. Mutation P1879S was detected for the first time in Plutella xylostella (Sonoda et al. 2008, Sonoda et al. 2010)—always in association with allele 1014F and/or other potential kdr mutations within the VGSC—and with strong support for its implication in PR (Rinkevich 2013; Sonoda et al. 2008). In mosquitoes this substitution has been detected by Clarkson et al (2021) in Anopheles coluzzii and another substitution in the same position (P1879L) has been detected in both, An. coluzzii and An. gambiae. Remarkably, also in these populations the mutation did not appear alone but in strong linkage disequilibrium with mutation L1014F, suggesting possible synergism between the two mutations.
Mutations in position 1014, situated in PyR2, are among the most well-known and widespread mutations not only in mosquitoes but also in other insect species (Dong et al. 2014). Mutation L1014F was the first kdr-mutation to be detected (in M. domestica; Williamson et al. 1993, 1996) and since then several other allelic variants were described for this position (L1014F/H/S/C/W, Rinkevich et al. 2013; Dong et al. 2014; Liu 2015; Scott et al. 2015; Du et al. 2016). Electrophysiological evidence for the impact of these mutations on pyrethroid susceptibility has been gathered since 1997 (Smith et al. 1997; Burton et al. 2011; Wang et al. 2015).
Among the different variants in position 1014, allele 1014F appears to be by far the most reported one in Cx. pipiens including reports from several European countries (Belgium, Greece, Italy, Romania, Turkey; Scott et al. 2015; ECDC 2023).
In the present study we observed in Italy three amino acid -variants for this position (1014F/C/S). In agreement with previous studies, allele 1014F appeared to be the most widespread (overall A.F. = 59%), with highest frequencies observed in Ferrara province (87%) where bioassays on samples from the same study site highlighted strongly reduced susceptibility to pyrethroids (mortality < 20%) in significant association with the presence of allele 1014F (Pichler et al. 2022).
The presence/distribution of the different kdr alleles in position 1014 across our sample may be explained by different insecticidal selective pressure or geographical connection between sites or both. For example, different selective pressure is supposed to promote the insurgence and spread of the two alleles 1014F and 1014S (first reported in Cx. pipiens in 1999; Martinez-torres et al. 1999; Scott et al. 2015): electrophysiological and bioassay data suggest indeed a different resistance phenotype of the two alleles with allele 1014F conferring resistance to both, pyrethroids and DDT and 1014S conferring resistance mainly to DDT (Martinez-torres et al. 1999; Burton et al. 2011).
For allele 1014C possibly both, geographical connections and differential selective pressure/fitness cost impact the observed distribution: in our study 1014C is the most frequent variant in the Greek sample (AF = 50%), in agreement with what observed by Fotakis et al. (2017, 2022) who detected this variant at high frequencies (> 70%) in several Greek populations. The exclusive presence of this allele in Italy in Bari province could thus be explained by the tight maritime connections with Greece. No clear data on the resistance phenotype and fitness cost of allele 1014C (first detected in Cx. pipiens in 2012; Wang et al. 2012) are available; anyway the allele is suggested to have similar relevance as the 1014S allele but with a higher contribution to pyrethroid resistance (Kim et al. 2007; Zhong et al. 2013; Taskin et al. 2016). It is also noteworthy that the insurgence of the 1014C allele (encoded by TGT) requires two mutational steps from the wildtype allele 1014L(TTA) to the kdr allele 1014F (TTT) and then 1014C (TGT), suggesting that at least in some circumstances allele 1014C has some selective advantage in terms of fitness cost or resistance phenotype compared to the 1014F allele (Taskin et al. 2016).
Further genetic variability is introduced by the 1014F allele being encoded by two different codons, i.e. TTT and TTC. This was described previously for Cx. pipiens by Taskin et al. (2016) who observed allele 1014FTTC at low frequencies. In the present study the 1014FTTC variant reached an overall frequency of 13% and was present in all Italian provinces at frequencies ranging from 10 to 20%.
Together with the presence of alleles 1014C and 1014S this represents an important diagnostic challenge: indeed, most studies apply the allele-specific-PCR approach by Martinez-torres et al. (1999) which is able to identify allele 1014FTTT but does not detect the variant 1014FTTC and fails to identify correctly alleles 1014C or 1014S, leading thus to the possible underreporting of these kdr variants. This can hamper the evaluation of the impact of different alleles on pyrethroid susceptibility and thus mosquito control activities.
Concerning the distribution of the described variants between the two Cx. pipiens biotypes the present study did not produce any support for significant differences, although this estimation is strongly limited by the small number of Cx. pipiens f. molestus included. There were instead some suggestions for differences in the circulation of alleles between Italy and Greece, with specimens from Greece showing a much higher frequency of allele 1014C compared to Italy (50% vs 5%) and the exclusive presence of allele 1879S. Anyway, due to the low number of specimens analysed and all of them coming from one single sampling site, no general conclusions about the Greek sample can be made.
Overall, this study provides a clearer picture of PR in Cx. pipiens by highlighting the circulation of multiple previously unknown potential kdr alleles as well as the circulation of the kdr allele 1014F in association with allele 918 T, which is analogous to the super-kdr haplotype (described in M. domestica). As emphasized by Lehane et al. (2024), it will be crucial to bridge gaps between knowledge about insecticide resistance and its actual impact on operational control measures. Only for allele 1014F data on its impact on pyrethroid susceptibility exist, including studies on populations from the same sampling sites examined in the present study (Pichler et al 2022). Further work is instead still required to confirm phenotypic impacts of the newly discovered alleles including toxicological and electrophysiological studies. Additionally, it will be necessary to clarify whether the observed kdr allele frequencies indicate an ongoing selective sweep or some balance between selective pressures on insecticide resistance and fitness cost (Kliot and Ghanim 2012) is essential for predicting future outcomes. While in presence of selective pressure the strength of the conferred resistance may lead to an increase in frequency of kdr alleles, associated fitness costs may counterbalance this (Kliot and Ghanim 2012). Information on both these aspects are thus necessary to understand the spread and rise of kdr alleles, as well as the possibility of a population to reverse to susceptibility in the absence of selective pressure. Altogether such information will provide valuable information to decision makers for the planning of mosquito control activities and the deployment of integrated vector management plans aiming at maintaining effective the currently only chemical tool available for the control of adult mosquito populations and arboviral disease outbreaks in Europe.
Author contribution
Verena Pichler, Kentaro Itokawa, Shinji Kasai, Alessandra della Torre contributed to conceptualization, Verena Pichler, Kentaro Itokawa, Noboru Minakawa, Paola Serini contributed to methodology; Beniamino Caputo, Romeo Bellini, Rodolfo Veronesi, Claudio De Liberato, Federico Romiti, Daniele Arnoldi, Annapaola Rizzoli, Riccardo Paolo Lia, Domenico Otranto, Antonios Michaelakis, Marina Bisia contributed to sampling; Kentaro Itokawa, Verena Pichler, Carlo Maria de Marco contributed to data analysis; Verena Pichler, Kentaro Itokawa, Shinji Kasai, Alessandra della Torre contributed to writing—original draft preparation; Verena Pichler, Noboru Minakawa, Shinji Kasai, Alessandra della Torre, Beniamino Caputo, Antonios Michaelakis contributed to funding acquisition. All authors commented on previous versions of the manuscript and read and approved the final manuscript.
Data availability
The datasets generated and analysed during the current study are available in the Sequence Read Archive (DRA) of DNA Data Bank of Japan (DDBJ) under accession number DRR528067–DRR528091 and DRR567865–DRR567921.
References
Arensburger P, Megy K, Waterhouse RM et al (2010) Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science 330:86–88. https://doi.org/10.1126/science.1191864
Bahnck CM, Fonseca DM (2006) Rapid assay to identify the two genetic forms of Culex (Culex) pipiens L. (Diptera: Culicidae) and hybrid populations. Am J Trop Med Hyg 75:251–5. https://doi.org/10.4269/ajtmh.2006.75.2.0750251
Becker N, Jöst A, Weitzel T (2012) The culex pipiens complex in Europe. J Am Mosq Control Assoc 28:53–67. https://doi.org/10.2987/8756-971X-28.4s.53
Ben Cheikh H, Ben Ali-Haouas Z, Marquine M, Pasteur N (1998) Resistance to organophosphorus and pyrethroid insecticides in Culex pipiens (Diptera: Culicidae) from Tunisia. J Med Entomol 35:251–260. https://doi.org/10.1093/jmedent/35.3.251
Bisia M, Jeffries CL, Lytra I et al (2020) A comparison of adult mosquito trapping methods to assess potential West Nile virus mosquito vectors in Greece during the onset of the 2018 transmission season. Insects 11:329. https://doi.org/10.3390/insects11060329
Brugman VA, Hern LM, Medlock JM, et al (2018) The role of Culex pipiens L . ( Diptera : Culicidae ) in virus transmission in Europe. Int J Environ Res Public Health 2018;15(2):389. https://doi.org/10.3390/ijerph15020389.
Burton MJ, Mellor IR, Duce IR et al (2011) Differential resistance of insect sodium channels with kdr mutations to deltamethrin, permethrin and DDT. Insect Biochem Mol Biol 41:723–732. https://doi.org/10.1016/j.ibmb.2011.05.004
Chen H, Li K, Wang X et al (2016) First identification of kdr allele F1534S in VGSC gene and its association with resistance to pyrethroid insecticides in Aedes albopictus populations from Haikou City, Hainan Island, China. Infect Dis Poverty 5:40–47. https://doi.org/10.1186/s40249-016-0125-x
Ciota AT (2017) West Nile virus and its vectors. Curr Opin Insect Sci 22:28–36. https://doi.org/10.1016/j.cois.2017.05.002
Clarkson CS, Miles A, Harding NJ et al (2021) The genetic architecture of target-site resistance to pyrethroid insecticides in the African malaria vectors Anopheles gambiae and Anopheles coluzzii. Mol Ecol 30:5303–5317. https://doi.org/10.1111/mec.15845
Danecek P, McCarthy SA (2017) BCFtools/csq: haplotype-aware variant consequences. Bioinformatics 33:2037–2039. https://doi.org/10.1093/bioinformatics/btx100
Davies TGE, Field LM, Usherwood PNR, Williamson MS (2007) A comparative study of voltage-gated sodium channels in the Insecta: Implications for pyrethroid resistance in Anopheline and other Neopteran species. Insect Mol Biol 16:361–375. https://doi.org/10.1111/j.1365-2583.2007.00733.x
Di Luca M, Toma L, Boccolini D et al (2016) Ecological Distribution and CQ11 genetic structure of Culex pipiens Complex (Diptera: Culicidae) in Italy. PLoS ONE 11:e0146476. https://doi.org/10.1371/journal.pone.0146476
Dong K, Du Y, Rinkevich F et al (2014) Molecular biology of insect sodium channels and pyrethroid resistance. Insect Biochem Mol Biol 50:1–17. https://doi.org/10.1016/j.ibmb.2014.03.012
Du Y, Nomura Y, Satar G et al (2013) Molecular evidence for dual pyrethroid-receptor sites on a mosquito sodium channel. Proc Natl Acad Sci U S A 110:11785–11790. https://doi.org/10.1073/pnas.1305118110
Du Y, Nomura Y, Zhorov BS, Dong K (2016) Sodium channel mutations and pyrethroid resistance in Aedes aegypti. Insects 7:1–11. https://doi.org/10.3390/insects7040060
ECDC (2012) Guidelines for the surveillance of invasive mosquitoes in Europe
ECDC (2020) Vector control practices and strategies against West Nile virus
ECDC (2023) Literature review on the state of biocide resistance in wild vector populations in the EU and neighbouring countries
EU Directive (2012) Biocidal Products Regulation 528/2012
EU Directive (1998) Biocidal Products Directive 98/8
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Farajollahi A, Fonseca DM, Kramer LD, Kilpatrick AM (2011) ‘“ Bird biting ”’ mosquitoes and human disease: a review of the role of Culex pipiens complex mosquitoes in epidemiology. Infect Genet Evol 11:1577–1585. https://doi.org/10.1016/j.meegid.2011.08.013
Fotakis EA, Chaskopoulou A, Grigoraki L et al (2017) Analysis of population structure and insecticide resistance in mosquitoes of the genus Culex, Anopheles and Aedes from different environments of Greece with a history of mosquito borne disease transmission. Acta Trop 174:29–37. https://doi.org/10.1016/j.actatropica.2017.06.005
Fotakis EA, Giantsis IA, Sierra JC et al (2020) Population dynamics, pathogen detection and insecticide resistance of mosquito and sand fly in refugee camps, Greece. Infect Dis Poverty 4:1–13. https://doi.org/10.1186/s40249-020-0635-4
Fotakis EA, Mavridis K, Kampouraki A et al (2022) Mosquito population structure, pathogen surveillance and insecticide resistance monitoring in urban regions of Crete Greece. Plos Negl Trop Dis 16:e0010186. https://doi.org/10.1371/journal.pntd.0010186
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing. 1–9
Guntay O, Yikilmaz MS, Ozaydin H et al (2018) Evaluation of pyrethroid susceptibility in Culex pipiens of Northern Izmir Province, Turkey. J Arthropod Borne Dis 12:370–377
Guz N, Cagatay NS, Fotakis EA et al (2020) Detection of diflubenzuron and pyrethroid resistance mutations in Culex pipiens from Muğla Turkey. Acta Trop 203:105294. https://doi.org/10.1016/j.actatropica.2019.105294
Gyure KA (2009) West Nile virus infections. J Neuropathol Exp Neurol 68:1053–1060. https://doi.org/10.1097/NEN.0b013e3181b88114
Haba Y, McBride L (2022) Origin and status of Culex pipiens mosquito ecotypes. Curr Biol 32:R237–R246. https://doi.org/10.1016/j.cub.2022.01.062
Harbach RE (2012) Culex pipiens: species versus species complex-taxonomic history and perspective. J Am Mosq Control Assoc 28:10–23. https://doi.org/10.2987/8756-971X-28.4.10
Hemingway J, Ranson H (2000) Insecticide resistance in insect vectors of human disease. Annu Rev Entomol 45:371–391
Itokawa K, Sekizuka T, Maekawa Y et al (2019) High-throughput genotyping of a full voltagegated sodium channel gene via genomic DNA using target capture sequencing and analytical pipeline MoNaS to discover novel insecticide resistance mutations. PLoS Negl Trop Dis 13:1–17. https://doi.org/10.1371/journal.pntd.0007818
Itokawa K, Furutani S, Takaoka A et al (2021) A first, naturally occurring substitution at the second pyrethroid receptor of voltage-gated sodium channel of Aedes aegypti. Pest Manag Sci 77:2887–2893. https://doi.org/10.1002/ps.6324
Kim H, Baek JH, Lee W-J, Lee SH (2007) Frequency detection of pyrethroid resistance allele in Anopheles sinensis populations by real-time PCR amplification of specific allele (rtPASA). Pestic Biochem Physiol 87:54–61. https://doi.org/10.1016/j.pestbp.2006.06.009
Kioulos I, Kampouraki A, Morou E et al (2013) Insecticide resistance status in the major West Nile virus vector Culex pipiens from Greece. Pest Manag Sci 70:623–627. https://doi.org/10.1002/ps.3595
Kliot A, Ghanim M (2012) Fitness costs associated with insecticide resistance. Pest Manag Sci 68:1431–1437. https://doi.org/10.1002/ps.3395
Komagata O, Kasai S, Tomita T (2009) Insecticide resistance in the mosquito culex pipiens complex: concerns about development of pyrethroid resistance. ACS Symp Ser 1014:27–37. https://doi.org/10.1021/bk-2009-1014.ch003
Kushwah RBS, Kaur T, Dykes CL et al (2020) A new knockdown resistance (kdr) mutation, F1534L, in the voltage-gated sodium channel of Aedes aegypti, co-occurring with F1534C, S989P and V1016G. Parasit Vectors 13:327. https://doi.org/10.1186/s13071-020-04201-3
Lee SH, Smith TJ, Knipple DC, Soderlund DM (1999) Mutations in the house fly Vssc1 sodium channel gene associated with super-kdr resistance abolish the pyrethroid sensitivity of Vssc1/tipE sodium channels expressed in Xenopus oocytes. Insect Biochem Mol Biol 29:185–194. https://doi.org/10.1016/S0965-1748(98)00122-2
Lehane Á, Parker-Crockett C, Norris E et al (2024) Measuring insecticide resistance in a vacuum: exploring next steps to link resistance data with mosquito control efficacy. J Med Entomol 61:584–594. https://doi.org/10.1093/jme/tjae029
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A et al (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Li T, Zhang L, Reid WR et al (2012) Multiple mutations and mutation combinations in the sodium channel of permethrin resistant mosquitoes, Culex quinquefasciatus. Sci Rep 2:1–9. https://doi.org/10.1038/srep00781
Liu N (2015) Insecticide resistance in mosquitoes: Impact, mechanisms, and research directions. Annu Rev Entomol 60:537–559. https://doi.org/10.1146/annurev-ento-010814-020828
Martinez-torres D, Chevillon C, Berge JB et al (1999) Voltage-dependent Na ‡ channels in pyrethroid- resistant Culex pipiens L mosquitoes. Pestic Sci 55:1012–1020
Moyes CL, Vontas J, Martins AJ et al (2017) Contemporary status of insecticide resistance in the major Aedes vectors of arboviruses infecting humans. PLoS Negl Trop Dis 7:1–20. https://doi.org/10.1371/journal.pntd.0005625
Paaijmans K, Brustollin M, Aranda C et al (2019) Phenotypic insecticide resistance in arbovirus mosquito vectors in Catalonia and its capital Barcelona (Spain). PLoS ONE 14:1–13. https://doi.org/10.1371/journal.pone.0217860
Peery ZM, Kirby R, Reid BN et al (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418. https://doi.org/10.1111/j.1365-294X.2012.05635.x
Pichler V, Giammarioli C, Bellini R et al (2022) First evidence of pyrethroid resistance in Italian populations of West Nile virus vector Culex pipiens. Med Vet Entomol 36:1–6. https://doi.org/10.1111/mve.12573
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. https://doi.org/10.1093/bioinformatics/btq033
Reiter P (2010) West Nile virus in Europe: Understanding the present to gauge the future. Eurosurveillance 15:4–10. https://doi.org/10.2807/ese.15.10.19508-en
Rinkevich FD, Du Y, Dong K (2013) Diversity and convergence of sodium channel mutations involved in resistance to pyrethroids. Pestic Biochem Physiol 106:93–100. https://doi.org/10.1016/j.pestbp.2013.02.007
Scott JG, Leichter CA, Rinkevihc FD et al (2013) Insecticide resistance in house flies from the United States: resistance levels and frequency of pyrethroid resistance alleles. Pestic Biochem Physiol 107:377–384. https://doi.org/10.1016/j.pestbp.2013.10.006
Scott JG, Hardstone M, Kasai S (2015) Pyrethroid resistance in Culex pipiens mosquitoes. Pestic Biochem Physiol 120:68–76. https://doi.org/10.1016/j.pestbp.2014.12.018
Ser O, Cetin H (2019) Investigation of susceptibility levels of Culex pipiens L. (Diptera: Culicidae) Populations to Synthetic Pyrethroids in Antalya Province of Turkey. J Arthropod Borne Dis 13:243–258
Smith TJ, Hyeock Lee S, Ingles PJ et al (1997) The L1014F point mutation in the house fly Vssc1 sodium channel confers knockdown resistance to pyrethroids. Insect Biochem Mol Biol 27:807–812. https://doi.org/10.1016/S0965-1748(97)00065-9
Soderlund DM, Knipple DC (2003) The molecular biology of knockdown resistance to pyrethroid insecticides. Insect Biochem Mol Biol 33:563–577. https://doi.org/10.1016/S0965-1748(03)00023-7
Su X, Guo Y, Deng J et al (2019) Fast emerging insecticide resistance in Aedes albopictus in Guangzhou China Alarm to the dengue epidemic. PLoS Negl Trop Dis. https://doi.org/10.1371/journal.pntd.0007665
Sun H, Tong KP, Kasai S, Scott JG (2016) Overcoming super-knock down resistance (super-kdr) mediated resistance: multi-halogenated benzyl pyrethroids are more toxic to super-kdr than kdr house flies. Insect Mol Biol 25:126–137. https://doi.org/10.1111/imb.12206
Sun H, Nomura Y, Du Y et al (2022) Characterization of two kdr mutations at predicted pyrethroid receptor site 2 in the sodium channels of Aedes aegypti and Nilaparvata lugens. Insect Biochem Mol Biol 148:1–5. https://doi.org/10.1016/j.ibmb.2022.103814
Taskin BG, Dogaroglu T, Kilic S et al (2016) Seasonal dynamics of insecticide resistance, multiple resistance, and morphometric variation in field populations of Culex pipiens. Pestic Biochem Physiol 129:14–27. https://doi.org/10.1016/j.pestbp.2015.10.012
Tmimi F-Z, Faraj C, Bkhache M et al (2018) Insecticide resistance and target site mutations (G119S ace-1 and L1014F kdr) of Culex pipiens in Morocco. Parasit Vectors 11:51. https://doi.org/10.1186/s13071-018-2625-y
Vais H, Atkinson S, Eldursi N et al (2000) A single amino acid change makes a rat neuronal sodium channel highly sensitive to pyrethroid insecticides. FEBS Lett 470:135–138. https://doi.org/10.1016/S0014-5793(00)01305-3
Vasquez MI, Violaris M, Hadjivassilis A, Wirth MC (2009) Susceptibility of Culex pipiens (Diptera: Culicidae) field populations in Cyprus to conventional organic insecticides, Bacillus thuringiensis subsp. israelensis, and methoprene. J Med Entomol 46:881–887. https://doi.org/10.1603/033.046.0421
Vereecken S, Vanslembrouck A, Kramer IM, Müller R (2022) Phenotypic insecticide resistance status of the Culex pipiens complex : a European perspective. Parasit Vectors 15:1–11. https://doi.org/10.1186/s13071-022-05542-x
Vontas J, Kioulos E, Pavlidi N et al (2012) Insecticide resistance in the major dengue vectors Aedes albopictus and Aedes aegypti. Pestic Biochem Physiol 104:126–131. https://doi.org/10.1016/j.pestbp.2012.05.008
Wang ZM, Li CX, Xing D et al (2012) Detection and widespread distribution of sodium channel alleles characteristic of insecticide resistance in Culex pipiens complex mosquitoes in China. Med Vet Entomol 26:228–232. https://doi.org/10.1111/j.1365-2915.2011.00985.x
Wang L, Nomura Y, Du Y et al (2015) A mutation in the intracellular loop III/IV of mosquito sodium channel synergizes the effect of mutations in Helix IIS6 on pyrethroid resistance. Mol Pharmacol 87:421–429. https://doi.org/10.1124/mol.114.094730
WHO (2012) Global plan for insecticide resistance management in malaria vectors.
WHO (2015) Investing to overcome the global impact of neglected tropical diseases.
WHO (2017) Global vector control repsonse 2017–2030. 2030
Williamson MS, Denholm I, Bell CA, Devonshire AL (1993) Knockdown resistance (kdr) to DDT and pyrethroid insecticides maps to a sodium channel gene locus in the housefly (Musca domestica). Mol Gen Genet 240:17–22. https://doi.org/10.1007/BF00276878
Williamson MS, Martinez-Torres D, Hick CA, Devonshire AL (1996) Identification of mutations in the housefly para-type sodium channel gene associated with knockdown resistance (kdr) to pyrethroid insecticides. Mol Gen Genet 252:51–60. https://doi.org/10.1007/BF02173204
Yan R, Zhou Q, Xu Z et al (2020) Three sodium channel mutations from Aedes albopictus confer resistance to Type I, but not Type II pyrethroids. Insect Biochem Mol Biol 123:103411. https://doi.org/10.1016/j.ibmb.2020.103411
Zayed ABB, Szumlas DE, Hanafi HA et al (2006) Use of bioassay and microplate assay to detect and measure insecticide resistance in field populations of Culex pipiens from filariasis endemic areas of Egypt. J Am Mosq Control Assoc 22:473–482. https://doi.org/10.2987/8756-971x(2006)22[473:uobama]2.0.co;2
Zhaonong H, Yuzhe D, Yoshiko N, Dong K (2011) A sodium channel mutation identified in Aedes aegypti selectively reduces cockroach sodium channel sensitivity to type I, but not type II pyrethroids. Insect Biochem Mol Biol 41:9–13. https://doi.org/10.1016/j.ibmb.2010.09.005
Zhong D, Lo E, Hu R et al (2013) Genetic analysis of invasive Aedes albopictus populations in Los Angeles County, California and its potential public health impact. PLoS ONE 8:1–9. https://doi.org/10.1371/journal.pone.0068586
Zhu F, Lavine L, O’Neal S et al (2016) Insecticide resistance and management strategies in urban ecosystems. Insects 7:1–26. https://doi.org/10.3390/insects7010002
Funding
Open access funding provided by Università degli Studi di Roma La Sapienza within the CRUI-CARE Agreement. This research was supported by EU funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT, Research Node 2), the grant BE-FOR-ERC 2021 by Sapienza University to VP, the Japan Agency for Medical Research and Development (AMED) (grant numbers JP23wm0225030, JP21jm0110024, JP20wm0125006, and JP21fk0108613) and Nagasaki University (Joint Usage/Research Center on Tropical Disease for General Joint Research). It was also supported by the project “Research project for the entomological surveillance of mosquitoes in Attica Region”, financed by the Region of Attica.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Communicated by Wannes Dermauw.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Fig. S1
Coverages (number of reads per exon, after exclusion of PCR duplicates) for each exon in all 82 mosquitoes analysed (PDF 314 KB)
Table S1
: Information on sampling sites per country and province. Table S2: Information on probe set Culicinae_VGSC_v2. Table S3: Information and position of 26 non-synonymous variants identified. Table S4: Number of homozygotes and heterozygotes per sampling province and biotype for each of the 5 loci possibly impacting pyrethroid susceptibility. Table S5: Genotyping results per specimen for the 5 loci possibly impacting pyrethroid susceptibility; in red kdr mutations in homo- or heterozygosis. Table S6: Frequency of the two allelic variants encoding allele 1014F, i.e. 1014FTTT and 1014FTTC, per sampling province. Table S7: Results for Fisher Exact Probability Test to evaluate differences between allele frequency between Cx. pipiens f. molestus and Cx. pipiens f. pipiens. (XLSX 37 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pichler, V., Itokawa, K., Caputo, B. et al. Unbiased sequence analysis of vgsc gene reveals circulation of novel and known knock-down resistance mutations in Culex pipiens, challenging vector control measures. J Pest Sci (2024). https://doi.org/10.1007/s10340-024-01818-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10340-024-01818-6