
Abstract
The invasive fly Drosophila suzukii is a pest that can infest a diverse range of intact, ripening fruits, using its serrated ovipositor. This constitutes a different niche compared to the rotting fruits its ancestors use, especially because these intact fruits have limited quantities of microbes and soluble nutrients for the developing larvae. To investigate the potential role of microbial associations in the niche expansion of this invasive fly, we characterized the bacterial and fungal communities of D. suzukii and various wild fruits from which they developed. To assess cross-generational microbial associations, we also lab-reared fly populations and characterized their microbial communities. Diversity metrics of microbial communities differed significantly between flies and fruits. Different fruit types varied substantially in microbial composition, while flies showed relatively uniform bacterial communities, irrespective of the fruit source they developed on. After approximately ten generations of lab-rearing, bacterial communities still showed considerable overlap with those of wild flies. Fungal communities of flies and fruits showed larger resemblance, with a substantial overlap between wild flies and the fruits on which they had developed. Our study thus reports that the fungal community structure in these pests largely reflects those on the breeding substrates, while these flies might have formed more persistent associations with some bacteria and transmit these across generations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The microbial communities associated with insects are highly diverse, and the dynamics within and between members of the microbiome can affect the fitness and behavior of insects in various ways (Gurung et al. 2019; Jing et al. 2020). Studies on insect microbiota have demonstrated that various factors shape the microbial community composition, ranging from life stages, environment, host genetics and diet (Yao et al. 2019). In some cases, microbes also contribute toward the pest status of invasive insects (Lu et al. 2016). When pest insects are (at least partially) dependent on microbes for their performance or fitness, these microbes have the potential of being utilized as pest management tools, and often one of the first steps is examining the microbiota profiles and their associations in the pests (Crotti et al. 2012; Lewis et al. 2019; Sacchetti et al. 2019; Yao et al. 2019; De Cock et al. 2020).
A pest of ripening fruits, Drosophila suzukii has its origin in Asia (Drosophila suzukii, Ioriatti 2019), and has successfully invaded several parts of America and Europe in the last decade (Calabria et al. 2012; Cini et al. 2014). The female lays eggs on intact ripening fruits and damages them, which is often accompanied by fruit tissue collapse, due to both feeding of the developing larvae inside the fruit, and the rot that is frequently induced by the infestation with D. suzukii (Ioriatti et al. 2018). Fruits become unsuitable for sale, resulting in huge economic losses for fruit growers (Goodhue et al. 2011; Atallah et al. 2014). Drosophila suzukii infests a large range of soft fruits, cherries, and different types of berries (Walsh et al. 2011; Rota-Stabelli et al. 2013; Boughdad et al. 2021; Kwadha et al. 2021). The ability of D. suzukii to colonize intact fruits is driven by its serrated ovipositor, which allows it to puncture the skin of fruits. This is a morphological innovation that enabled the species to expand from its ancestral niche of rotting and decomposing fruit to also infest fresh and ripening fruits (Atallah et al. 2014).
Ripening fruits have fewer nutrients (proteins and sugars) (Silva-Soares et al. 2017) and also harbor a more limited number of microbes than fermenting fruits. This poses possible dietary challenges to the larvae of D. suzukii that develop mostly inside the fruit host. A number of Drosophila species depend on yeasts during their larval stage for essential nutrients, and feed on these yeasts when they are developing on fruits (Starmer and Fogleman 1986; Carvalho et al. 2010). In-line with this, earlier research demonstrated that germ-free D. suzukii larvae failed to develop on protein-poor and fruit-based artificial diets, but this could be rescued with the supplementation of microbiota (Bing et al. 2018). If the microbial communities on intact fruits are indeed insufficient to sustain larval development, then during oviposition D. suzukii females may possibly inoculate the fruits with a set of microbes to supply their developing offspring with the required microbiota (Ben-Yosef et al. 2015; Deans and Hutchinson 2021). Association with some microbes may thus have an essential impact on the life history of these flies. Furthermore, microbes vectored by the flies may have plant pathogenic potential, causing fruit collapse (Hamby and Swett 2015). The question as to what kind of microbial associations these pests might have can thus provide us with a better understanding of microbial contribution in the niche expansion of this invasive pest, in its broad life history, as well as in its pest status.
One of the earliest studies on the bacterial community composition in field-caught D. suzukii revealed Tatumella to be a dominant bacterium across larval and adult stages (Chandler et al. 2014). Although their exact role on insect host fitness is not yet known, the species T. ptyseos reportedly showed plant pathogenic traits (Marín‐Cevada V et al. 2010). Furthermore, bacteria belonging to the families of Acetobacteriaceae, Enterobacteriaceae and Firmicutes have also been reported in D. suzukii. Some of these bacteria are associated with Drosophila in general and provide fitness benefits (Bing et al. 2018). Also yeasts such as Hanseniasporum, Pichia, and Issatchenkia, impact various life history traits of D. suzukii (Lewis and Hamby 2019). Such reports on various microbial associations, point out a question whether the pest associates with microbes through exposure on their breeding site, or whether they might be retaining and transmitting microbiota in a more persistent association.
To investigate the microbial associations of this invasive fly, we characterized the similarities and differences among the bacterial and fungal communities of D. suzukii and various wild fruits from which they developed. The microbial communities that are typically associated with surfaces of various fruits and vegetables can vary considerably, both within and between species (Leff and Fierer 2013). When developing larvae rely mostly on the microbial communities that they encounter on these fruits, we would expect that emerging flies from different fruits would exhibit high variations in their community patterns and align with those of the corresponding fruits. Alternatively, when D. suzukii forms more persistent associations with some of the microbes that they inoculate into the fruits while ovipositing, we would expect flies emerging from different fruit types to harbor similar microbiota. To explore whether emerged flies exhibit consistent microbial associations across various fruits, or whether fruit source plays an important role in determining the flies' microbiome, we sampled the microbiome composition (bacteria and fungi) of fruits and the corresponding emerging D. suzukii flies, collected at four different locations in the Netherlands. Additionally, to identify members that potentially have a more intimate relationship with D. suzukii, and are vertically or horizontally transmitted, we subjected some of these wild flies to lab-rearing conditions and evaluated the changes in their microbiome after several generations.
Methods
Sampling wild flies and fruits
We collected fruits that were infested with D. suzukii in The Netherlands in 2018, across four different locations (between 7.5 and 200 km apart) and five fruit types (Summary in Table 1). In three of these locations, we sampled different fruit types or cultivars. Infestation was determined by identifying the dented spots on the fruits that resulted from egg laying. A large number of collected fruits were individually placed in plastic cups, parafilm-sealed and brought into the laboratory to further incubate them in bottles with autoclaved sawdust (saw dust soaks up unwanted moisture from decomposing fruits) until the adult flies emerged. Incubation conditions were 20 °C, 65% relative humidity and 16 h:8 h light:dark cycle. The emerging flies (referred as “wild flies”) from each fruit and the corresponding fruits were collected and stored at −80 °C until DNA extraction. For each fruit type per sampling site, five individual fruit samples that yielded at least five female flies were randomly chosen for analyses. An additional batch of flies that emerged from a subset of fruits was subsequently lab-reared (referred as “lab-reared flies”). In total, this resulted in 105 samples for analysis, comprising of 45 fruit samples (9 site/fruit combinations × 5 biological replicates) and the corresponding 45 wild fly samples, and an additional 15 lab fly samples (3 site/fruit combinations × 5 biological replicates).
Lab rearing
We started three lab cultures from wild flies that emerged from (1) a cherry cultivar collected in Randwijk, (2) strawberry collected in Dirksland, and (3) elderberry collected in Hoogeveen. These strains were reared in the lab on artificial food (see supplementary file for the fly food composition), for approximately ten generations, before being collected and stored at −80 °C for microbiome characterization. The fly food contained sucrose, glucose, agar, cornmeal, wheatgerm, soy flour, molasses, yeast, ethanol, and was supplemented with antimicrobial agents (propionic acid and nipagin) to reduce mold contamination. The founding population of flies was composed of 8–10 individuals per fruit type. Each successive generation, at least fifty individual flies were transferred to the fresh medium.
DNA extraction and sequencing
Each fly sample consisted of five female flies (wild and lab-reared flies) that emerged from a single fruit, and approximately 1 g of fruit was excised with sterilized bladed for the corresponding fruit samples. DNA extractions were performed using Qiagen Soil DNA extraction kit according to manufacturer's protocol. Prior to extraction, pooled flies in 2 mL centrifugation tubes were washed once in sterile water by vortexing for approximately 10 s to remove saw dust. Finally, in order to check for any kit-associated contamination (Glassing et al. 2016), five extraction controls with no samples were subjected to DNA extraction and further sequencing.
Following extractions, we quantified DNA using NanoDrop2000 (Thermo Fisher Scientific, MA, USA). Sequencing was outsourced to the University of Minnesota Genomics Center (USA), on Illumina MiSeq platform (V3 Chemistry and 2 × 300 paired end run), targeting the V4-V6 region of the 16S rRNA gene and ITS1 region for bacteria and fungi, respectively (Primer sequence information in supplementary file).
Sequence analyses
We processed the sequences in QIIME2 platform (version 2019.10, Bolyen et al. 2019). To avoid losing a substantial amount of reads after pairing (due to non-overlapping coverage of the amplicon), we chose to analyze only the forward reads for both bacteria and fungi (note: a preliminary analysis showed that the community pattern post pairing remained similar to that of the forward reads, and example figure of Weighted UniFrac is provided in the supplementary file). For bacteria, we trimmed the primers using the ‘Cutadapt’ plugin (Martin 2011). We denoised and chimera filtered the sequences using DADA2 plugin with its default settings at Phred score of at least 25 and truncation length of 220 bp (Callahan et al. 2016). Taxonomic assignment was done using RDP classifier (Wang et al. 2007). We removed sequences belonging to archaea, eukaryotes, chloroplast and mitochondria. For fungi, we used ITSxpress plugin (version 1.7.2, Rivers et al. 2018) to extract the fungal ITS1 region. Denoising and chimera filtering was performed using DADA2 with a truncation length of zero. Taxonomic assignment was performed using the UNITE database (version 8_99_04.02.2020, Abarenkov et al. 2010) that was trained and classified. We used the feature table and taxonomy table for both bacteria and fungi and additionally phylogenetic tree for bacteria generated from the QIIME2 workflow in R (version 3.6.3, R Core Team 2020). The reads in the extraction controls were subtracted and removed from the read abundances in the samples prior to downstream analysis (taxa found in controls are provided in the supplementary file as contam.pdf).
Statistical analysis
After excluding the five negative controls, there were a total of 105 samples that comprised 45 fruit samples, 45 wild fly samples and 15 lab-reared fly samples. This whole dataset was used to provide an overview of the microbial communities associated with flies and fruits. Comparative analyses were done on subsets (see Table 1): for the fruit-wild fly comparisons (90 samples, bacterial and fungal community) and for the lab-reared fly-wild fly comparisons (30 samples, bacterial community). We did not include the analyses from fungal communities of lab-reared and wild fly subsets as we used antifungal agents which could be a highly confounding factor in their community pattern, but we have added the fungal community data in the supplementary file.
For the fruit-wild fly comparisons, the data were rarefied to a sequencing depth of 4500 for bacteria and 7800 for fungi, resulting in the loss of seven and six samples for bacterial and fungal communities, respectively. For the lab-reared fly-wild fly comparison, the reads were rarefied to a sequencing depth of 4500 for bacteria with no loss of samples. Analysis on alpha and beta diversity were performed on rarefied data, while remaining analyses were performed on unrarefied data.
For bacterial alpha diversity we calculated richness in the form of observed ASVs (Amplicon Sequence Variants) and Faith’s phylogenetic diversity using packages phyloseq (version 1.30.0, McMurdie and Holmes 2013) and picante (version 1.8.1, Kembel et al. 2010). Fungal alpha diversity was calculated based on richness and Shannon index using phyloseq. We used the Kruskal–Wallis test (Kruskal and Wallis 1952) for comparing the alpha diversity between different sample types (across different combinations of fruit type x site sources, for the fruit samples and the emerging wild flies), followed by post hoc Dunn tests with Benjamini–Hochberg p-adjustment method (Dunn 1961; Benjamini and Hochberg 1995) using phyloseq and Dunn test (version 1.3.5).
We determined the beta diversity by PCoA based on UniFrac distance metrics for bacterial communities (Lozupone and Knight 2005) and Bray Curtis metric for fungal communities (Beals 1984) using the packages phyloseq and vegan (2.5–6, Oksanen 2015). We tested for statistically significant differences among all the fruits-wild flies, and between lab-reared flies-wild flies using PERMANOVA (Anderson 2014). Differences in community dispersion was checked by PERMDISP (Anderson 2006) for the significant p-values resulting from PERMANOVA.
Using procrustes analysis, we further tested the degree of association of the bacterial and fungal communities based on the Bray Curtis metrics, between the fruits-wild fly subset as well as between the wild and lab-reared fly subset (Jackson 1995), with codes adapted from van Veelen et al. (2020).
We assessed taxonomic relative abundances across fruits-wild flies and in the lab-reared flies-wild flies using package microbiome (version 1.12.0, Lahti and Shetty 2017), with default parameters and transformation set to ‘compositional’.
Results
Microbial diversity of fruits and associated flies
We observed 8634 ASVs across the wild flies and their corresponding fruits. The alpha diversity of bacterial communities across the fruits and wild flies differed by fruit types in terms of observed ASVs (X2 = 25.829, p-value < 0.001, Fig. 1a) and Faith’s phylogenetic diversity (X2 = 12.085, p-value < 0.001, Fig.S1a). In general, alpha diversity was higher in wild flies than in fruits, except for the flies originating from raspberries in both locations Winssen and Dirksland (Dunn test, p-value = 0.05).

Alpha diversity (number of observed ASVs) of bacterial a and fungal b communities associated with fruits and wild D. suzukii flies, arranged by fruit type and collection site. The richness of both bacterial and fungal communities across fruits and the emerging wild flies differed significantly (p-values < 0.1 and < 0.05, respectively)
We observed 696 fungal ASVs across the wild flies and their corresponding fruits. Richness and Shannon diversity index of fungal communities also differed significantly in fruits and wild flies according to the fruit x site sources (X2 = 22.702, p-value < 0.001, Fig. 1b; X2 = 48.429, p-value < 0.001, Fig. S1b; Dunn test, p-value = 0.05), but contrary to richness for bacterial communities, the fungal communities on fruits generally showed higher alpha diversity than the flies.
Microbial community patterns in fruits and associated flies
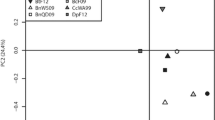
To assess whether the microbiota of the flies would reflect those of the fruits, we compared the microbiome composition of the fruit sources and wild flies (Fig. 2). The bacterial communities of wild flies clustered together and away from their corresponding fruit sources, thereby indicating a strong separation across these two sample types. For bacterial communities, PERMANOVA based on weighted UniFrac distance revealed significant differences between the wild fly and their corresponding fruits (R2 = 0.2688, p-value = 0.001, Fig. 2a and b). Additionally, the communities were homogeneous in their dispersion (PERMDISP, p-value = 0.755). Based on unweighted UniFrac, PERMANOVA (R2 = 0.115, p-value = 0.001, Figure S2) showed significant difference and dispersion was not homogenous (PERMDISP, p-value = 0.001, Figure S2). While replicate samples for the same fruit type per location clustered mostly together, they showed some variance both within and among fruit types (Figure S2).

Beta diversity of bacterial (a and b) and fungal (c and d) communities associated with fruits (triangles) and wild D. suzukii (dots). a PCoA of weighted UniFrac distance of bacterial communities differed significantly between fruits and flies (p-value = 0.001). b Individual facets of PCoA using weighted UniFrac for every fruit type in every collection site. c PCoA of Bray Curtis dissimilarity index for fungal communities, which differed significantly between fruits and flies (p-value = 0.021). d Individual facets of PCoA using Bray Curtis by fruit type in every collection site
The beta diversity of fungal communities, assessed using PERMANOVA on Bray–Curtis, also showed differences between wild flies and their corresponding fruits (R2 = 0.022, p-value = 0.021, Fig. 2c and d). Unlike the bacterial communities, however, the fungal communities showed no clear clustering into either fruit or fly samples (Fig. 2c and d); rather the fruit and fly samples overlapped considerably within fruit type, but fruit types differed substantially from each other. The dispersion was heterogeneous (PERMDISP, F-value = 8.30, p-value = 0.005), revealing differences in within-group variances. We observed considerable overlap of the fungal communities between the wild fly and its corresponding fruit source, as shown from the clustering by fruit source in most of the combinations (Fig. 2d). Additionally, considerable clustering occurred between different fruits and flies from similar locations (as noted for the three cherry cultivars from Randwijk, and among strawberries, raspberries and blackberries from Dirksland).
We further performed procrustes analysis to assess if the microbial communities across sample subsets covaried (Table 2). For the bacterial communities, we did not observe concordance between the wild flies and the corresponding fruit subset (M2 = 0.934, p-value = 0.197). In contrast, association between fungal communities of wild flies and corresponding fruits was relatively high (M2 = 0.554, p-value = 0.001), confirming the observed overlap in Fig. 2d.
Bacterial community across wild and lab-reared flies
We observed 2827 ASVs across the wild flies and their respective lab fly samples. Bacterial richness in lab-reared flies did not differ significantly from their wild counterparts (p-value = 0.40, Fig. 3a). Also, the Faith’s phylogenetic diversity indices did not differ significantly across the sample types (p-value = 0.101, figure S3).

Alpha and beta diversity (of bacterial communities across wild and lab-reared flies Alpha diversity (observed ASVs) of a) did not differ significantly between wild and lab-reared flies (p-value = 0.40). Beta diversity c) based on Weighted UniFrac distances for bacterial communities differs significantly between the lab-reared and wild flies (p-value = 0.001)
We observed significant differences in beta diversity of the bacterial communities between lab-reared flies and their respective wild flies in composition (R2 = 0.06, p-value = 0.011, Fig. 3b), but only 6% of the variation could be explained by the rearing environment. We did not observe any differences in the dispersion (PERMDISP, F-value = 3.15, p-value = 0.66). Further, the correspondence in bacterial communities between wild flies and lab-reared flies was significant (M2 = 0.5298, p-value = 0.003, Table 2).
Community composition and core microbes among different sample types
We profiled the relative abundances of the top ten most abundant bacteria and fungi across the different samples (Fig. 4). At the genera level, we observed Candidatus Carsonella, Candidatus Scalindua, Gluconobacter, Minicystis, Omnitrophica genera incertae sedis, Pantoea, Pelospora, Stella, Tatumella, and Tephidisphaera as the top ten abundant bacterial genera across the wild flies and fruit samples. Some of these genera were limited to only the flies, or were recorded at a higher abundance in flies than in the corresponding fruits (Fig. 4a). We further identified four bacteria that were found in at least 95% of wild fly samples at an abundance threshold of 0.001 (Fig. 4b). These were Succinatimonas, Pelospora, Ca. Carsonella and Ca. Omnitrophica. In the fruit samples, we were unable to identify any bacterial genera that were common to all the fruit samples, even at lower prevalence. Furthermore, these fly core bacteria were only observed in a few fruit samples and in much lower abundances than in wild flies, except for one fruit sample (Figure S4). Three of the core bacteria were also observed in the lab-reared flies (Fig. 4C). Besides, in addition to Gluconobacter, we also observed Acetobacter in most of the lab-reared flies. Hence, almost all wild flies shared a subset of bacteria, and occurrence of some of these bacteria was rare in fruits.

Top ten abundant bacteria across wild flies and their source fruits (a). Four core bacteria were observed across the wild flies (b). Top ten abundant bacteria across lab-reared flies and wild flies, which also comprised three of the core bacteria (c). Top ten abundant fungi across wild flies and their corresponding fruits (d)
Both yeasts and molds made up the fungal composition across the fruit and wild fly subsets, mostly belonging to the genera Aureobasidium, Saturnispora, Botrytis, Candida, Cladosporium, Saccharomycopsis, Gibberella, Metschnikowia, Penicillium, and Aspergillus, with the latter three also still present in some of the lab-reared flies (Fig. 4d and S5c). We did not find any core fungal ASVs at 95% prevalence across either fruits or wild flies, or across both combined. Furthermore, there was no core fungal ASV common in the wild fly subset even with relaxed criteria of lower prevalence and detection threshold. In the fruit samples, however, we did find Mycosphaerella tassiana at a prevalence of 70% and 0.0001 abundance threshold. Hence, in contrast to the findings for bacteria, fungi were not commonly shared among the wild flies.
Discussion
In this study we characterized the bacterial and fungal communities associated with wild D. suzukii flies, an invasive pest insect originating from Asia, which causes severe damage in the production of soft fruits around the globe (Walsh et al. 2011). The success of this species lies in its ability to perforate the fruit skin to lay eggs on ripening fruits, as well as its capacity to develop in a large variety of these fruits. This new niche of ripening fruits may lack essential nutrients that microbes could be providing (Bing et al. 2018). Our primary focus was to study the microbiome in D. suzukii, as well as its associated fruit substrates, to assess whether the flies acquired their associated microbiota mostly from exposure to the microbial communities of the fruits on which they developed—in this case, we expected substantial overlap of the fruit and fly microbiota with differences in microbial composition for flies emerging from different fruit types—or whether they have formed more persistent associations with some microbiota that they retain irrespective of their fruit hosts.
We observed significant differences in bacterial and fungal diversity among different fruit types, but the flies that emerged from these fruits had a remarkably uniform bacterial community composition. In contrast, the fungal community distribution in the flies largely resembled those in their host fruits. Flies that were lab-reared for ten generations still harbored some of the bacterial genera that largely resembled those of their wild ancestors. We observed no significant difference in alpha diversity metrics between bacterial communities of lab-reared and wild flies. For fungal communities, although we observed no significant difference in beta diversity between the lab-reared and wild flies, we did see a severe reduction in alpha diversity for lab-reared flies, but this is likely attributable to the use of anti-fungal agents (i.e., proprionic acid and nipagin) in the artificial diet (Figure S5a and b). Thus, the bacterial communities in emerging flies were largely shaped irrespective of the fruit type and were distinct from the bacterial communities on the fruits itself, while fruit source most likely determined the fungal community distribution in the flies. At the level of ASVs, several of the microbes present in the wild flies also seem to be present in the lab-reared samples, suggesting that they were vertically or horizontally transmitted.
It is important to note that in our study we did not analyze the larval microbiome, as we were interested specifically in the associated microbes of the adults. In holometabolous insects like Drosophila, metamorphosis induces changes at morphological and physiological level, which restructures several tissues and might also result in shedding of microbes (Tissot and Stocker 2000; Johnston and Rolff 2015). However, sometimes, depending on the family or life history of the insects, a few selected microbes might be retained (Johnston and Rolff 2015). In order to verify whether such restructuring during development results in microbial shifts for D. suzukii, future studies should also focus on determining the microbiome of wild D. suzukii larvae as well as pupae collected from fruits across a broader geographical range. In line with our findings, a study on associated microbes of the tephritid fruit flies Bactrocera showed difference in bacterial community pattern for fruits and larvae (Majumder et al. 2019). In Drosophila species, Martinson et al. (2017) reported the bacterial community composition in flies to be distinct from their food source. Although we do not yet know how this separation of fruit and fly bacterial communities can persist, the continuation of the bacterial associations for ten generations on artificial diet suggest that perhaps ovipositing females can transmit bacteria across generations to their offspring. And in addition to diet, host type may also have been a factor driving the community composition in our fly samples.
Drosophila suzukii flies spend a substantial amount of time in close association with the fruits from which they emerge as adults. We therefore expected to observe a subset of microbes commonly shared across both fruits and flies. At the genera level, we observed Gluconobacter, Pantoea, and Tatumella across the fruits and some of the wild flies, as reported by previous studies (Chandler et al. 2014; Vacchini et al. 2017; Solomon et al. 2019). We also observed bacteria Ca. Carsonella, Ca. Scalindua, Succinatimonas, etc., to be more abundant in the flies, and Acetobacter to be also present in most of the lab flies. Some of the prevalent fungal members of the genera Penicillium and Metschnikowia were common among wild flies and fruits, with the former being dominant in the lab-reared flies as well (perhaps they might be resistant to the anti-fungal agents we used). In addition, few other fungi like Pichia, Botrytis, and Cladosporium were also noted in some of the fly samples. However, no fungal genera were consistently shared among wild flies or fruits. The fungal community composition varied among fruit types, and those of the emerged flies showed substantial overlap. Similar to our findings, previous studies on coleopteran insects also suggested a strong role of the immediate environment in structuring the fungal communities in the insects (Kudo et al. 2019; Rassati et al. 2019).
Although our findings are limited to the Netherlands, the community composition that we reported, to some extent aligns with the samplings done in a number of other countries across USA, Europe and Asia (Vacchini et al. 2017; Martinez‐Sañudo et al. 2018; Fountain et al. 2018; Jiménez-Padilla et al. 2020). Among the top ten abundant bacteria, Gluconobacter, Pantoea, Tatumella observed in our studies are commonly associated with D. suzukii (Chandler et al. 2014; Vacchini et al. 2017; Martinez‐Sañudo et al. 2018). These bacteria have been reported as beneficial for insect health and growth, by providing nutritional benefits under certain conditions (Crotti et al. 2010; Medrano and Bell 2016). Fungi like Metschnikowia, Candida, Saccharomycopsis, etc., observed in our study, influence the fitness traits of D. suzukii (Bellutti et al. 2018; Spitaler et al. 2020). The core bacteria that we observed in our samples have been reported from diverse habitats but not in D. suzukii, and engage in carbohydrate and amino acid metabolic pathways (Riley et al. 2017; Morotomi et al. 2010; Stackebrandt and Hespell 2006; Matthies et al. 2000). The bacteria Ca. Carsonella rudii reportedly contributes to the amino acid metabolism of the psyllids, hemipteran insect pests that feed on low-nutritional plant sap (Thao et al. 2000; Riley et al. 2017). The genus Succinatimonas has been reported in GI tract of mammals and is associated with the metabolism of carbohydrates by generating succinates and acetates (Stackebrandt and Hespell 2006; Morotomi et al. 2010). The etiology of Omnitrophica_genera_incertae_sedis, which belongs to the phylum Ca. Omnitrophica, remains unknown (Rivas-Marín and Devos 2018). The genus Pelospora is associated with the fermentation of glutarate, and under in vitro conditions, this genus produces butyrates and CO2 (Matthies et al. 2000; Kucuk 2019). Although we did not observe any core fungi in our study, we would not like to rule out the possibility that the observed fungal members, despite differing in taxonomic identities, might have similar functional capabilities of benefit to their insect host—something which remains a matter of future testing (Estrada-Peña et al. 2020).
Determining the composition of these communities, may reveal unknown aspects on their fundamental biology, and sometimes also provides insight into dependencies and vulnerabilities that can be exploited in pest management strategies, e.g., by introducing metabolites and disrupting the insect fitness or by symbiont mediated RNA interference (Usta 2013; Ruiu 2015; Whitten and Dyson 2017; Raza et al. 2020). Culture-based and sequencing approaches have revealed the influence of environmental factors on the bacterial community composition of pest insects, such as diet, climate and geographic location (Rizzi et al. 2013; Jones et al. 2019; Colman et al. 2012). To understand the processes by which associations between pest insects and microbiota arise, it is important to investigate the microbial composition on their food source (Behar and Jurkevitch 2008; Beaulieu et al. 2017; Deans and Hutchinson 2021), which has been reported as a potential source from which insects can acquire their associated microbiota (Jones et al. 2019). It is also important to realize that specific insect-microbe associations can arise that transcend the intermediate of a food source.
The ability of D. suzukii to grow on such a wide range of fruits makes it an interesting system to explore whether fruit source plays an important role in determining its microbiome. Microbial communities vary considerably among various fruits and vegetables (Leff and Fierer 2013), as we corroborated in the analyses of microbial communities on our fruit samples. We speculated that flies developing on different fruits would exhibit high variations in their community patterns and align with those of the corresponding fruits when they relied on their fruit hosts for acquiring microbes. Our results, however, showed that the bacterial communities of the wild flies were largely distinct from those of the fruits, and showed large similarities irrespective of fruit type. Some of the microbial members that were found in wild flies from different fruits were also retained after ten generations of lab-rearing.
Understanding the interactions between microbiome and insects is interesting from an evolutionary perspective, as it may facilitate niche expansions and adaptations to challenging environmental conditions. We propose that D. suzukii may have formed persistent associations with some bacteria, and we hypothesize that this might have facilitated its niche expansion to fresh fruits. Earlier studies showed that D. suzukii larvae failed to develop on diets that were based on fresh fruits when the larvae were germ-free, but this could be rescued with the supplementation of the microbiota from adult flies (Bing et al. 2018). The persistent associations with some bacteria across generations, which were acquired and maintained largely independent of the fruits or food source, may hint at these bacteria being an ally for the flies that may aid them in overcoming certain challenges associated with this particular niche. In order to investigate what precisely fosters the host-microbiome interactions in this pest, studies similar to Ben-Yosef et al. (2015) could likely answer whether indeed there is any particular microbial associations that drives the pest status of D. suzukii. The fungal communities seemed to be less persistent, and here we did see more alignment in community patterns between fruits and emerging flies. This does not necessarily imply that these fungi are unimportant in the niche expansion, as they could still be providing essential nutrients, e.g., sterols or proteins (Carvalho et al. 2010; Lewis and Hamby 2019; Belluti et al. 2018). However, it does appear that the associations with fungi are more transient than those with bacteria.
Concluding, our study reports the microbial community structure of Drosophila suzukii across a range of fruits. By characterizing the microbial communities of both the flies and the fruits from which they emerged, we could assess the similarity and differences between these communities across the fruits and the flies, in several different fruit sources. While the fungal community structure in these pests largely reflected those on the breeding substrates, D. suzukii might have formed more persistent associations with some bacteria and transmit these across generations rather than obtaining them from their food source. The characterization of these microbial communities represented, thus, an important step to identify any potential symbiotic relationship between these flies and bacterial or fungal species.
Author contribution
KG, JFS, BW conceived the experimental plan. KG performed research and data analysis. SNV and JFS provided support with sequence analysis. BW provided support with statistical analysis. KG wrote the original draft. KG, SNV, JFS and BW reviewed and edited subsequent drafts.
Data availability
Raw sequence reads are available in NCBI SRA database under accession PRJNA842670. Additional files and data available in the link: https://figshare.com/s/4eebffffaab656b540f8
Change history
02 July 2022
The original online version of this article was revised: the figure 4 caption is not aligned under figure 4 in Springer link.
References
Abarenkov K, Nilsson RH, Larsson KH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R (2010) The UNITE database for molecular identification of fungi–recent updates and future perspectives. New Phytol 186(2):281–285. https://doi.org/10.1111/j.1469-8137.2009.03160.x
Anderson MJ (2006) Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62(1):245–253. https://doi.org/10.1111/j.1541-0420.2005.00440.x
Anderson MJ (2014) Permutational multivariate analysis of variance (PERMANOVA). Wiley Statsref Stat Ref Online 14:1–5. https://doi.org/10.1002/9781118445112.stat07841
Atallah J, Teixeira L, Salazar R, Zaragoza G, Kopp A (2014) The making of a pest: the evolution of a fruit-penetrating ovipositor in Drosophila suzukii and related species. Proc R Soc Lond [biol] 281(1781):20132840. https://doi.org/10.1098/rspb.2013.2840
Beals EW (1984) Bray-Curtis ordination: an effective strategy for analysis of multivariate ecological data. Adv Ecol Res 14:1–55. https://doi.org/10.1016/S0065-2504(08)60168-3
Beaulieu M, Franke K, Fischer K (2017) Feeding on ripening and over-ripening fruit: interactions between sugar, ethanol and polyphenol contents in a tropical butterfly. J Exp Biol 220(17):3127–3134. https://doi.org/10.1242/jeb.162008
Behar A, Jurkevitch E, Yuval B (2008) Bringing back the fruit into fruit fly–bacteria interactions. Mol Ecol 17(5):1375–1386. https://doi.org/10.1111/j.1365-294X.2008.03674.x
Bellutti N, Gallmetzer A, Innerebner G, Schmidt S, Zelger R, Koschier EH (2018) Dietary yeast affects preference and performance in Drosophila suzukii. J Pest Sci 91(2):651–660. https://doi.org/10.1007/s10340-017-0932-2
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57(1):289–300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x
Ben-Yosef M, Pasternak Z, Jurkevitch E, Yuval B (2015) Symbiotic bacteria enable olive fly larvae to overcome host defences. R Soc Open Sci 2(7):150170. https://doi.org/10.1098/rsos.150170
Bing X, Gerlach J, Loeb G, Buchon N (2018) Nutrient-dependent impact of microbes on Drosophila suzukii development. MBio. https://doi.org/10.1128/mBio.02199-17
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37(8):852–857. https://doi.org/10.1038/s41587-019-0209-9
Calabria G, Máca J, Bächli G, Serra L, Pascual M (2012) First records of the potential pest species Drosophila suzukii (Diptera: Drosophilidae) in Europe. J Appl Entomol 136(1–2):139–147. https://doi.org/10.1111/j.1439-0418.2010.01583.x
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13(7):581–583. https://doi.org/10.1038/nmeth.3869
Carvalho M, Schwudke D, Sampaio JL, Palm W, Riezman I, Dey G, Gupta GD, Mayor S, Riezman H, Shevchenko A, Kurzchalia TV (2010) Survival strategies of a sterol auxotroph. Development 137(21):3675–3685. https://doi.org/10.1242/dev.044560
Chandler JA, James PM, Jospin G, Lang JM (2014) The bacterial communities of Drosophila suzukii collected from undamaged cherries. PeerJ 2:e474. https://doi.org/10.7717/peerj.474
Cini A, Anfora G, Escudero-Colomar LA, Grassi A, Santosuosso U, Seljak G, Papini A (2014) Tracking the invasion of the alien fruit pest Drosophila suzukii in Europe. J Pest Sci 87(4):559–566. https://doi.org/10.1007/s10340-014-0617-z
Colman DR, Toolson EC, Takacs-Vesbach CD (2012) Do diet and taxonomy influence insect gut bacterial communities? Mol Ecol 21(20):5124–5137. https://doi.org/10.1111/j.1365-294X.2012.05752.x
Crotti E, Balloi A, Hamdi C, Sansonno L, Marzorati M, Gonella E, Favia G, Cherif A, Bandi C, Alma A, Daffonchio D (2012) Microbial symbionts: a resource for the management of insect-related problems. Microb Biotechnol 5(3):307–317. https://doi.org/10.1111/j.1751-7915.2011.00312.x
Crotti E, Rizzi A, Chouaia B, Ricci I, Favia G, Alma A, Sacchi L, Bourtzis K, Mandrioli M, Cherif A, Bandi C (2010) Acetic acid bacteria, newly emerging symbionts of insects. Appl Enviro Microbiol 76(21):6963–6970. https://doi.org/10.1128/AEM.01336-10
De Cock M, Virgilio M, Vandamme P, Bourtzis K, De Meyer M, Willems A (2020) Comparative microbiomics of tephritid frugivorous pests (Diptera: Tephritidae) from the field: a tale of high variability across and within species. Front Microbiol. https://doi.org/10.3389/fmicb.2020.01890
Kwadha CA, Okwaro LA, Kleman I, Rehermann G, Revadi S, Ndlela S, Khamis FM, Nderitu PW, Kasina M, George MK, Kithusi GG, Becher PG (2021) Detection of the spotted wing drosophila, Drosophila suzukii, in continental sub-Saharan Africa. J Pest Sci 94(2):251–259. https://doi.org/10.1007/s10340-021-01330-1
Boughdad A, Haddi K, El Bouazzati A, Nassiri A, Tahiri A, El Anbri C, Eddaya T, Zaid A, Biondi A (2021) First record of the invasive spotted wing Drosophila infesting berry crops in Africa. J Pest Sci 94(2):261–271. https://doi.org/10.1007/s10340-020-01280-0
Deans C, Hutchison WD (2021) The protein paradox: elucidating the complex nutritional ecology of the invasive berry pest, spotted-wing drosophila (Diptera: Drosophila suzukii). Front Insect Sci 21. https://doi.org/10.3389/finsc.2021.787169
Dunn OJ (1961) Multiple comparisons among means. J Am Stat Assoc 56(293):52–64. https://doi.org/10.1080/01621459.1961.10482090
Fountain MT, Bennett J, Cobo-Medina M, Conde Ruiz R, Deakin G, Delgado A, Harrison R, Harrison N (2018) Alimentary microbes of winter-form Drosophila suzukii. Insect Mol Biol 27(3):383–392. https://doi.org/10.1111/imb.12377
Estrada-Peña A, Cabezas-Cruz A, Obregón D (2020) Behind taxonomic variability: the functional redundancy in the tick microbiome. Microorganisms 8(11):1829. https://doi.org/10.3390/microorganisms8111829
Glassing A, Dowd SE, Galandiuk S, Davis B, Chiodini RJ (2016) Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog 8(1):1–2. https://doi.org/10.1186/s13099-016-0103-7
Goodhue RE, Bolda M, Farnsworth D, Williams JC, Zalom FG (2011) Spotted wing drosophila infestation of California strawberries and raspberries: economic analysis of potential revenue losses and control costs. Pest Manag Sci 67(11):1396–1402. https://doi.org/10.1002/ps.2259
Gurung K, Wertheim B, Falcao Salles J (2019) The microbiome of pest insects: it is not just bacteria. Entomol Exp Appl 167(3):156–170. https://doi.org/10.1111/eea.12768
Hamby KA, Swett CL (2015) Elucidating symbioses between Drosophila suzukii and fungal communities for improved insect and disease management in raspberry production. North American Bramble Growers Research Foundation Funding. https://raspberryblackberry.com/wp-content/uploads/Elucidating-symbioses-between-Drosophila-suzukii-and-fungal-communities-for-improved.pdf
Ioriatti C, Guzzon R, Anfora G, Ghidoni F, Mazzoni V, Villegas TR, Dalton DT, Walton VM (2018) Drosophila suzukii (Diptera: Drosophilidae) contributes to the development of sour rot in grape. J Econ Entomol 111(1):283–292. https://doi.org/10.1093/jee/tox292
Ioriatti C, Stacconi M, Anfor G (2019) Drosophila suzukii (spotted wing drosophila). https://www.cabi.org/isc/datasheet/109283
Jackson DA (1995) PROTEST: a PROcrustean randomization TEST of community environment concordance. Ecoscience 2(3):297–303. https://doi.org/10.1080/11956860.1995.11682297
Jiménez-Padilla Y, Esan EO, Floate KD, Sinclair BJ (2020) Persistence of diet effects on the microbiota of Drosophila suzukii (Diptera: Drosophilidae). Can Entomol 152(4):516–531. https://doi.org/10.4039/tce.2020.37
Jing TZ, Qi FH, Wang ZY (2020) Most dominant roles of insect gut bacteria: digestion, detoxification, or essential nutrient provision? Microbiome 8(1):1–20. https://doi.org/10.1186/s40168-020-00823-y
Johnston PR, Rolff J (2015) Host and symbiont jointly control gut microbiota during complete metamorphosis. PLoS Pathog 11(11):e1005246. https://doi.org/10.1371/journal.ppat.1005246
Jones AG, Mason CJ, Felton GW, Hoover K (2019) Host plant and population source drive diversity of microbial gut communities in two polyphagous insects. Sci Rep 9(1):1–1. https://doi.org/10.1038/s41598-019-39163-9
Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO (2010) Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26(11):1463–1464. https://doi.org/10.1093/bioinformatics/btq166
Kruskal WH, Wallis WA (1952) Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47(260):583–621. https://doi.org/10.1080/01621459.1952.10483441
Kucuk RA (2019) Bacterial Diversity of the Gut of Cotinis nitida. Doctoral dissertation, Clemson University. https://tigerprints.clemson.edu/all_theses/3218
Kudo R, Masuya H, Endoh R, Kikuchi T, Ikeda H (2019) Gut bacterial and fungal communities in ground-dwelling beetles are associated with host food habit and habitat. ISME J 13(3):676–685. https://doi.org/10.1038/s41396-018-0298-3
Lahti L, Shetty S (2017) Tools for microbiome analysis in R Version 1 1 10013. http://microbiome.github.com/microbiome
Leff JW, Fierer N (2013) Bacterial communities associated with the surfaces of fresh fruits and vegetables. PLoS ONE 8(3):e59310. https://doi.org/10.1371/journal.pone.0059310
Lewis MT, Hamby KA (2019) Differential impacts of yeasts on feeding behavior and development in larval Drosophila suzukii (Diptera: Drosophilidae). Sci Rep 9(1):1–2. https://doi.org/10.1038/s41598-019-48863-1
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71(12):8228–8235. https://doi.org/10.1128/AEM.71.12.8228-8235.2005
Lu M, Hulcr J, Sun J (2016) The role of symbiotic microbes in insect invasions. Annu Rev Ecol Evol Syst 47:487–505. https://doi.org/10.1146/annurev-ecolsys-121415-032050
Majumder R, Sutcliffe B, Taylor PW, Chapman TA (2019) Next-generation sequencing reveals relationship between the larval microbiome and food substrate in the polyphagous Queensland fruit fly. Sci Rep 9(1):1–12. https://doi.org/10.1038/s41598-019-50602-5
Marín-Cevada V, Caballero-Mellado J, Bustillos-Cristales R, Muñoz-Rojas J, Mascarúa-Esparza MA, Castañeda-Lucio M, López-Reyes L, Martínez-Aguilar L, Fuentes-Ramírez LE (2010) Tatumella ptyseos, an unrevealed causative agent of pink disease in pineapple. J Phytopathol 158(2):93–99. https://doi.org/10.1111/j.1439-0434.2009.01575.x
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17(1):10–12. https://doi.org/10.14806/ej.17.1.200
Martinez-Sañudo I, Simonato M, Squartini A, Mori N, Marri L, Mazzon L (2018) Metagenomic analysis reveals changes of the Drosophila suzukii microbiota in the newly colonized regions. Insect Sci 25(5):833–846. https://doi.org/10.1111/1744-7917.12458
Martinson VG, Douglas AE, Jaenike J (2017) Community structure of the gut microbiota in sympatric species of wild Drosophila. Ecol Lett 20(5):629–639. https://doi.org/10.1111/ele.12761
Matthies C, Springer N, Ludwig W, Schink B (2000) Pelospora glutarica gen nov, sp nov, a glutarate-fermenting, strictly anaerobic, spore-forming bacterium. Int J Syst Evol Microbiol 50(2):645–648. https://doi.org/10.1099/00207713-50-2-645
McMurdie PJ, Holmes S (2013) phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8(4):e61217. https://doi.org/10.1371/journal.pone.0061217
Medrano EG, Bell AA (2016) An opportunistic Pantoea sp. Isolated from a cotton fleahopper that is capable of causing cotton (Gossypium hirsutum L.) Bud Rot. Agric Sci 8(1):64–76. https://doi.org/10.4236/as.2017.81006
Morotomi M, Nagai F, Watanabe Y, Tanaka R (2010) Succinatimonas hippei gen nov, sp nov, isolated from human faeces. Int J Syst Evol Microbiol 60(8):1788–1793. https://doi.org/10.1099/ijs.0.015958-0
Oksanen J (2015) Vegan: an introduction to ordination. 8:19. https://cran.r-project.org/web/packages/vegan/vignettes/intro-vegan.pdf
R Core Team (2020) R: a language and environment for statistical computing R Foundation for Statistical Computing. Austria. Vienna. https://www.r-project.org/
Rassati D, Marini L, Malacrinò A (2019) Acquisition of fungi from the environment modifies ambrosia beetle mycobiome during invasion. PeerJ 7:e8103. https://doi.org/10.7717/peerj.8103
Raza MF, Yao Z, Bai S, Cai Z, Zhang H (2020) Tephritidae fruit fly gut microbiome diversity function and potential for applications. Bull Entomol Res 110(4):423–437. https://doi.org/10.1017/S0007485319000853
Riley AB, Kim D, Hansen AK (2017) Genome sequence of “Candidatus Carsonella ruddii” strain BC, a nutritional endosymbiont of Bactericera cockerelli. Genome Announc. https://doi.org/10.1128/genomeA.00236-17
Rivas-Marín E, Devos DP (2018) The Paradigms They Are a-Changin’: past, present and future of PVC bacteria research. Antonie Van Leeuwenhoek 111(6):785–99. https://doi.org/10.1007/s10482-017-0962-z
Rivers AR, Weber KC, Gardner TG, Liu S, Armstrong SD (2018) ITSxpress: software to rapidly trim internally transcribed spacer sequences with quality scores for marker gene analysis. F1000Research 7:1418. https://doi.org/10.12688/f1000research.15704.1
Rizzi A, Crotti E, Borruso L, Jucker C, Lupi D, Colombo M, Daffonchio D (2013) Characterization of the bacterial community associated with larvae and adults of Anoplophora chinensis collected in Italy by culture and culture-independent methods. BioMed Res Int. https://doi.org/10.1155/2013/420287
Rota-Stabelli O, Blaxter M, Anfora G (2013) Drosophila suzukii. Curr Biol 23(1):R8-9. https://doi.org/10.1016/j.cub.2012.11.021
Ruiu L (2015) Insect pathogenic bacteria in integrated pest management. Insects 6(2):352–367. https://doi.org/10.3390/insects6020352
Sacchetti P, Pastorelli R, Bigiotti G, Guidi R, Ruschioni S, Viti C, Belcari A (2019) Olive fruit fly rearing procedures affect the vertical transmission of the bacterial symbiont Candidatus Erwinia dacicola. BMC Biotechnol 19(2):1–13. https://doi.org/10.1186/s12896-019-0582-y
Silva-Soares NF, Nogueira-Alves A, Beldade P, Mirth CK (2017) Adaptation to new nutritional environments: larval performance, foraging decisions, and adult oviposition choices in Drosophila suzukii. BMC Ecol 17(1):1–3. https://doi.org/10.1186/s12898-017-0131-2
Solomon GM, Dodangoda H, McCarthy-Walker T, Ntim-Gyakari R, Newell PD (2019) The microbiota of Drosophila suzukii influences the larval development of Drosophila melanogaster. PeerJ 7:e8097. https://doi.org/10.7717/peerj.8097
Spitaler U, Bianchi F, Eisenstecken D, Castellan I, Angeli S, Dordevic N, Robatscher P, Vogel RF, Koschier EH, Schmidt S (2020) Yeast species affects feeding and fitness of Drosophila suzukii adults. J Pest Sci 93(4):1295–1309. https://doi.org/10.1007/s10340-020-01266-y
Stackebrandt ER, Hespell R The family succinivibrionaceae (2006) The prokaryotes. Springer Berlin, Heidelberg 3:419–429. https://doi.org/10.1007/0-387-30743-5_20
Starmer WT (1986) Fogleman JC Coadaptation of Drosophila and yeasts in their natural habitat. J Chem Ecol 12(5):1037–1055. https://doi.org/10.1007/BF01638995
Sudarshan AS, Leo L (2020) Microbiomeutilities: utilities for microbiome analytics. https://microsud.github.io/microbiomeutilities/
Thao ML, Moran NA, Abbot P, Brennan EB, Burckhardt DH, Baumann P (2000) Cospeciation of psyllids and their primary prokaryotic endosymbionts. Appl Environ Microbiol 66(7):2898–2905. https://doi.org/10.1128/AEM.66.7.2898-2905.2000
Tissot M, Stocker RF (2000) Metamorphosis in Drosophila and other insects: the fate of neurons throughout the stages. Prog Neurobiol 62(1):89–111. https://doi.org/10.1016/S0301-0082(99)00069-6
Usta C (2013) Microorganisms in biological pest control—a review (bacterial toxin application and effect of environmental factors). Current Prog Biol Res 13:287–317. https://doi.org/10.5772/55786
Vacchini V, Gonella E, Crotti E, Prosdocimi EM, Mazzetto F, Chouaia B, Callegari M, Mapelli F, Mandrioli M, Alma A, Daffonchio D (2017) Bacterial diversity shift determined by different diets in the gut of the spotted wing fly Drosophila suzukii is primarily reflected on acetic acid bacteria. Environ Microbiol Rep 9(2):91–103. https://doi.org/10.1111/1758-2229.12505
van Veelen HP, Salles JF, Matson KD, van der Velde M, Tieleman BI (2020) Microbial environment shapes immune function and cloacal microbiota dynamics in zebra finches Taeniopygia guttata. Anim Microbiome 2:1–7. https://doi.org/10.1186/s42523-020-00039-3
Walsh DB, Bolda MP, Goodhue RE, Dreves AJ, Lee J, Bruck DJ, Walton VM, O’Neal SD, Zalom FG (2011) Drosophila suzukii (Diptera: Drosophilidae): invasive pest of ripening soft fruit expanding its geographic range and damage potential. J Integr Pest Manag 2(1):G1-7. https://doi.org/10.1603/IPM10010
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267. https://doi.org/10.1128/AEM.00062-07
Whitten M, Dyson P (2017) Gene silencing in non-model insects: overcoming hurdles using symbiotic bacteria for trauma-free sustainable delivery of RNA interference: sustained RNA interference in insects mediated by symbiotic bacteria: applications as a genetic tool and as a biocide. BioEssays 39(3):1600247. https://doi.org/10.1002/bies.201600247
Yao Z, Ma Q, Cai Z, Raza MF, Bai S, Wang Y, Zhang P, Ma H, Zhang H (2019) Similar shift patterns in gut bacterial and fungal communities across the life stages of Bactrocera minax larvae from two field populations. Front Microbiol 10:2262. https://doi.org/10.3389/fmicb.2019.02262
Acknowledgements
We thank Herman Helsen who provided the details of infestation and the growers. We are grateful to Tom Groot (Dirksland), Maria Buitenkamp and Edo Biewinga (Hoogeveen—Tiendeveen); Gijs Gerritse (Randwijk); Jeroen Spitzen (Winssen) who provided access to their garden and orchards. KG was supported by the Adaptive Life scholarship program (2017), University of Groningen. We thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high performance computing cluster. This research has been carried out in the groups of Evolutionary Genetics and Microbial Ecology at the Groningen Institute for Evolutionary Life Sciences (GELIFES) according to the requirements of the Graduate School of Science and Engineering (Faculty of Science and Engineering, University of Groningen; Groningen, the Netherlands).
Funding
Adaptive Life Program, University of Groningen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial (or any other) interests.
Ethical approval
None required.
Additional information
Communicated by Antonio Biondi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: the figure 4 caption is not aligned under figure 4 in Springer link.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gurung, K., Vink, S.N., Salles, J.F. et al. More persistent bacterial than fungal associations in the microbiota of a pest insect. J Pest Sci 96, 785–796 (2023). https://doi.org/10.1007/s10340-022-01524-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10340-022-01524-1