
Abstract
A rapid reversed-phase gradient method employing a 50 mm × 1 mm i.d., C18 microbore column, combined with ion mobility and high-resolution mass spectrometry, was applied to the metabolic phenotyping of urine samples obtained from rats receiving different diets. This method was directly compared to a “conventional” method employing a 150 × 2.1 mm i.d. column packed with the same C18 bonded phase using the same samples. Multivariate statistical analysis of the resulting data showed similar class discrimination for both microbore and conventional methods, despite the detection of fewer mass/retention time features by the former. Multivariate statistical analysis highlighted a number of ions that represented diet-specific markers in the samples. Several of these were then identified using the combination of mass, ion-mobility-derived collision cross section and retention time including N-acetylglutamate, urocanic acid, and xanthurenic acid. Kynurenic acid was tentatively identified based on mass and ion mobility data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metabolic phenotyping (metabotyping) [1,2,3], using methods based on liquid chromatography (LC), of the type routinely undertaken during metabonomic/metabolomic investigations faces a number of challenges connected with both throughput and metabolite identification. The tension between maximizing throughput versus obtaining the most comprehensive metabolite coverage possible is particularly acute when large numbers of samples are to be analysed, or there are constraints on the availability of instrumentation. As a partial solution to some of these problems we recently introduced the concept of rapid microbore metabolic profiling (RAMMP) using reversed-phase ultra (high) performance liquid chromatography (U(H)PLC) on a 1 mm i.d. column, coupled to mass spectrometric detection (MS) [4] and, more recently, similar rapid HILIC and RP-lipidomic methods have been described [5, 6]. Reducing the separation time via a rapid gradient enables much higher throughput, whilst the use of a 1 mm i.d. column also brings advantages of increased sensitivity and reduced solvent consumption. However, and as would be expected, the peak capacity of the RAMMP method was reduced (from 150 to 50) compared to analysis on a conventional 2.1 mm i.d., and the number of measured features was decreased from ca. 19,000 to ca. 6000 [4]. Notwithstanding the loss of peak capacity and reduced number of features the RAMMP methodology was still able to achieve similar levels of group discrimination by PCA, and this was based largely on the same features seen using conventional analysis [4].
More recently we have examined the effect of adding ion mobility (IM) separations and spectrometry (IMS) to provide an added dimension of both separation and compound characterisation to rapid methods of LC–MS-based metabotyping [5,6,7]. This study showed that the use of IM increased the number of features detected significantly from ca. 16,000 by LC–MS to nearly 20,000 for LC–IM–MS, with similar increases seen as the run time and column length were reduced [7]. As IM would not compensate for ion suppression effects, this increase in detected features presumably resulted from the improved resolution of the ions formed in the ion source, enabling easier feature detection. However, as well as this welcome increase in features, incorporation of IM can result in improved MS data as a result of the separation of co-eluting compounds and can also be used to determine the rotationally averaged collision cross section (CCS) of analytes. These CCS values, which represent a characteristic property of a molecule, provide a further means of improving the identification of metabolites detected in metabolic phenotyping studies in addition to MS [8,9,10,11,12].
Here, we have coupled the 2.5 min gradient RAMMP–UPLC–MS method with ion mobility and employed this combination to analyze a subset of rat urine samples obtained as part of a study examining the effects of a methionine-choline deficient (MCD) diet [13] on the resulting induced liver pathology in rats related to non-alcoholic steatosis hepatitis (NASH), a severe stage of non-alcoholic fatty liver disease (NAFLD). The resulting metabolic phenotypes (metabotypes) were compared with those obtained using a conventional, 12.5 min gradient, UPLC–MS analysis [14] to highlight the advantages and limitations of the RAMMP–UPLC–IM–MS approach.
Experimental Section
Chemicals and Reagents
Water, methanol, 0.1% formic acid in water and 0.1% formic acid in acetonitrile were purchased from Fisher Scientific Ltd (Loughborough, UK). Standards used for instrument calibration were from the “Waters Major Mix IMS/ToF Calibration Kit for IMS” (Product Number 186008113, (Waters Corp., Milford, USA). Leu Enk (Sigma, Dorset, UK) was used to calibrate the QTof mass spectrometer.
Study Design
Full details of the study design are provided in Ref. [13]. Briefly, the urine samples used here were collected from ten male Sprague Dawley rats, of which half were maintained on a control diet whilst the other half received a methionine-choline deficient (MCD) diet for 8 weeks to model non-alcoholic steatohepatitis (NASH) as part of a study to explore methotrexate-induced liver toxicity [13]. Samples were collected from animals that had been fed with either control or MCD diets for 8 weeks for four 6 h periods over 2 days (− 6 to 0 h, 6–12 h, 18–24 h and 36–48 h). The urine samples were collected from animals forming the study control group after placing the rats into metabolism cages. Urine was collected on ice and mixed with sodium azide (1 mL 1% w/v in H2O) to prevent the unwanted growth of bacteria. Urine samples were stored at – 80 °C until analysis. This investigation was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Arizona. The work was undertaken within the NIH guidelines on the care and use of experimental animals as previously described [13].
Sample Preparation
Urine samples were prepared as previously described [4]. Briefly, 20 µL of urine were mixed with 60 µL of MeOH and stored at − 20 °C overnight for protein removal. The samples were then centrifuged (15,000g, 5 min, 4 °C) and 25 µL of the clear supernatant transferred into a 350 µL 96 well-plate. To provide a quality control (QC) sample [15, 16] 30 µL of each sample was taken and mixed to prepare a bulk pooled sample, followed by thorough vortex mixing (3 s). To each of the samples, including the QCs, 225 µL of water was then added. The plates were centrifuged (700g, 5 min, room temperature) and then placed into the auto sampler at 4 °C. The column was conditioned by the injection of 20 QC samples prior to the beginning of the run. To monitor the quality of the analysis a QC was injected every 11 samples.
UPLC–MS
A Waters Acquity UPLC system and a HSS T3 1.8 µm column (2.1 × 150 mm) were used for sample analysis using the conventional profiling method [14]. The temperature of the column was set at 45 °C with the autosampler temperature at 5 °C. The mobile phases used were water (solvent A) and acetonitrile (solvent B), both modified with 0.1% v/v of formic acid with an initial solvent composition of 99% of A and a flow rate of 600 µL min−1. For gradient chromatography, the proportion of solvent B was increased from 1% at 0.10 s to 55% at 10 min after which solvent B was increased by 10% every 15 s until it reached 100% (at a flow rate of 0.8 mL min−1) before a wash step for 1 min. The wash step was followed by a re-equilibration period of 1 min with 99% solvent A (total run time 12.65 min). The volume of the sample injected was 5 µL and the purge solvent used was water. Mass spectrometry was performed using a Synapt G2-S mass spectrometer employing electrospray ionization in positive mode (ESI +). The capillary voltage was 1.5 kV and the source temperature were set at 120 °C. The cone gas flow was 50 L h−1 and the gas used was nitrogen. The desolvation gas temperature was 450 °C, the desolvation gas flow was 900 L h−1 and the nebuliser gas flow was 6 bars. The acquisition was carried out over the m/z range 50–1200. To obtain fragmentation data, centroid mode with MSe acquisition was used with a low collision energy of 4 eV and the high collision energy ramped from 15 to 45 eV. Leucine encephalin (MW = 555.62) was used for mass accuracy with a scan collected every 60 s and a cone voltage fixed at 30 V. The data were collected using MassLynx V 4.1 (Waters Corp., Milford, USA).
RAMMP–IM–MS
A Waters Acquity UPLC system was used for sample analysis using the rapid gradient method with the separation performed on a HSS T3 1.8 µm column (1 × 50 mm). The temperature of the column was set at 40 °C with the autosampler temperature at 5 °C. The mobile phases were those used for the conventional method. The initial solvent composition 99% of A, at a flow rate of 400 µL min−1. For gradient chromatography, the proportion of solvent B was increased from 15% at 0.42 min to 50% at 0.83 min, 95% at 1.25 min and 99% at 1.51 min, before a wash step of 1 min (total run time 2.5 min). The volume of the sample injected was 5 µL and the purge solvent used was water. Ion mobility and mass spectrometry were also performed using a Synapt G2-S mass spectrometer in ESI + mode with a capillary voltage of 3.0 kV and a source temperature set at 120 °C. The cone gas flow was 50 L/h and the gas used was nitrogen. The desolvation gas temperature was 450 °C, the desolvation gas flow was 1000 Lh−1 and the nebuliser gas flow was 6 bar. Following ionisation, the ions were passed into the ion mobility cell. The helium cell gas flow used to fill the ion mobility cell was fixed at 180 mL min−1 and the IMS gas flow was fixed at 90 mL min−1. A wave velocity of 650 m/s, a wave height of 40 V, a EDC delay coefficient of 1.41 V, a bias of 3, a mobility RF offset of 250, a Rf offset of 300, an IMS wave delay of 1000 µs and a DC entrance of 20 were applied. The IMS was calibrated using the Waters Major Mix IMS/ToF Calibration Kit for IMS. Following IM the ions were subjected to MS with data acquired over the m/z range 50–1200. Continuum mode was applied to the experiment with a low collision energy of 15 eV and an elevation for the high collision energy of 45 eV. Leucine encephalin (MW = 555.62) was used for mass accuracy with a scan collected every 15 s and a cone voltage fixed at 30 V. The data were collected using MassLynx V 4.1 (Waters Corp., Milford, USA).
Data Pre-processing
The conventional UPLC–MS raw dataset was converted into the mzML format using ProteoWizard 3.0 [17] and pre-processed using XCMS software 1.52.0 [18] as a package into RStudio. The peak detection was done using the centWave method, with a peak width going from 1 to 15 s, a mass accuracy of 25 ppm, a signal to noise threshold cut-off of 3, a retention time error of 12 s, a m/z error of 0.05 Da and a prefilter defining a minimal number of 8 scans per peak with a minimal intensity of 1000. Peak grouping was done according to the nearest method. A minimum fraction filter was applied before the normalization to conserve only the features present at a minimal level of 30% in at least one of the sample groups. Median fold change normalization was then applied as well as a coefficient of variation filter, based only on QCs and with a threshold of 0.2.
The RAMMP–IM–MS raw dataset was firstly centroided using “Accurate Mass Measure” in MassLynx according to the “Automatic Peak Detection” process type. As with the conventional UPLC–MS raw dataset, the RAMMP–IM–MS data were converted into the mzMLformat using ProteoWizard and pre-processed using the same XCMS script, but using the following parameters for the dataset: maximum peak width—20 s, mass accuracy—15 ppm, retention time error—6 s and minimum number of scans considered—7.
Data Visualization and Analysis
The output compound measurements were imported into SIMCA 14.1 (Umetrics). The datasets were log-transformed with an offset of 20 and mean-centred. Then PCA score plots were used to assess the general distribution of the samples and the QCs, and cross-validated OPLS-DA models were used to select the features of interest.
Feature Selection
To select the features impacting the most on the class separation between the animals fed with a control diet or those fed with the MCD diet an S-plot was created from the OPLS-DA model for each method. A p(corr) value of 0.05 was used to select the features showing the greatest influence on the cross-validated OPLS-DA model from the conventional UPLC–MS method whilst for the RAMMP–IM–MS method, a p(corr) value of 0.1 was used.
Feature Annotation
For the RAMMP–IMS–MS dataset metabolite annotation was performed using UNIFI® Scientific Information System software 1.9, service release 4 (Waters Corp., Milford, MA, USA), which matched these data with an in-house database constructed using the compounds provided in the IROA Mass Spectrometry Metabolite Library of Standards (MSLMS) [12]. The database was searched using accurate mass m/z values (± 10 ppm) and CCS values (± 2% the mean direct infused (DI)-IMS), including protonated or sodiated adducts within 0.2 min of the expected retention time. Database-matching was performed on the entire dataset and, from the annotations that were obtained, the features selected as driving the separation between the controls and those fed the MCD diet, and that were present at least two-thirds of the samples in one of these classes were annotated where possible.
Results and Discussion
As indicated in the experimental section, the urine samples obtained from these animals were analysed using both conventional UPLC–MS [14] and RAMMP–IM–MS. Following peak picking and peak grouping ca. 24,000 retention time/mass features were found using the standard UPLC–MS method and ca. 3300 using the RAMMP–IM–MS method, respectively (Table 1). In the first instance, the metabolic profile of the RAMMP–IM–MS dataset was compared with that obtained by conventional UPLC–MS, to confirm that the outputs of both methods were comparable in terms of group discrimination, as previously shown for RAMMP–MS vs UPLC–MS [4]. Application of the minimum fraction filter resulted in the number of features being divided by 3.55 for the conventional UPLC–MS method and by 3.93 for the RAMMP–IM–MS method. Finally, after filtering the data based on a coefficient of variation of 0.2, the final number of features detected for each method were 3,414 and 479 by UPLC–MS and RAMMP–IM–MS methods, respectively (summarized in Table 1).
Metabolite Profiling
As previously described [13], following 8 weeks on the MCD diet designed to mimic NASH, a severe state of non-alcoholic fatty liver disease (NAFLD) was successfully induced in the five rats to which it was administerd, which all exhibited liver pathology. Following metabolic phenotyping of the urine samples from this study by UPLC–MS and RAMMP–IM–MS, as described above, unsupervised multivariate statistical analysis of the data (principal components analysis, PCA) showed a clear separation along principal component 1 (PC1), with a similar distribution of the urine samples seen for both LC methods, which clustered according to the dietary regime (Fig. 1). The QCs used to determine the reproducibility of the measurements were all well clustered in the PCA score plots for both UPLC–MS and RAMMP–IMS–MS datasets, with all 479 features in the latter showing a CV of 20% or less (indeed the vast majority were 15% or better) (see Supplementary Table S1).

PCA score plots showing the distribution of the urine samples according to their diet (control diet in black and MCD diet in red) at the different time points after 8 days on the two diets. The PCA score plots showing the distribution of the urine samples according to diet with control diet in black and MCD diet in red at the different time points (i.e., 8 weeks + 0, + 12, + 24 and + 48 h, respectively, shown with the shade becoming lighter with time) and QCs (blue). Left upper: PCA score plots obtained when the samples were analysed by UPLC–MS (4 principal components, N = 56 samples, R2X(cum) = 0.667 and Q2(cum) = 0.561). Right upper: PCA score plot obtained when the samples were analysed using RAMMP–IMS–MS (2 principal components, N = 56 samples, R2X(cum) = 0.543 and Q2(cum) = 0.495). The PCA plots in the two lower panels show the results for the − 6 to 0 h samples analysed by either UPLC–MS (Lower left) or RAMMP–IMS–MS (Lower right)
The PCA score plots provided very similar R2X(cum) values for both the UPLC–MS (0.667) and RAMMP–IMS–MS (0.543) methods, showing a good fit of the model to the dataset. Both methods also gave similar Q2(cum) results (0.561 for conventional UPLC–MS and 0.495 for RAMMP–IMS–MS method), indicating that models derived from both methods provided good predictions of the datasets after cross-validation. With both analytical methods, a slight time-related effect was observed along PC2 for both diets showing that, even with the reduced feature detection for the RAMMP–IMS–MS vs conventional UPLC–MS, the discrimination of metabolic phenotypes between the two groups of rats was conserved.
To simplify further comparisons, only the data for the 0 h time point samples obtained for animals on each diet were selected for further analysis (Fig. 1, lower panels).
Feature Selection
Cross-validated OPLS-DA models were generated from the 0 h time point urine samples for each diet (Fig. 2). Similar results in terms of group separation were obtained for the data generated by both methods, as demonstrated by the comparable values of R2X(cum), R2Y(cum) and Q2(cum) which were for the conventional method, 1 predictive and 1 orthogonal component, based on N = 10 samples, R2X(cum) = 0.540, R2Y(cum) = 0.997 and Q2(cum) = 0.944). The equivalent figures for the RAMMP/IM/MS method were, for1 predictive and 1 orthogonal component, R2X(cum) = 0.669, R2Y(cum) = 0.998 and Q2(cum) = 0.966). The OPLS-DA loading S plots were then used to identify the features driving the separation between the control and MCD diet-fed animals. To select these features, a p(corr) of 0.05 was used for the conventional UPLC–MS method, which resulted in 35 features being selected. However, as discussed and expected, the number of features detected with the RAMMP–IM–MS method that passed through the various data filters was much lower than for the conventional method (Table 1). The number of features found to be significant in terms of driving the discrimination between the two groups using a p(corr) of 0.05 on the S plot of the RAMMP–IM–MS model gave 11 features. Increasing the p(corr) value to 0.1 for the RAMMP/IM/MS method increased the number of features selected to 47.

Upper: cross-validated OPLS-DA score plots representing the distribution of the urine samples at the 0 h time point coloured according to control (black) or MCD (red) diets. Upper left: OPLS-DA score plot for the conventional UPLC–MS method.Upper right: OPLS-DA score plot for the RAMMP–IM–MS method. Lower: OPLS-DA loading S plots comparing features between the control and the MCD diet with the features selected for metabolite identification coloured in red (p(corr) value of 0.05 for the conventional UPLC–MS (left) and 0.1 for the RAMMP–IM–MS method (right)
Comparison of the Profiles from Conventional and RAMMP–IM–MS Methods.
Based on the OPLS-DA loading S plots, a comparison of the most discriminating signals from each group (control and MCD diets) was made according to both UPLC–MS and RAMMP–IMS–MS methods. Features were selected according to a p(corr) value of 0.05 for UPLC–MS (N = 35) and 0.1 for RAMMP–IM–MS N = 47. Out of the 35 discriminating features in the conventional UPLC–MS method and the 47 discriminating features in the RAMMP–IM–MS method detected in this preliminary analysis of the data, there were very few in common. However, despite the lack of overlap between the two methods the class separations observed in Fig. 1 are clear. This result is interesting given that in a previous study where RP-RAMMP–MS was compared with conventional UPLC–MS the discriminating features were similar. However, such an outcome is perhaps not unexpected as the introduction of an IM-based separation effectively changes the selectivity of the separation system.
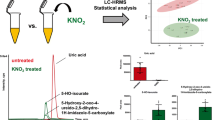
The data for the 47 features driving the separation between the control rats and those fed the MCD diet and measured with the RAMMP–IMS–MS method were matched against a previously described in-house database, constructed using the compounds in the IROA Mass Spectrometry Metabolite Library of Standards (see Ref. [12]). This database includes retention time, m/z and the CCS values for ca. 530 compounds. Preliminary annotations on the features that it contained that might represent compounds for which data were available were initially selected on the basis of CCS and m/z values, and these were then compared against retention time. These preliminary annotations were therefore made on the basis of all three of these properties wherever possible using retention matching of ± 0.2 min, CCS of ± 2% and mass accuracy of ± 10 ppm, as indicated in the experimental section. In addition, exclusion based the compound not being detected in a sufficient number of samples, or “biochemical plausibility” were also used to screen out e.g., xenobiotics etc. As a result, three metabolites, N-acetylglutamate, urocanic acid, and the tryptophan catabolite xanthurenic acid were identified based on tR, m/z and CCS values match based on the UPLC–IM–MS database [12]. Kynurenic acid, another tryptophan metabolite, was more tentatively identified based only on matches to mass and CCS values. Other potential compounds, initially selected on the basis of the CCS and m/z filters, including hypoxanthine, salicyamide, methyl-β-d-galactoside, galacterate and 10-hydroxydecanoate were eliminated from consideration based on tR, poor abundance across the sample set, or insufficient biological plausibility. The data for N-acetylglutamate, which was found to be more abundant in the urine of control compared to MCD diet-fed rats, are shown in Fig. 3 whilst the equivalent results for urocanic, xanthurenic and kynurenic acids are shown in Supplementary Figures S1–S3 and Tables S2–S5.

Representative data for RAMMP–IM–MS results illustrated by the example of N-acetyl-glutamate which was omnipresent in control samples but not MCD-diet-derived urines
Since its early beginnings in LC–MS-based metabolic phenotyping [19] the potential of IM, especially when combined with the CCS values derived from it, has been obvious, and both are increasingly being used in metabotyping (e.g., [5, 6, 8,9,10,11,12]). However, whilst this trend is welcome, and the results obtained here employing the combination of tR, CCS are gratifying, the relatively low number of positive identifications does once again highlight a major problem for LC–MS-based metabolic phenotyping in that it is much easier to detect potential biomarkers than to actually convincingly identify them. The in-house database that we have constructed based on the IROA MSMLS is obviously limited by the number of compounds that it contains and the fact that many of them are not found in urine. Indeed, of the 300 compounds in the library that were detected using the RAMMP method in + ve ESI for profiling rat urine only 63 metabolites (some present in only 1 sample/biological replicate) were positively identified using all three criteria [12]. The development of more comprehensive, matrix-specific, libraries of compounds is an obvious approach that could be developed, as is prediction of e.g. CCS and tR values. Indeed, there is much interest and indeed progress in the calculation of CCS values based on compound structures using both molecular modelling [20] or machine learning-based approaches [21, 22], and significant inroads have been made in this area. In the case of the IROA compounds, our previous study showed that calculated and measured CCS values were generally within 2% (or better) of each other [12] offering hope that CCS can be calculated as routinely as accurate mass, without the need for prior experimental determination. The remaining challenge, if productivity is to be maximized in metabolic profiling is, therefore, one for separation scientists in solving the problem(s) inherent in the prediction of chromatographic properties to the point where they can be used with confidence in the absence of a readily available authentic standard.
Conclusion
Comparison of conventional untargeted UPLC–MS method with RAMMP–IMS–MS has demonstrated that the results of both methods are complementary. The RAMMP approach, whilst detecting fewer features, had similar discriminating power to the conventional method and could be used as a means of rapid fingerprinting/screening samples prior to a more in-depth analysis using e.g., the conventional UPLC–MS method (perhaps supplemented with IM). The use of the database, containing m/z, CCS and separation-specific tR to identify analytes highlights both the benefits and the limitations of such approaches, and points to the need for improved methods for calculating/predicting properties such as CCS and tR.
References
Gavaghan-McKee CL, Wilson ID, Nicholson JK (2006) Metabolic phenotyping of nude and normal (Alpk:ApfCD, C57BL10J) mice. J Proteome Res 5:378–384
Nicholson JK, Lindon JC (2008) Systems biology: metabonomics. Nature 455:1054–1056
Hillesheim E, Brennan L (2019) Metabotyping and its role in nutrition research. Nutr Res Rev. https://doi.org/10.1017/S0954422419000179(Epub ahead of print)
Gray N, Adesina-Georgiadis K, Chekmeneva K, Plumb RS, Wilson ID, Nicholson JK (2016) Development of a rapid microbore metabolic profiling ultraperformance liquid chromatography–mass spectrometry approach for high-throughput phenotyping studies. Anal Chem 88:5742–5751
King AM, Mullin LG, Wilson ID, Coen M, Rainville PD, Plumb RS, Gethings LA, Maker G, Trengove R (2019) Development of a rapid profiling method for the analysis of polar analytes in urine using HILIC–MS and ion mobility enabled HILIC–MS. Metabolomics 15:17. https://doi.org/10.1007/s11306-019-1474-9
King AM, Trengove RD, Mullin LG, Rainville PD, Isaac G, Plumb RS, Gethings LA, Wilson ID (2019) Rapid profiling method for the analysis of lipids in human plasma using ion mobility enabled-reversed phase-ultra high performance liquid chromatography/mass spectrometry. J Chromatogr A. https://doi.org/10.1016/j.chroma.2019.460597(Epub ahead of print)
Rainville PD, Wilson ID, Nicholson JK, Isaac G, Mullin L, Langridge JI, Plumb RS (2017) Ion mobility spectrometry combined with ultra performance liquid chromatography/mass spectrometry for metabolic phenotyping of urine: effects of column length, gradient duration and ion mobility spectrometry on metabolite detection. Anal Chim Acta 982:1–8
Mairinger TJ, Causon S, Hann S (2018) The potential of ion mobility–mass spectrometry for non-targeted metabolomics. Curr Opin Chem Biol 42:9–15
Zhang X, Quinn K, Cruickshank-Quinn C, Reisdorph R, Reisdorph N (2018) The application of ion mobility mass spectrometry to metabolomics. Curr Opin Chem Biol 42:60–66
Szykula KM, Neurs J, Turner MA, Creaser CS, Reynolds JC (2019) Combined hydrophilic interaction liquid chromatography-scanning field asymetric waveform spectrometry-time of flight mass spectrometry for untargeted metabolomics. Anal Bioanal Chem 411:6309–6317
Nichols CM, Dodds JN, Rose BS, Picache JA, Morris CB, Codreanu SG, Sherrod MJCS, D, McLean JA, (2018) Untargeted molecular discovery in primary metabolism: collision cross section as a molecular descriptor in ion mobility-mass spectrometry. Anal Chem 90:14484–14492
Nye LC, Williams JP, Munjoma NC, Letertre MPM, Coen M, Bouwmeester M, Martens L, Swann JR, Nicholson JK, Plumb RS, McCullagha M, A. Gethings LA, Lai S, I. Langridge J, Vissers JPC, Wilson ID, (2019) A comparison of collision cross section values obtained via travelling wave ion mobility-mass spectrometry and ultra high performance liquid chromatography-ion mobility-mass spectrometry: application to the characterisation of metabolites in rat urine. J Chromatogr 1602:386–396
Kyriakides M, Hardwick RN, Jin Z, Goedken MJ, Holmes E, Cherrington NJ, Coen M (2014) Systems level metabolic phenotype of methotrexate administration in the context of non-alcoholic steatohepatitis in the rat. Toxicol Sci 142:105–116
Lewis MR, Pearce JTM, Spagou K, Green M, Dona AC, Yuen AHY, David M, Berry DJ, Chappell K, Horneffer-van der Sluis V, Shaw R, Lovestone S, Elliott P, Shockcor J, Lindon JC, Cloarec O, Takats Z, Holmes E, Nicholson JK, (2016) Ultra-performance liquid chromatography-TOF MS for precision large scale urinary metabolic phenotyping. Anal Chem 88:9004–9013
Sangster T, Major H, Plumb R, Amy J, Wilson AJ, Wilson ID (2006) A pragmatic and readily implemented quality control strategy for HPLC–MS and GC–MS-based metabonomic analysis. Analyst 131:1075–1078
Gika HG, Theodoridis GA, Wingate JE, Wilson ID (2007) Within-day reproducibility of an HPLC–MS-based method for metabonomic analysis: application to human urine. J Proteome Res 6:3291–3303
Holman JD, Tabb DL, Mallick P (2014) Employing ProteoWizard to convert raw mass spectrometry data. Curr Protoc Bioinform 46:1–9
Tautenhahn R, Patti GJ, Rinehart D, Gary Siuzdak G (2012) XCMS Online: a web-based platform to process untargeted metabolomic data. Anal Chem 5(84):5035–5039
Harry EL, Weston DJ, Bristow AWT, Wilson ID, Creaser CS (2008) An approach to enhancing coverage of the urinary metabonome using liquid chromatography-ion mobility-mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci 871:357–361
Colby SM, Thomas DG, Nuñez JR, Baxter DJ, Glaesemann KR, Brown JM, Pirrung M, Govind N, Teeguarden JG, Metz TO, Renslow RS (2018) ISiCLE: a molecular collision cross section calculation pipeline for establishing large in silico reference libraries for compound identification. https://arxiv.org/abs/1809.08378
Zhou Z, Shen X, Tu J, Zhu ZJ (2016) Large-scale prediction of collision cross-section values for metabolites in ion mobility-mass spectrometry. Anal Chem 88:11084–11091
Heinonen M, Shen H, Zamboni N, Rousu J (2012) Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 28:2333–2341
Funding
Stratigrad is gratefully acknowledged for financial support of a Ph.D. studentship to Martine Letertre NIHR Imperial BRC is acknowledged for support to Muireann Coen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no conflicts of interest with respect to the research, authorship, and/or publication of this article. This investigation was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Arizona. The work was undertaken within the NIH guidelines on the care and use of experimental animals as previously described [13].
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Letertre, M., Munjoma, N.C., Slade, S.E. et al. Metabolic Phenotyping Using UPLC–MS and Rapid Microbore UPLC–IM–MS: Determination of the Effect of Different Dietary Regimes on the Urinary Metabolome of the Rat. Chromatographia 83, 853–861 (2020). https://doi.org/10.1007/s10337-020-03900-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-020-03900-4