
Abstract
A sensitive and specific liquid chromatography electrospray ionisation–tandem mass spectrometry method for determination of new non-imidazole histamine H3 receptor antagonist 1-[3-(4-tert-butylphenoxy)propyl]piperidine (DL76) in rat serum has been developed and validated. Chromatography was performed on a XBridge™ C18 analytical column (2.1 × 30 mm, 3.5 µm, Waters, Ireland) with gradient elution using a mobile phase containing acetonitrile and water with an addition of 0.1% of formic acid. Detection was achieved by an Applied Biosystems MDS Sciex (Concord, Ontario, Canada) API 2000 triple quadrupole mass spectrometer. Electrospray ionization (ESI) was used for ion production. The limit of detection in the SRM mode was found to be 0.5 ng mL−1. The limit of quantification was 1 ng mL−1. The precision and accuracy for both intra- and inter-day determination of DL76 ranged from 1.65 to 15.09% and from 88.74 to 113.43%. The results of this analytical method validation allow to carry out pharmacokinetic studies in rats. The method was used for the pilot study of the pharmacokinetic behavior of DL76 in rats after intravenous administration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The biogenic amine histamine is implicated in the wide range of physiological and pathophysiological functions. It is a ligand for four histaminergic receptors subtypes, from which receptor H3 is acting as presynaptic auto- and heteroreceptor mainly in the CNS controlling the synthesis and release of histamine. Its activation modulates also the release of various important neurotransmitters, such as dopamine, acetylcholine, norepinephrine, serotonin, GABA, glutamate and substance P. Therefore, histaminic neurons and the cross-linkage between major neurotransmitter systems are involved in many brain functions including vigilance, memory processes, feeding behavior and locomotor activity.
Blockade of the H3 autoreceptors by antagonists interrupts the negative feedback mechanism and leads to increased levels of histamine and other neurotransmitters. These effects suggest its potential therapeutic role for the treatment of several diseases and neurological disorders, such as schizophrenia, depression, sleep-wake disorders, dementia or epilepsy.
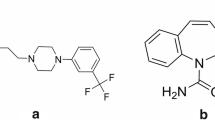
Known compounds, H3 receptor antagonists, which are imidazole derivatives make poor candidates for drugs due to their tendency to inhibit numerous mammalian Cyt 450 isozymes and in case of a CNS drugs, their low brain penetration. Therefore, a significant effort was undertaken to identify non-imidazole-derived H3 antagonists [1–4]. The example of such compound is DL76 (1-[3-(4-tert- butylphenoxy)propyl]piperidine) which in preliminary pharmacological studies showed good affinity for human H3 receptor stably expressed in CHO-K1 cells (hKi equal 22±3 nM) [5].
Preliminarily, both pharmacodynamic as well as pharmacokinetic characteristics of any new compound, potential drug, require availability of a validated analytical method, therefore, the purpose of this study was to develop and validate a high performance liquid chromatography–tandem mass spectrometry method (LC–MS–MS) for quantification of DL76 compound in rat serum samples. The new method was successfully applied to a pilot study of pharmacokinetics of DL76 in rats.
Experimental
Chemicals and Materials
The 1-[3-(4-tert-butylphenoxy)propyl]piperidine was supplied from the Department of Technology and Biotechnology of Drugs (Faculty of Pharmacy, Jagiellonian University Medical College, Krakow, Poland). Pentoxifylline (3,7-dimethyl-1-(5-oxohexyl)-3,7-dihydro-1H-purine-2,6-dione, PTX), used as an internal standard, was obtained from Sigma (St. Louis, MO, USA). HPLC grade acetonitrile, water, formic acid and ethyl acetate were purchased from Merck (Darmstadt, Germany). Control serum was obtained from adult male Wistar rats Krf:(WI)WV (Charles River Laboratory, Germany). Rats were injected intraperitoneally with thiopental (70 mg kg−1), and blood was collected after decapitation. The serum samples were separated by centrifugation (3,000 rpm, 10 min) and stored at −30 °C until used.
Instrumentation and Operating Conditions
Liquid Chromatographic Conditions
Liquid chromatography was performed using an Agilent 1100 HPLC (Agilent Technologies, Waldbronn, Germany) system consisting of a degasser, a binary pump, a column oven and an autosampler. Chromatographic separation was carried out on XBridge™ C18 analytical column (2.1 × 30 mm, 3.5 µm, Waters, Dublin, Ireland) with the oven temperature set at 30 °C. The autosampler temperature was maintained at 10 °C. The mobile phase of acetonitrile (A) and water (B) with an addition of 0.1% of formic acid was set at a flow rate of 0.3 mL min−1. Initial mobile phase composition was 100% B with a linear gradient to 30% B in the first 8 min, then isocratic mode for 1 min with the following rapid change back to 100% B in 0.1 min. The remaining time of elution was set at 100% B. The whole HPLC operation lasted 20 min. A sample volume of 10 μL was injected into LC–MS–MS system.
Mass Spectrometric Conditions
Mass spectrometric detection was performed on an Applied Biosystems MDS Sciex (Concord, Ontario, Canada) API 2000 triple quadrupole mass spectrometer equipped with an electrospray ionization (ESI) interface. ESI ionization was performed in the positive ion mode. The tandem mass spectrometer was operated at unit resolution in the selected reaction monitoring mode (SRM), monitoring the transition of the protonated molecular ions m/z 276 and 279 to the product ions m/z 98 and 181 for DL76 and PTX, respectively.
The mass spectrometric conditions were optimized for DL76 by continuous infusion of the standard solution at the rate of 10 μL min−1 using a Harvard infusion pump. The ion source temperature was maintained at 450 °C. The ionspray voltage was set at 5,500 V. The curtain gas (CUR) was set at 10 and the collision gas (CAD) at 2. The optimal collision energy (CE) was 50 V. The following parameters of ion path were used as the most favorable ones: declustering potential (DP) at 45 V, focusing potential (FP) at 300 V and entrance potential (EP) at 12 V. The quantification was performed via peak area ratio. Data acquisition and processing were accomplished using the Applied Biosystems Analyst version 1.4.2 software.
Preparation of Stock Solutions
The stock solutions of DL76 and PTX (IS) were prepared at 1.0 mg mL−1 in water. The stock solution of DL76 was further serially diluted with water to make working solutions at concentrations of 20, 10, 5, 2.5, 1, 0.5, 0.1 and 0.01 μg mL−1. Internal standard working solution was prepared by diluting the stock solution of PTX with water to a concentration of 2.5 μg mL−1. All stock and working solutions were stored at 4 °C.
Calibration Curve
Calibration curve was generated to confirm the linear relationship between the peak area ratio and the concentration of DL76 in the test samples. Pooled blank serum was spiked with an appropriate working solution to produce the calibration curve points equivalent to 2,000, 1,000, 500, 250, 100, 50, 10 and 1 ng mL−1 of DL76 compound. Samples were made in five replicates and each of them also contained a 10 μL of the IS working solution. The results (peak area of analyte/peak area of IS) versus concentration were fitted to the linear equation. The quality of fit was evaluated by comparing back-calculated concentrations to the nominal ones.
Quality Control Samples
The quality control samples used during the validation and in the pharmacokinetic studies have been prepared in the same way as the calibration curve. Namely, they were prepared by spiking blank serum with DL76 and IS and produced at different concentrations along the calibration range (LLOQ at 1 ng mL−1, low at 40 ng mL−1, medium at 400 ng mL−1 and high at 1,800 ng mL−1 of DL76).
Extraction Procedures
Serum Samples
All analyzed samples were extracted employing a liquid–liquid extraction technique into an ethyl acetate without any modification of pH. A 100 μL aliquot of rat serum was pipetted out into a centrifuge tube and the working IS solution (10 μL) was added. Then the samples were briefly mixed and 1 mL of ethyl acetate was added. The mixture was shaken on a mechanical shaker for 20 min. Afterwards, samples were centrifuged at 3,000 rpm for 15 min and the organic layer (0.5 mL) was transferred to the conical tubes and evaporated to dryness at 37 °C under the gentle stream of nitrogen gas in the TurboVap evaporator (Zymark, Hopkinton, MA, USA). The dry residue was reconstituted with 100 μL of the acetonitrile/water (50/50, v/v) mixture and an aliquot of 10 μL was injected onto the LC–MS–MS system.
Method Validation
The analytical method was validated within the guidelines established by the FDA for bioanalytical method validation.
Assay Specificity
The specificity of the method was evaluated by analyzing blank serum samples from six rats. Each blank sample was tested for any interference, also the ones associated with thiopental, using the proposed extraction procedure and chromatographic/mass spectrometric conditions and compared with those obtained with an aqueous solution of the analyte at a concentration near the lower limit of quantification (LLOQ). An assessment of absolute matrix effect was done using matrix-free (solvent) samples and samples containing extracted blank serum with an addition (post-extraction) of an appropriate concentration of the DL76 compound. Three different concentration levels of DL76 were evaluated (40, 400, and 1,800 ng mL−1) by analyzing five samples at each level. For evaluation of the relative matrix effect, five different sources of rat serum were used [6, 7]. The relative ME was expressed as RSD (%).
The limit of detection (LOD) was estimated as a signal to noise ratio (3:1). The lower limit of quantification (LLOQ) defined in this assay was based on the expected range for DL76 in the samples, as opposed to minimum criteria of response or precision.
Precision and Accuracy
Precision of the assay was determined from the QC samples by replicate analysis of four concentration levels of DL76 (1, 40, 400 and 1,800 ng mL−1). Accuracy (percent of recovery) was evaluated as [mean found concentration/theoretical concentration] × 100. Precision was given by the percent relative standard deviations (RSD, CV%). The criteria for acceptability of the data included accuracy within ±15% deviation from the nominal values and a precision within 15% RSD except for LLOQ, where it should not exceed ±20% of CV.
Within-day precision and accuracy were determined by repeated analysis (n = 5) of the samples at different QC levels (1, 40, 400 and 1,800 ng mL−1 of DL76 in serum) on the same day. Between days precision and accuracy were determined by repeated analysis on three consecutive days. The concentration of each sample was determined using the calibration curve prepared and analyzed on the same day.
Extraction Recovery
Extraction recoveries of DL76 from serum were determined at low, medium and high concentrations. Recoveries were calculated by comparing the mean peak areas obtained for extracted QC samples (1, 40, 400 and 1,800 ng mL−1) with those of blank extracts reconstituted in the mobile phase with standards added at appropriate concentrations which represented the 100% recovery value. The recovery value of the IS was determined in the same way at a single concentration of 250 ng mL−1.
Stability
Freeze and Thaw Stability
QC serum samples at four concentration levels were stored at the storage temperature (−30 °C) for 24 h and thawed unassisted at room temperature, then the samples were refrozen for 24 h. After three freeze–thaw cycles samples were analyzed.
Short-Term Stability
QC serum samples at four concentration levels were kept at room temperature for a period that exceeded the routine sample preparation time (about 5 h).
Long-Term Stability
QC serum samples at four concentration levels were kept at the temperature of −30 °C for the period of 2 months and then analyzed.
Post-Preparative Stability
The autosampler stability was conducted by reanalyzing extracted QC samples kept under autosampler conditions (10 °C) for 24 h.
Samples were considered to be stable where 85–115% of the initial concentrations were found.
Application of the Assay
Study in Rats—Drug Administration and Sampling
Male Wistar rats, 13–15 weeks of age and weighting between 200 and 220 g were used for this study. They were kept under conditions of constant temperature (21–25 °C) and relative humidity of approximately 40–65% with standard light/dark cycle. Animals were housed in stainless steel cages with suspended wire-mesh floors (maximum of 5 rats per cage). Animals were fasted overnight and then weighed. Rats had free access to water throughout the experimental period. DL76 compound dissolved in 0.9% sterile isotonic saline at a dose of 3 mg kg−1 was administered intravenously. A total number of 30 rats were used in this experiment. Rats were anaesthetized with thiopental at a dose of 70 mg kg−1 and blood samples were collected by decapitation to microfuge tubes at 5, 10, 30, 60, 120 and 240 min after dosing (n = 5). Serum was harvested by centrifuging at 3,000 rpm for 10 min and stored at −30 °C until bioanalysis. Serum samples (100 μL) were spiked with IS and processed as described above. The study was approved by the Institutional Animal Care and Ethics Committee of the Jagiellonian University Medical College, Krakow, Poland.
Non-Compartmental Pharmacokinetic Analysis
Pharmacokinetic parameters were calculated by employing a non-compartmental approach from the average concentration values, using WinNonlin 3.3 software (Pharsight, USA). The area under the mean serum concentration versus time curve extrapolated to infinity (AUC0-inf) was estimated using the log/linear trapezoidal rule. AUMC was estimated by the calculation of total area under the first-moment curve by combining trapezoid calculation of AUMC0-t and extrapolated area. The mean residence time (MRT) was calculated from AUMC0-inf/AUC0-inf. The terminal rate constant (λ z) was calculated by log-linear regression of the drug concentration data in the terminal phase and the half-life (t 1/2) was calculated as 0.693/λ z. The systemic clearance (Cl) was estimated from the administered dose divided by AUC0-inf. The volume of distribution at steady state (V ss) was calculated from D i.v.·AUMC/AUC2, where D i.v stands for the dose administered intravenously in mg kg−1 of body weight.
Results and Discussion
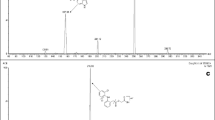
In the mass spectra of DL76 and IS dominated protonated molecules [M+H]+ at m/z 276 and m/z 279, respectively. The product ion spectra of these analytes and proposed fragmentation paths are presented in Fig. 1. After collision-induced dissociation, the major fragment ions observed in each product ion spectrum were at m/z 98 for DL76 and 181 for IS, so the SRM transitions m/z 276→98 and m/z 279→181 were selected for quantitative analysis.

Product ion mass spectra of [M+H+]+ ions of PTX (a) and DL76 (b) and proposed fragmentation paths
The mobile phase consisting of acetonitrile and water provided low background noise and fast separation. The running time of each sample was 20 min. The gradient used resulted in retention times of 5.45 and 7.67 min for IS and DL76, respectively, and the remaining time provided for column equilibration and clean up to avoid carry over between samples. Presence of formic acid in the mobile phase improved the sensitivity by promoting ionization of the analyte. The representative extracted ion chromatograms (XIC) of a blank sample and a rat serum sample with the concentration of DL76 determined at 67.82 ng mL−1 are shown in Fig. 2a and c, respectively. The number of theoretical plates is 16,000 and the retention factor 21.55.

The representative extracted ion chromatograms (XIC) of blank serum (a), serum sample spiked with the concentration of 1 ng mL−1 of DL76 (b), rat serum sample after i.v. administration of DL76 in the dose of 3 mg kg−1, with the concentration of DL76 determined at 67.82 ng mL−1 (c) and IS (d)
The lower limit of quantification (LLOQ) for DL76 was 1 ng mL−1, and the limit of detection (LOD) was 0.5 ng mL−1 with the noise to signal ratio equal 1:3. The lowest concentration on the calibration curve was chosen based on the expected concentrations in the biological matrices. At LLOQ the precision did not exceed 15%.
The absolute matrix effect for DL76 was 30%, and the absolute recovery was 60% giving low process efficiency (mean peak area obtained for the analyte added to the matrix before the extraction to the corresponding area for the pure solution), but because of high ionization of the analyte in the ESI source and its good retention on analytical column the LLOQ was sufficient for the purpose of conducting pharmacokinetic studies. No relative matrix effect for studied compound was observed for five different serum pools tested, the RSD values did not exceed 5%.
Calibration curve for DL76 compound in serum was linear over the proposed concentration range (1–2,000 ng mL−1). The calibration curve was generated by weighted (1/x) linear regression analysis.
As seen in Table 1 for each point on the calibration curve for DL76, the concentration back-calculated from the equation of the linear regression analysis was always within the deviation of 3% from the nominal concentrations and the relative standard deviations did not exceed ±15%.
Data for within day and between days precision and accuracy of this method are presented in Table 2. Values of precision of the method ranged from 1.65 to 15.9% and for accuracy ranged from 88.74 to 113.43%, generally accepted for bioanalytical methods.
The stability of DL76 in rats serum was investigated under a variety of storage and process conditions as described in previous section. Tables 3 and 4 summarize these results. Compound DL76 showed good stability during all these tests and there were no stability related problems during the routine analysis of samples for pharmacokinetic studies.
Application to Pharmacokinetic Study
The method described above was applied to pharmacokinetic study, in which serum concentrations of DL76 compound were determined for 4 h after intravenous administration to rats.
The plot of time course for mean serum concentrations is shown in Fig. 3. A non-compartmental pharmacokinetic model was used to describe the pharmacokinetics of DL76 following intravenous administration. DL76 was quickly eliminated with systemic clearance of 13 L h−1 kg−1. The elimination half-life was 84.32 min. The volume of distribution at the central compartment was 20 L kg−1 suggesting that there is wide distribution in peripheral tissues. The detailed pharmacokinetics and tissue distribution of DL76 will be presented in a separate paper.

Mean plasma concentrations ±SD (n = 5) versus time profile obtained after a single i.v. dose (3 mg kg−1) of DL76 compound
Conclusion
In this manuscript, we described a sensitive and selective high performance liquid chromatography–tandem mass spectrometry method for the analysis of DL76 compound in rat serum. Validation of the method in selected conditions shows that the method is selective and precise with linear response of mass spectrometer. The liquid–liquid extraction procedures produced clean chromatograms and high and reproducible recovery was obtained for investigated compound. The method has been successfully employed to support a number of pilot pharmacokinetic studies.
References
Sander K, Kottke T, Stark H (2008) Histamine H3 receptor antagonists go to clinics. Biol Pharm Bull 31:2163–2181
von Coburg Y, Kottke T, Weizel L, Ligneau X, Stark H (2009) Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg Med Chem Lett 19:538–542
Esbenshade TA, Browman KE, Bitner RS, Strakhova M, Cowart MD, Brioni JD (2008) The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br J Pharmacol 154:1166–1181
Gemkow MJ, Davenport AJ, Harich S, Ellenbroeck BA, Cesura A, Hallett D (2009) The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov Today 14:509–515
Lazewska D, Ligneau X, Schwartz JC, Schunack W, Stark H, Kiec-Kononowicz K (2006) Ether derivatives of 3-piperidinopropan-1-ol as non-imidazole histamine H3 receptor antagonists. Bioorg Med Chem 14:3522–3529
Taylor PJ (2005) Matrix effects: the Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin Biochem 38:328–334
Matuszewski BK, Constanzer ML, Chavez-Eng CM (2003) Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC–MS–MS. Anal Chem 75:3019–3030
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Szafarz, M., Szymura-Oleksiak, J., Lazewska, D. et al. LC–MS–MS Method for the Analysis of New Non-Imidazole Histamine H3 Receptor Antagonist 1-[3-(4-tert-Butylphenoxy)propyl]piperidine in Rat Serum—Application to Pharmacokinetic Studies. Chromatographia 73, 913–919 (2011). https://doi.org/10.1007/s10337-011-1983-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-011-1983-9