
Abstract
To prevent threats from pathogens such as Phytophthora species from international plant trade, molecular identification techniques are needed for rapid, accurate quarantine inspection. Here, for quarantine control in Japan, we developed a simple DNA extraction for plants and a practical detection method that combines multiplexed PCR using primers specific for Phytophthora species, for P. nicotianae, which is the only non-quarantine Phytophthora species, and as internal controls, for plants. For the new genus-level primer set, we modified previously reported genus-specific primers to improve detectability. The new primers were able to detect mycelial DNA of 155 taxa among Phytophthora clades 1–10, with a sensitivity of 100 fg/µL for three representative species, P. ramorum, P. kernoviae and P. nicotianae. In the PCRs using DNA from non-target species, amplification was observed for only three taxa, and for some strains, four taxa in a closely related genus. Duplex and triplex PCR of the genus-specific primers combined with previously reported plant primers verified the success of DNA extraction and PCR detection from diseased plant samples, and in the triplex PCR, whether the pathogen was diagnosed as P. nicotianae or not by the species-specific primer. The new method detected the pathogen in naturally infected and inoculated plants. The amplicons using the genus-specific primer have enough variation to be sequenced to identify the species. This new method can be used immediately for detecting Phytophthora species and for quarantine control in Japan.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The oomycete genus Phytophthora includes many plant pathogens that cause destructive diseases and severe commercial losses, not only to agricultural crops, but also to forest trees and nursery plants. Many new species of Phytophthora have been identified from diseased nursery trees and plants in natural ecosystems, and new species continue to be found (Brasier 2009). With the expansion of global plant trade, Phytophthora species have also spread globally (Brasier 2009). These pathogens usually cause little damage to their host plants in their native habitats and have coevolved with their hosts, achieving a natural balance. However, when introduced by human activity into other regions with a favorable environment that lacks natural enemies, they can be highly virulent and cause serious damage on genetically susceptible host plants (Brasier 2008).
More precise phytosanitary measures are required to prevent economic loss of crops on a global scale and to conserve endemic biodiversity. In Japan, P. ramorum was isolated during quarantine inspection from discolored leaves of rhododendrons imported from the United Kingdom in 2015 (Sakoda et al. 2017). In Japan, P. ramorum and P. kernoviae are considered the most important quarantined species, and only P. nicotianae is exempt from quarantine, since it is already a major pathogen. Molecular identification techniques are required for simultaneous detection of Phytophthora species; therefore, we developed a simple identification technique using loop-mediated isothermal amplification (LAMP) with a quenching probe (QProbe) (Hieno et al. 2020) that was designed for a sequence specific to P. nicotianae in the amplified region of a LAMP primer specific for Phytophthora. The total time from sampling to detection is approximately 2 h; thus, it is a time-saving technique for import/export quarantine. Other advantages include high tolerance to inhibitors from biological sources, and thus, easy DNA extraction methods are applicable (Hieno et al. 2019). However, the multiplex reaction in the LAMP method presents difficulties in that the strong amplification of more abundant DNA may overwhelm the amplification of less abundant DNA, and the sensitivity of multiple targets may be reduced (Hieno et al. 2021). In contrast, PCR can achieve high sensitivity in a simultaneous detection method (Li et al. 2011). In this study, we aimed to develop a multiplex PCR method using primers specific for Phytophthora at the genus level and primers specific for P. nicotianae (Li et al. 2011) as an accurate and time-saving method to detect multiple targets. This multiplex PCR can be used to inspect samples for the broader Phytophthora genus and for P. nicotianae whether the pathogens are present in the sample or subject to quarantine or not. Furthermore, the genus-specific primers are expected to be used for sequencing to identify the Phytophthora species detected in the sample. In previous studies on PCR techniques using primers specific for Phytophthora species, most primers were designed for the ITS region, but identical ITS sequences have been identified for 16 pairings of species from molecular phylogenetic clades 1, 5, 6, 7 or 8, making identification of the species difficult (Yang and Hong 2018). The introns of the Ypt1 gene are sufficiently polymorphic to discriminate all Phytophthora species and are located near the conserved coding regions, which is suitable for designing primers specific to the genus Phytophthora (Schena and Cooke 2006). In the present study, we designed genus-specific primers for Ypt1 by modifying the primers reported by Schena et al. (2008). The modified forward and reverse primers have been used for multiplex PCR detection of the kiwifruit pathogens Phytophthora cactorum, P. cinnamomi and P. lateralis (Bi et al. 2019). The modified primers have the potential to be used for detecting additional species of Phytophthora but have not been tested with enough strains of Phytophthora and with other genera, including closely related Pythium and Phytopythium.
In this study, we reevaluated the applicability of our modified genus-specific primers by using a sufficient number of strains, then developed a duplex PCR using the Phytophthora genus-specific primers and plant primers (Martin et al. 2004) as an internal control. We also developed a triplex PCR using additional P. nicotianae-specific primers (Li et al. 2011) for effective quarantine control in Japan. By combining this multiplex PCR method with a simple method to extract DNA from diseased plants, we aimed to establish a more practicable and user-friendly protocol for quarantine inspections.
Materials and methods
Isolates and mycelial DNA extraction
Isolates of Phytophthora spp., Phytopythium spp., Pythium spp. and other pathogens used in this study are listed in Table 1. For the mycelial DNA extraction, isolates were grown on V8 juice agar plates [Miller (1955) with the following modifications: 1 l including 162 mL V8 juice (Campbell Japan), 20 g agar, pH modified with CaCO3] at 25 °C until the mycelium reached the edge of the plates. The mycelium was scraped from the plates with inoculation needles into 1.5-ml Eppendorf tubes containing 100 µl of 50% PrepMan Ultra Reagent (Thermo Fischer Scientific, Waltham, MA, USA) and incubated at 100 °C for 10 min. After 3 min at room temperature, the sample was centrifuged at 15,000 rpm for 3 min. The supernatant was transferred to a new 1.5-ml tube. The DNA concentration was measured using the QuantiFluor dsDNA System (Promega, Madison, WI, USA) and adjusted to 100 pg/µl with Tris–EDTA buffer (TE buffer; 10 mM Tris–HCl, 1 mM EDTA, pH 8.0). The samples were stored at 4 °C until further use.
DNA preparations from inoculated and from naturally infected plants
The host–pathogen combinations used in the inoculation tests are shown in supplementary Table S1. P. nicotianae (GK10Eg1 and 13ASP1-1), P. capsici (CH01CUCU10 and CH02UE0202), P. hedraiandra (TGTA1-1) and P. melonis (CH00ME21-21) were grown on V8 juice agar plates at 25 °C until mycelial growth reached the edge of the plate, and 6- or 10-mm-diameter mycelial disks were taken from actively growing colonies. For tomato, eggplant, pumpkin and cucumber, the smaller mycelial discs were placed on the fruit. Inoculated materials were placed on wet paper towels in plastic trays, which were covered with polyethylene bags to maintain high humidity and incubated in a growth chamber (12 h light/12 h dark) at 25 °C for 5–7 days until symptoms were obvious. For hydrangea in pots, the larger mycelial disks were placed on a needle wound on the basal stem, then wrapped in parafilm. The whole plant was covered with a polyethylene bag to maintain humidity and kept at room temperature (approximately 25 °C) in the laboratory for 10 days until symptoms were seen. For ivy, P. citrophthora (CH94HE11) was cultured in V8 juice broth in a 6 cm Petri dish until the entire surface was covered with hyphae (~ 4 days). The broth was then removed with a filter paper, and the mycelia were homogenized with 100 mL of sterilized distilled water at 3,000 rpm for 5 min. Young ivy leaves were then detached and placed in the mycelial suspension for 7–10 days at 20 °C until symptoms were obvious.
Detached leaves of rhododendron, Japanese andromeda and camellia were inoculated with one or two mycelial disks (7 mm) P. ramorum (CBS 101553), P. kernoviae (P19875) and P. lateralis (P3361) in a plastic box as described by Hieno et al. (2021) with 12 h light at 20 °C/12 h dark at 15 °C for 3–5 days until symptoms were seen.
Symptomatic stems of periwinkle from Gifu, Japan and tobacco plants from Java island, Indonesia were used as naturally infected samples.
For DNA extraction from symptomatic leaves or stems, a 5 × 5 mm leaf piece or 0.2 g of epidermis shaved from the stem was shredded with a blade, then all material for the test plant was placed in a 1.5-ml Eppendorf tube. DNA was extracted using the Kaneka Easy DNA Extraction Kit version 2 (Kaneka, Tokyo, Japan) and the manufacturer’s protocol. All extracts were diluted 20 times with TE buffer and stored at 4 °C until further use.
Primers specific for Phytophthora
In our previous study (Bi et al. 2019), we designed a primer pair specific for Phytophthora (Yph1F_mod2: CGACCATKGTGGACTTTG, Yph2R_mod2: ACGTTCTCRCAGGCGTATCTG) based on the Phytophthora-specific primers (Yph1F and Yph2R) of Schena et al. (2008). In the present study, we further tested the specificity and applicability of our genus-specific primer pair (Bi et al. 2019) using 222 Phytophthora isolates that represented 155 taxa and 104 isolates that belonged to other genera (Table 1). We then used this primer pair to develop the multiplex PCR assay (described later) to detect Phytophthora species from infected plants.
Simplex PCR using genus-specific primers for Phytophthora
The reaction mixture contained 0.5 µM of each primer (Yph1F_mod2, Yph2R_mod2), 0.625 U Taq HS DNA polymerase (Takara Bio, Kusatsu, Shiga, Japan), 0.2 mM dNTP mixture, 1 × PCR buffer (10 mM Tris–HCl pH 8.9, 50 mM KCl and 1.5 mM MgCl2), 10 ng of bovine serum albumin (Merck KGaA, Darmstadt, Germany), and 0.1 ng of DNA template, in a total volume of 25 µl. The PCR was run in a BioRad T100 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) at 95 °C for 2 min; 40 cycles of denaturation at 94 °C for 30 s, annealing at 62 °C for 45 s, an extension at 72 °C for 30 s; and a final extension at 72 °C for 10 min.
Applicability of the primers was evaluated in simplex PCR for a wide range of strains with different molecular phylogenetic groups (André Lévesque and De Cock 2004; Abad et al. 2022) as shown in Table 1. The sensitivity of the simplex PCR was tested with serial dilutions (10 pg–1 fg per reaction) of mycelial DNA of P. ramorum (Pr-1), P. kernoviae (P19875) and P. nicotianae (CBS 305.29). The simplex PCR was also tested for detectability of DNA from inoculated and naturally infested plants (Supplementary Table S1).
Duplex and triplex PCR
Plant primer pair (FMPl-2b and FMPl-3b) reported by Martin et al. (2004), which amplifies cox1, was used as an internal control to determine the success or failure of DNA extraction and PCR of the test samples. The plant species used to test primer detectability are shown in Table 2. We extracted DNA from plant samples and used it in PCR tests as described for the simplex PCR. The same DNA thermal cycler was used, but thermocyling conditions were 95 °C for 8.5 min; 40 cycles at 95 °C for 30 s, 60 °C for 30 s, 72 °C for 1 min; and 72 °C for 10 min.
Duplex PCR for plant–Phytophthora spp. was also done as described for the simplex PCR above except for the concentrations of MgCl2 and plant primers. MgCl2 and the plant primers were tested in combinations of 1.5, 2.0, 2.5 mM and 0.05, 0.125, 0.25, 0.5 µM, respectively, to determine the optimal concentration of each.
Triplex PCR for plant–Phytophthora spp.–P. nicotianae was performed as described above for the duplex PCR, but using the primer pair specific for P. nicotianae, Nic-F1/Nic-R1 reported by Li et al. (2011). The primers were tested at 0.05, 0.1, 0.25, 0.5, 1.0 μM to determine the optimal concentration.
To evaluate the sensitivity of the duplex and triplex PCRs, we used mixtures of plant DNA extracted from aseptically cultivated plants (tomato and cucumber) and mycelial DNA of P. nicotianae (CBS 305.29), P. capsici (P1319) or P. melonis (P6870), which was serially diluted (10 pg–1 fg per reaction). Tomato and cucumber seeds were soaked in 10% v/v H2O2 solution for 20 min and then washed three times with sterile distilled water. The sterilized seeds were sown in 75 × 75 × 10 mm plant boxes on Murashige-Skoog agar (Fujifilm Wako, Osaka, Japan; 0.8% agar). The plants were grown at 25 °C for 2 weeks in a growth chamber. For aseptically grown plants, DNA was extracted from 5-mm lengths of the hypocotyl for tomato and 5 × 5 mm piece of the main leaf for cucumber as described above.
The DNA detectability of the duplex or triplex PCR was tested using DNA extracted as described above from inoculated and naturally infected plants (Supplementary Table S1).
PCR products were separated by electrophoresis in 2.5% Agarose S (Fujifilm Wako, Osaka, Japan). Gels were stained with Gel Red (10,000 × , Biotium, Fremont, CA, USA) and photographed under ultraviolet light. All experiments were done at least twice.
Results
Specificity and sensitivity of Phytophthora genus-specific primers
The modified primers were tested with more genera, species, and strains to examine the reliability of the detection of the genus Phytophthora. The simplex PCR assay enabled the amplification of mycelial DNA of all 222 Phytophthora isolates among 155 taxa (Table 1). The amplification efficiency varied among species, and P. aquimorbida was the most difficult to detect (detectable in three of five replicate reactions using at least two DNA extractions from the same isolate).
None of the DNA was amplified from the 97 non-target isolates, representing 55 species of Phytopythium, Pythium, Globisporangium, Elongisporangium, and one isolate each of seven species of soil-borne pathogens (Table 1). However, DNA from some closely related species was nonspecifically amplified including Pythium adhaerens and Py. plurisporium, one of three isolates of Py. periilum, and six of seven isolates of Globisporangium heterothallicum, Phytopythium cucurbitacearum, two of nine isolates of Pp. vexans and one of three isolates of Pp. mercuriale (Table 1).
The detection limit of the modified primer pair using mycelial DNA of P. ramorum, P. kernoviae, and P. nicotianae was determined to be 100 fg (Supplementary Fig. S1).
Application of simplex PCR using diseased plants
The genus-specific simplex PCR assay using the modified primer pair was tested in symptomatic plants (ivy, tomato, hydrangea, rhododendron, Japanese andromeda, and camellia) that had been inoculated with or naturally infected (periwinkle and tobacco) with various Phytophthora species. Phytophthora was detected in all inoculated and naturally infected plants (Supplementary Table S1), and no amplicons were obtained using healthy plant tissues.
Development of duplex PCR assay
As an internal control to determine the success or failure of DNA extraction and PCR detection, we developed a duplex PCR to combine our genus-specific primers with the plant primers reported by Martin et al. (2004). The plant primers amplified DNA from all 41 tested plant species of 33 families belonging to 21 orders (Table 2). In the tests for optimal concentrations of magnesium and each primer, in the assay, the higher the magnesium concentration, the higher the activity of the polymerase and the higher the amplification efficiency, but excessive polymerase activity resulted in a loss of specificity and non-target amplification. When we tested several combinations of magnesium concentrations (1.5, 2.0, 2.5 mM) and plant primer concentrations (0.05, 0.0625, 0.125, 0.025, 0.5 µM) using the same concentrations of genus-specific primers as in the simplex PCR reaction solution, DNA extracted from several diseased plant species were amplified stably using 2.5 mM MgCl2 and 0.05 µM of each plant primer (1/10 the concentration of the genus-specific primers) (data not shown). The detection limit of the duplex PCR with the mixture of DNA from aseptically grown plants and from mycelia was 10 to 100 fg for plants and 1 pg for Phytophthora spp. (Supplementary Fig. S2).
Development of the triplex PCR assay
Because P. nicotianae is already present in Japan and thus exempt from quarantine inspections, we needed a triplex PCR assay to simultaneously detect Phytophthora species and P. nicotianae in plant samples. In the tests of concentrations for the specific primers for P. nicotianae in the triplex PCR, the DNA for the three targets was amplified almost equally by using P. nicotianae specific primers at 1/10 the concentration of the genus-specific primers, as found for the plant primers. The detection limit of this triplex PCR using the mixture of DNA from aseptically grown plants and mycelia, was 1 pg for Phytophthora spp. and 100 fg for plants and P. nicotianae (Supplementary Fig. S3).
Detection of Phytophthora species in diseased plants using duplex and triplex PCR
The results of the PCRs using plants that were inoculated (eggplant, tomato, and pumpkin) or naturally infected (periwinkle and tobacco) are shown in Supplementary Table S1, with representative agarose gels of the amplicons in Figs. 1 and 2. The Phytophthora species were detected in all the diseased plant samples, and amplification of the plant DNA (cox1) was confirmed in all samples. These results indicate that DNA was extracted using the Kaneka Easy DNA Extraction Kit version 2 from diseased samples, and the presence or absence of Phytophthora was accurately determined. Triplex PCR results showed that the P. nicotianae-specific primers detected P. nicotianae from eggplant and tomato plants that were inoculated with the pathogen and from naturally infected periwinkle and tobacco; no amplicons were obtained from pumpkins inoculated with P. capsici. Thus, the duplex and triplex PCR tests confirmed the success of DNA extraction and PCR detection and determined whether the cause was, in fact, Phytophthora, and if the pathogen was determined to be in the triplex PCR, whether or not it was P. nicotianae, using only one tube.

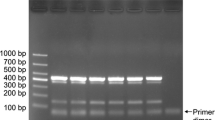
Duplex PCR detection of Phytophthora spp. from various plant species inoculated with different pathogens. Plant–pathogen combinations: Eggplant 1 and tomato, P. nicotianae GK10Eg1; Eggplant 2, P. nicotianae 13Asp1-1; Pumpkin 1, cucumber 1 and 2, P. capsici CH01CUCU10; Pumpkin 2, P. capsici CH02UE0202; ivy, P. citrophthora CH94HE11; Hydrangea 1 and 2, P. hedraiandra TGTA1-1; Cucumber 3 and 4, P. melonis CH00ME21-21. PC: positive control, mycelial DNA of P. nicotianae CBS 305.29, P. capsici P0253. P. citrophthora P3693, P. hedraiandra P11725 and P. melonis P6870. NC: negative control, sterile distilled water. Black arrowhead: amplicons using Phytophthora genus-specific primers (around 470 bp). White arrowhead: amplicons using plant primers (approximately 140 bp)

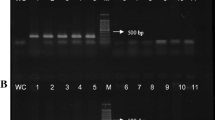
Triplex PCR detection of Phytophthora spp. and P. nicotianae using DNA from symptomatic plants that were inoculated or naturally infected. a, Inoculated plants. Eggplant 1 and tomato were inoculatsed with P. nicotianae GK10Eg1. Eggplant 2 was inoculated with 13Asp1-1. Pumpkin 1 and 2 were inoculated with P. capsici CH01CUCU10 and CH02UE0202, respectively. b, Naturally infected plants. PC, positive controls using mycelial DNA of P. nicotianae CBS 305.29 (PC 1) or P. capsici P0253 (PC 2). NC, negative control using sterile distilled water. Black arrowhead: amplification with Phytophthora genus-specific primers (around 470 bp). Gray arrowhead amplicons using P. nicotianae-specific primers (267 bp). White arrowhead: amplicons using plant primers (approximately 140 bp)
Discussion
Here we tested our previously designed primer pair to detect Phytophthora genus-specific, which were based on the primers of Schena et al. (2008) and Bi et al. (2019), using mycelial DNA from 155 taxa (including subspecies, varieties and hybrids), representing members from all Phytophthora clades (1–10; Abad et al. 2022). All the tested taxa were detected (Table 1). When non-targets of the primer pair were tested, among 97 isolates representing 55 species of closely related genera, DNA was amplified from only three species and from a small portion of the isolates tested for four species. Thus, our improved primers detected all species of Phytophthora tested, although a few closely related species of Pythium, Globisporangium and Phytopythium yielded false positives (Table 1). Many more taxa were tested than in previous reports of genus-specific detection: 101 species (Bilodeau et al. 2014), 45 species (Scibetta et al. 2012), 35 species (Schena et al. 2008), and 136 taxa (Miles et al. 2015). Therefore, our primers will benefit quarantine efforts, where strict border control is required and detection without omission is of utmost importance.
The detection limit of the simplex PCR using mycelial DNA of P. ramorum, P. kernoviae, and P. nicotianae was 100 fg for all three species (Supplementary Fig. S1), the same detection limit as that of the nested PCR of Schena et al. (2008). We were able to achieve the same level of sensitivity as that of Schen et al. (2008) in one round of amplification. In a comparison of the electrophoretic results for the 155 taxa in the specificity test, the intensity of the amplicon band varied among species, even though 100 pg of DNA was used in all cases (data not shown). Amplification failed only for a few PCR replicate tests for P. aquimorbida (2 of 5 tests) and P. macilentosa (1 of 4 tests), both minor pathogens. Thus, the detection results for plant samples that are potentially infected with those two species should be evaluated carefully. However, for other species, positive results were obtained in all repeated tests.
For the duplex and triplex PCR for diseased plants, the composition of the reaction solution, including the concentrations of primers and magnesium, were optimized so that differences in amplification between primers due to competition between polymerase and substrates could be reduced. The detection limits for Phytophthora DNA in the multiplexed PCRs using mixtures containing plant DNA were 10 times higher than those of simplex PCR using only mycelial DNA (Supplementary Figs. S1–S3). Knowing that detection sensitivity is better with higher quality DNA, we modified the extraction method of Kageyama et al. (2003) to obtain higher quality DNA and increased the sensitivity to 100 fg (data not shown). For the present study, considering the huge number of samples tested during quarantine inspections, we selected the Kaneka Easy DNA Extraction Kit version 2 method for its simple, rapid extraction of DNA (Hieno et al. 2019). Thus, users should choose DNA extraction and detection methods (simplex PCR or multiplex PCR) that are best for their situation and objectives.
Our newly designed triplex PCR was able to simultaneously determine the presence of a Phytophthora species and P. nicotianae, the only Phytophthora species not subject to quarantine in Japan, in a one-tube reaction. In addition, Ypt1 regions have accumulated sufficient mutations to be highly discriminative of species so that the genus-specific primer can be used sequencing the amplicons to identify the species. In addition, the simplex PCR amplicons from DNA extracted from diseased plants (naturally infected tobacco sample no. 1 with P. nicotianae and rhododendron inoculated with P. kernoviae or P. lateralis; Supplementary Table S1) were subjected to a sequencing analysis, and these species were identified (data not shown). Although we previously developed a LAMP assay using a QProbe to simultaneously detect Phytophthora spp. and P. nicotianae (Hieno et al. 2020) that is a rapid, accurate highly applicable method for import/export quarantine inspections, it cannot be used to identify the actual species of Phytophthora, except for P. nicotianae. By comparing the advantages of each detection method, it is possible to select the method that best meets the conditions required by the user.
Molecular detection methods can also be used for practical inspection of nonsymptomatic plants. Fichtner et al. (2012) pointed out that symptomless infections by P. kernoviae in North American native plants may thwart pathogen detection and underscore the importance of implementing a proactive and adaptive biosecurity plan. Harris and Webber (2016) also pointed out that symptomless infections of larch by P. ramorum can lead to an underestimation of infection plants. We are confident that our optimized detection method can contribute to more effective quarantine control.
References
Abad ZG, Burgess T, Bienapfl JC, Redford AJ, Coffey M, Knight L (2022) IDphy: molecular and morphological identification of Phytophthora based on the types. USDA APHIS PPQ S&T Beltsville Lab, USDA APHIS PPQ S&T ITP, Centre for Phytophthora Science and Management, and World Phytophthora Collection. https://idtools.org/id/phytophthora
André LéVesque C, De Cock AWAM (2004) Molecular phylogeny and taxonomy of the genus Pythium. Mycol Res 108:1363–1383
Bi X, Hieno A, Otsubo K, Kageyama K, Liu G, Li M (2019) A multiplex PCR assay for three pathogenic Phytophthora species related to kiwifruit diseases in China. J Gen Plant Pathol 85:12–22
Bilodeau GJ, Martin FN, Coffey MD, Blomquist CL (2014) Development of a multiplex assay for genus- and species-specific detection of Phytophthora based on differences in mitochondrial gene order. Phytopathology 104:733–748
Brasier CM (2008) The biosecurity threat to the UK and global environment from international trade in plants. Plant Pathol 57:792–808
Brasier C (2009) Phytophthora biodiversity: How many Phytophthora species are there? In: Proceedings of the 4th meeting of the international union of forest research organizations (JUFO) working party s07.02.09: Phythophthoras in forests and natural ecosystems. Goheen EM, Frankel SJ (tech. coords.) Gen Tech Rep PSW-GTR-221, U.S. Department Agriculture Forest Service, Pacific Southwest Research Station, Albany, CA, USA pp 101–105
Fichtner EJ, Rizzo DM, Kirk SA, Webber JF (2012) Infectivity and sporulation potential of Phytophthora kernoviae to select North American native plants. Plant Pathol 61:224–233
Harris AR, Webber JF (2016) Sporulation potential, symptom expression and detection of Phytophthora ramorum on larch needles and other foliar hosts. Plant Pathol 65:1441–1451
Hieno A, Li M, Afandi A, Otsubo K, Suga H, Kageyama K (2019) Rapid detection of Phytophthora nicotianae by simple DNA extraction and real-time loop-mediated isothermal amplification assay. J Phytopathol 167:174–184
Hieno A, Li M, Afandi A, Otsubo K, Suga H, Kageyama K (2020) Detection of the genus Phytophthora and the species Phytophthora nicotianae by LAMP with a QProbe. Plant Dis 104:2469–2480
Hieno A, Li M, Otsubo K, Suga H, Kageyama K (2021) Multiplex LAMP detection of the genus Phytophthora and four Phytophthora species P. ramorum, P. lateralis, P. kernoviae, and P. nicotianae, with a plant internal control. Microbes Environ 36:ME21019
Kageyama K, Komatsu T, Suga H (2003) Refined PCR protocol for detection of plant pathogens in soil. J Gen Plant Pathol 69:153–160
Li M, Asano T, Suga H, Kageyama K (2011) A multiplex PCR for the detection of Phytophthora nicotianae and P. cactorum, and a survey of their occurrence in strawberry production areas of Japan. Plant Dis 95:1270–1278
Martin FN, Tooley PW, Blomquist C (2004) Molecular detection of Phytophthora ramorum, the causal agent of sudden oak death in California, and two additional species commonly recovered from diseased plant material. Phytopathology 94:621–631
Miles TD, Martin FN, Coffey MD (2015) Development of rapid isothermal amplification assays for detection of Phytophthora spp. in plant tissue. Phytopathology 105:265–278
Miller PM (1955) V-8 juice agar as a general-purpose medium for fungal and bacteria. Phytopathology 45:461–462
Sakoda T, Goto H, Kanno T, Hiyama T, Hirakawa T, Nakanishi Y, Hirata T (2017) Ramorum blight of Rhododendron sp. caused by Phytophthora ramorum intercepted in plant quarantine inspection in Japan. Res Bull Plant Prot Serv Jpn 53:75–81
Schena L, Cooke DEL (2006) Assessing the potential of regions of the nuclear and mitochondrial genome to develop a “molecular tool box” for the detection and characterization of Phytophthora species. J Microbiol Methods 67:70–85
Schena L, Duncan JM, Cooke DEL (2008) Development and application of a PCR-based “molecular tool box” for the identification of Phytophthora species damaging forests and natural ecosystems. Plant Pathol 57:64–75
Scibetta S, Schena L, Chimento A, Cacciola SO, Cooke DEL (2012) A molecular method to assess Phytophthora diversity in environmental samples. J Microbiol Methods 88:356–368
Uzuhashi S, Tojo M, Kakishima M (2010) Phylogeny of the genus Pythium and description of new genera. Mycoscience 51:337–365
Yang X, Hong C (2018) Differential usefulness of nine commonly used genetic markers for identifying Phytophthora species. Front Microbiol 9:2334
Acknowledgements
We thank Emeritus Prof. E. M. Hansen, Prof. M. D. Coffey, Dr. P. W. Tooley, Prof. C. Hong, Dr. B. S. Weir, Prof. T. I. Burgess, Dr. T. Jung, Mr. S. Uematsu, and Dr. H. Watanabe for providing many important isolates and Dr. S. Subandiyah and Dr. A. Wibowo for providing plant DNA samples. We also thank Mr. T. Sakoda and Mr. K. Ueda for technical assistance.
Funding
Open Access funding provided by Gifu University. The present study was supported by a Grant-in-Aid for “Development of detection and identification techniques of pests” under “Research and development of adaptation measures for global warming and abnormal weather” from the Ministry of Agriculture, Forestry, and Fisheries of Japan.
Author information
Authors and Affiliations
Contributions
KO, HS, KK and AH contributed to conceiving and designing the study. KO, ML and AA prepared materials and performed the experiments. KO and KK analyzed the data. KO, KK and AH wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Human and animal rights
This article does not contain any studies involving human participants or experimental animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file 1. Supplementary Table S1
: Detection of Phytophthora spp. from naturally infected or inoculated plants using simplex, duplex and triplex PCR with plant-, Phytophthora- and P. nicotianae-specific primers. (XLSX 13 kb)
Supplementary file 2. Supplementary Fig. S1
: Detection limits of simplex PCR for P. ramorum, P. kernoviae and P. nicotianae using Phytophthora genus-specific primers. (XLSX 77 kb)
Supplementary file 3. Supplementary Fig. S2
: Detection limits of duplex PCR using Phytophthora-specific primers and plant primers. (XLSX 208 kb)
Supplementary file 4. Supplementary Fig. S3
: Detection limits of triplex PCR using Phytophthora-specific primers, P. nicotianae-specific primers, and plant-specific primers. (XLSX 55 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Otsubo, K., Li, M., Afandi, A. et al. Multiplex PCR specific for genus Phytophthora and P. nicotianae with an internal plant DNA control for effective quarantine of Phytophthora species in Japan. J Gen Plant Pathol 90, 201–216 (2024). https://doi.org/10.1007/s10327-024-01179-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10327-024-01179-z