
Abstract
Current guidelines advocate to treat refractory status epilepticus (RSE) with continuously administered anesthetics to induce an artificial coma if first- and second-line antiseizure drugs have failed to stop seizure activity. A common surrogate for monitoring the depth of the artificial coma is the appearance of a burst-suppression pattern (BS) in the EEG. This review summarizes the current knowledge on the origin and neurophysiology of the BS phenomenon as well as the evidence from the literature for the presumed benefit of BS as therapy in adult patients with RSE.
Zusammenfassung
In aktuellen Leitlinien wird empfohlen, einen refraktären Status epilepticus (RSE) mit kontinuierlich verabreichten Anästhetika zu behandeln, um ein künstliches Koma herbeizuführen, sofern durch Erst- und Zweitlinien-Antiepileptika die Anfallsaktivität nicht gestoppt werden konnte. Ein gängiges Surrogat zur Überwachung der Tiefe des künstlichen Komas ist das Auftreten eines Burst-Suppressions-Musters (BS) im Elektroenzephalogramm (EEG). In der vorliegenden Übersichtsarbeit werden sowohl der aktuelle Wissensstand über den Ursprung und die Neurophysiologie des BS-Phänomens als auch die Belege aus der Literatur für den vermuteten Nutzen der BS als Therapie bei erwachsenen Patienten mit RSE zusammengefasst..
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Status epilepticus (SE) bears a high risk of significant morbidity and mortality [1]. With an annual frequency of 10–40 per 100,000 it represents the second most frequent neurological emergency following stroke [1, 2].
Status epilepticus becomes refractory (RSE) if it persists after the sufficient administration of benzodiazepines as first-line and one or more second-line antiseizure drugs (ASDs). Current guidelines recommend aggressive treatment for RSE with convulsions or impaired consciousness using continuously administered anesthetic drugs to induce a deep coma with the aim of terminating RSE [2,3,4]. In addition, a recent two-center study revealed that early administration of anesthetics immediately after first-line treatment may bring more benefits than risks in specific subgroups [5].
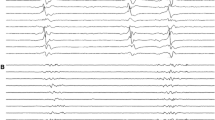
The most frequently used anesthetics in this context are midazolam, propofol, pentobarbital, and thiopental. Less frequently used substances are the halogenated ether isoflurane and ketamine [2,3,4, 6, 7]. The main target of these agents, except ketamine, is the facilitation of gamma-aminobutyric acid type A (GABAA) receptor effect and each substance may induce a generalized slowing, burst-suppression (BS) and eventually generalized suppression in the surface electroencephalography (EEG) in a dose-dependent manner ([8, 9]; Fig. 1).
To treat RSE with anesthetics, experts suggested close monitoring of anesthetized patients with EEG, either aiming for the cessation of electrographic seizures, the emergence of a BS pattern, or isoelectricity-specific EEG patterns that can be established with increasing doses [10,11,12,13,14]. Monitoring with EEG also helps to avoid a too-deep anesthesia, especially in the context of barbiturate coma. Consequently, national and international guidelines adopted this approach for maintaining such artificial coma for at least 24 h, followed by gradual withdrawal, concurrently acknowledging the lack of evidence for the ideal titration goal [3, 4, 15]. Based on similar weak evidence regarding BS, experts consider achieving an inter-burst interval of about 10 s for 24 h to be reasonable [2]. An international survey from 2003 among epileptologists and critical care neurologists in Austria, Germany, and Switzerland revealed that two thirds of physicians considered a BS pattern as the suitable titration target for proper treatment of RSE [16]. However, larger prospective studies concerning the achievement of such a goal and its effect on outcome are lacking. Further, concerns regarding the adverse effects and complications that can accompany prolonged and deep anesthesia, including prolonged postictal mechanical ventilation, infections, and severe arterial hypotension, propofol infusion syndrome, and cardiotoxicity or paralytic ileus from barbiturates raise doubts about this therapeutic step [7, 17,18,19,20,21,22].
This narrative review is based on a literature search of PubMed and in the references lists of selected studies. It summarizes the current knowledge on the origin and neurophysiology of the EEG BS phenomenon and aims to compile and revise the evidence from the literature for the benefit of BS as therapy in adult patients with RSE.
Origin and neurophysiology of the burst-suppression phenomenon
The BS phenomenon describes an EEG pattern consisting of alternating epochs of high-voltage broad-spectrum oscillations (bursts) and electrical suppression. According to the current American Clinical Neurophysiology Society’s (ACNS) Standardized Critical Care EEG Terminology, bursts must average ≥ 0.5 s and have at least four phases and may last up to 30 s, whereas suppression is either < 10 μV or ≥ 10 μV and < 50% of the higher voltage background activity (while the latter is formally an attenuation, the principle of the BS pattern remains). Finally, the proportion of suppression must comprise of 50–99% of the recording [23].

Different stages of coma in a patient with refractory status epilepticus under continuous anesthetic treatment. A superficial stage of artificial coma displays a diffuse generalized slowing and absence of the posterior dominant rhythm (a). An increase in anesthetic dose leads to a typical burst-suppression pattern. In this 15‑s epoch, the cumulative duration of burst (dashed lines) is approximately 4.5 s (b). A further increase in anesthetic dose results in an isoelectric surface EEG pattern (c). All EEGs are shown with an anterior–posterior bipolar montage with a low-pass filter of 0.5 Hz and a high-pass filter of 70 Hz; scale bar: horizontal 1 s, vertical 100 µV
The BS phenomenon was first described in 1936 in the cat cortex [24], whereas the term itself was first introduced in 1949 by Swank and Watson [25].
In humans, BS is physiologically present during sleep in preterm babies and neonates in form of the so-called tracé discontinu and tracé alternant and might most likely represent immature brain structures, unstable neuronal circuits, and hypofunctional neurotransmitter receptors and ion channels [26]. Beyond the perinatal period, BS is mainly encountered in several pathological conditions, such as infantile epilepsy syndromes including Ohtahara syndrome, early myoclonic encephalopathy, and Aicardi syndrome [27, 28], as well as comatose states due to various pathological states, including space-occupying lesions, toxic or metabolic causes, infection, trauma, stroke, or hypoxic–ischemic brain injury [8, 9, 29, 30]. Furthermore, iatrogenic hypothermia [31] and deep general anesthesia may also elicit a BS pattern [32].
Surface EEG suggested a synchronous onset of BS [33]. Further, the EEG appearance of BS in functionally or anatomically disconnected cortex supported deafferented cortical neurons as its source [34,35,36]. However, both observations were recently challenged. Intracranial EEG recordings from patients under propofol anesthesia as well as calcium imaging in rat cortices revealed a substantial asynchrony of cortical bursts that are spatially inhomogeneously distributed [37, 38]. In addition, animal experiments suggested a subcortical source for BS modulation by observing persisting hippocampal neuronal oscillations in the presence of cortical isoelectric EEG and identifying thalamic activity as a modulator of the cortical suppression-to-burst transition [38, 39].
Currently, there are mainly two hypotheses discussed regarding the neurophysiological mechanisms underlying BS. The first is the cortical hypersensitivity hypothesis that is based on observations from isoflurane-anesthetized cats in BS, where mechanical stimuli triggered bursts, followed by a stimulus-refractory suppression caused by depletion of extracellular calcium levels [40]. Further studies suggested concurrent suppression of inhibitory cortical signals [41]; thus, hypersensitivity during BS may be the result of reduced inhibition rather than increased cortical excitation. The second proposition is the metabolic hypothesis that explains the occurrence of suppression by relative intracellular depletion of adenosine triphosphate (ATP), which enhances opening of ATP-regulated potassium channels until intracellular ATP levels are restored and bursts may re-emerge, which in turn rapidly deplete ATP [29].
Evidence for burst-suppression to treat refractory status epilepticus
The most prominent challenge in treating SE is the time-dependent development of pharmacoresistance against ASDs and anesthetics [42]. In vitro and in vivo animal experiments indicate that ongoing neuronal discharges result in an internalization of the postsynaptic GABAA receptors as early as 1 h after the onset of epileptic seizures [43,44,45] as well as alterations in GABAA receptor-associated scaffold proteins [46]. More recently, α‑amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor plasticity [47] and intracellular accumulation of chloride ions [48] were suggested to contribute to SE perpetuation. Consequently, early seizure control appears to be key in preventing these molecular modifications. It is conceivable that once these molecular modifications have already taken place, a supersaturated therapy with continuously administered anesthetics may be needed to break the self-sustaining vicious cycle of RSE.
To reach a BS pattern is a common EEG surrogate for adequate seizure-terminating anesthetic dosage [2,3,4]. However, this approach and the known risks of adverse events and clinical complications that may emerge during such prolonged and deep artificial coma raise further questions, such as whether the achievement of BS is truly beneficial or whether seizure suppression following a more superficial artificial coma might be equivocal, and whether the morphology of BS may contain relevant information about the success of the treatment per se.
Depths of artificial coma until burst-suppression
The first study that specifically analyzed the depth of artificial coma on outcome in adult patients with RSE included 35 patients on continuously administered barbiturates [49]. The proportion of surviving patients varied among subgroups showing different EEG patterns. While among 20 patients with an isoelectric EEG curve 12 survived, three of 12 with a BS pattern and three of three patients with seizure suppression only survived. From these results, the authors concluded that a deeper suppression was associated with better outcome [49]. However, multivariable models adjusting for other important confounders regarding mortality were not performed. This study was followed by a systematic review of 28 studies between 1980 and 2001 with a total of 193 adult patients with RSE treated by midazolam, propofol, or pentobarbital [6]. Compared to seizure suppression, patients treated with the aim of generalized EEG background suppression (i.e., BS or isoelectric curve) had a lower frequency of breakthrough epileptic seizures but also a higher need for continuously administered vasopressor treatment due to severe arterial hypotension. These findings are limited by the fact that most patients with the goal of EEG background suppression were treated with pentobarbital but none with midazolam. However, mortality was not associated with any specified titration goals. A retrospective analysis including 49 episodes with RSE in adults could not demonstrate a benefit in outcome by achievement of BS [50]. As a limitation, the presence of BS was retrospectively identified from EEG reports and, thus, the duration of BS was not quantified and multivariable models adjusting for potential confounders in this context were lacking. Another retrospective study of 53 patients with RSE not following hypoxic–ischemic encephalopathy with almost 90% qualifying as super-RSE and a high mortality of nearly 1 in 3 found worse outcome correlated with BS or isoelectric EEG. On the other hand, seizure suppression alone was significantly associated with good outcome [51]. By contrast, further retrospective RSE studies excluding hypoxic–ischemic encephalopathy could not reproduce any association between BS and poor outcome [52, 53].
How long a BS pattern should be maintained remains unknown. While guidelines advocate to keep a BS pattern for at least 24 h [3, 4, 15], a shorter duration might be equally sufficient in selected cases [54].
Unfortunately, only few studies regarding RSE treatment have quantified BS in any way. A recent investigation computationally calculated the burst suppression ratio (QBRS) in EEG recordings of 17 patients with RSE. On adjusted multivariable analysis, there was no association between QBSR and outcome, thus depth or duration of BS ratio were not associated with outcome [55].
Morphology of bursts in burst-suppression
Overall, data on burst morphology and outcome on RSE are scarce. The ACNS Terminology of Critical Care EEG defined “highly epileptiform bursts” if two or more epileptiform discharges are seen within most (> 50%) bursts and occur at an average of ≥ 1 Hz within a single burst, or if a rhythmic, potentially ictal-appearing pattern occurs within > 50% bursts [23]. A retrospective single-center study identified a BS pattern with “identical bursts,” characterized by identical first 0.5 s of each burst, in 20% of adult patients with diffuse hypoxic–ischemic encephalopathy, all with a fatal course. Such identical bursts were absent in patients who had an artificial BS pattern due to isoflurane or propofol anesthesia, although the indication for the anesthesia was not further specified [56]. In addition, it remains unclear whether nonsurvivors died of uncontrolled RSE or from an underlying potentially fatal etiology. A study of 19 adult patients with RSE not caused by hypoxic–ischemic encephalopathy studied whether burst characteristics within the 12 h of continuous EEG monitoring prior to a weaning attempt could predict successful weaning. Predictors for successful weaning were bursts with absence of monomorphic sharp waves within the bursts, amplitudes of < 125 μV, and containing < 50% epileptiform activity. However, the results did not indicate any benefit of inter-bursts intervals of ≥ 10 s or any role of the lengths of bursts [57]. Another study with 24 adult patients with a BS not caused by hypoxic–ischemic encephalopathy showed that highly epileptiform discharges within the bursts were associated with subsequent emergence of epileptic seizures [58]. A novel approach was recently demonstrated by the analysis of functional connectivity measures that revealed successful anesthetic weaning in association with larger, more densely connected, and more highly clustered spatial functional networks [59].
The struggle to maintain a burst-suppression pattern
As a general limitation, the methods to assess BS in all the aforementioned studies were heterogeneous, limiting the generalizability of the discussed findings and urgently calling for studies applying more systematic and standardized assessments.
There is a discrepancy between the common recommendation to achieve a BS with inter-burst suppression of about 10 s [2], and the daily struggles mirrored by the fact that such BS in retrospective studies was only achieved in approximately 40–60% of cases but the overall duration of BS remained unspecified [6, 50]. A study that quantitatively assessed BS in 35 adult RSE patients revealed a remarkable inter-patient and intra-patient variability of suppression proportions despite an uninterrupted and constant administration rate of intravenous anesthetics [60].
Our own preliminary retrospective EEG analysis of 147 patients with RSE treated with anesthetics between 2011 and 2019 in a Swiss tertiary medical care center underscores this issue by revealing a low number of achieved EEG BS pattern (Table 1) despite having continuous EEG monitoring units and the same consulting neurologists and epileptologists during the entire study period. As a limitation to this overview, the titration target might not have been specified as a BS in all 147 patients (preliminary results).
Conclusion
In summary, the limited data and small evidence from the literature suggests that EEG burst-suppression (BS) might be a useful surrogate for the titration of anesthetics to achieve deep artificial coma in patients suffering from refractory status epilepticus (RSE).
Despite the importance of optimal RSE treatment in clinical practice, prospective clinical trials in this context are lacking and the evidence for EEG BS pattern as a surrogate for sufficient anesthesia and persistent seizure suppression are anecdotal case reports or retrospective studies describing heterogeneous patient populations and a variety of different methods to quantify and qualify the EEG BS. The difficulty to maintain BS in daily practice, even in tertiary care centers, is in contrast with the current guidelines and represent a further limitation of the evidence supporting BS for the treatment of RSE.
Aside from several early studies raising concerns regarding the risks and complications associated with continuously administered anesthetics to induce deep coma, recent studies provide limited evidence that such artificial coma may probably not markedly increase the overall complication rate especially in RSE patients with underlying potentially life-threatening conditions. By contrast, limited data further suggest that patients with rather benign etiologies of SE are at risk of adverse events and complications that may alter their outcome. Thus, until better data are available, clinicians are urged to tailor antiseizure treatment to the different patients’ conditions and to carefully balance the potential risks against the benefits of artificial coma with EEG BS when managing patients in RSE.
References
Sutter R, Kaplan PW, Rüegg S (2013) Outcome predictors for status epilepticus—what really counts. Nat Rev Neurol 9:525–534
Rossetti AO, Lowenstein DH (2011) Management of refractory status epilepticus in adults: still more questions than answers. Lancet Neurol 10:922–930
Brophy GM, Bell R, Claassen J, Alldredge B, Bleck TP, Glauser T et al (2012) Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 17:3–23
Meierkord H, Boon P, Engelsen B, Gocke K, Shorvon S, Tinuper P et al (2010) EFNS guideline on the management of status epilepticus in adults. Eur J Neurol 17:348–355
De Stefano P, Baumann SM, Semmlack S, Rüegg S, Marsch S, Seeck M et al (2021) Safety and efficacy of coma induction following first-line treatment in status epilepticus: a 2‑center study. Neurology 97:e564–e576
Claassen J, Hirsch LJ, Emerson RG, Mayer SA (2002) Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia 43:146–153
Sutter R, Marsch S, Fuhr P, Kaplan PW, Rüegg S (2014) Anesthetic drugs in status epilepticus—risk or rescue? A six-year cohort study. Neurology 82:656–664
Shanker A, Abel JH, Schamberg G, Brown EN (2021) Etiology of burst suppression EEG patterns. Front Psychol 12:673529
Sutter R, Kaplan PW (2012) Electroencephalographic criteria for nonconvulsive status epilepticus: synopsis and comprehensive survey. Epilepsia 53(Suppl 3):1–51
Kaplan PW (2003) Nonconvulsive status epilepticus. Neurology 61:1035–1036
Lowenstein DH, Aminoff MJ, Simon RP (1988) Barbiturate anesthesia in the treatment of status epilepticus: clinical experience with 14 patients. Neurology 38:395–400
Rashkin MC, Youngs C, Penovich P (1987) Pentobarbital treatment of refractory status epilepticus. Neurology 37:500–503
Van Ness PC (1990) Pentobarbital and EEG burst suppression in treatment of status epilepticus refractory to benzodiazepines and phenytoin. Epilepsia 31:61–67
Osorio I, Reed RC (1989) Treatment of refractory generalized tonic-clonic status epilepticus with pentobarbital anesthesia after high-dose phenytoin. Epilepsia 30:464–471
Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J et al (2016) Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American epilepsy society. Epilepsy Curr 16:48–61
Holtkamp M, Masuhr F, Harms L, Einhäupl KM, Meierkord H, Buchheim K (2003) The management of refractory generalised convulsive and complex partial status epilepticus in three European countries: a survey among epileptologists and critical care neurologists. J Neurol Neurosurg Psychiatry 74:1095–1099
Sutter R, De Marchis GM, Semmlack S, Fuhr P, Rüegg S, Marsch S et al (2017) Anesthetics and outcome in status epilepticus: a matched two-center cohort study. CNS Drugs 31:65–74
Kowalski RG, Ziai WC, Rees RN, Werner JK, Kim G, Goodwin H et al (2012) Third-line antiepileptic therapy and outcome in status epilepticus: the impact of vasopressor use and prolonged mechanical ventilation. Crit Care Med 40:2677–2684
Marchi NA, Novy J, Faouzi M, Stähli C, Burnand B, Rossetti AO (2015) Status epilepticus: impact of therapeutic coma on outcome. Crit Care Med 43:1003–1009
Opić P, Sutter R (2020) The unease when using anesthetics for treatment-refractory status epilepticus: still far too many questions. J Clin Neurophysiol 37:399–405
Baumann SM, Semmlack S, Rybitschka A (2021) Prolonged mechanical ventilation in patients with terminated status epilepticus and outcome: an observational cohort study. Epilepsia 62:3042–3057
Newey CR, Wisco D, Nattanmai P, Sarwal A (2016) Observed medical and surgical complications of prolonged barbiturate coma for refractory status epilepticus. Ther Adv Drug Saf 7:195–203
Hirsch LJ, Fong MWK, Leitinger M, LaRoche SM, Beniczky S, Abend NS et al (2021) American clinical neurophysiology society’s standardized critical care EEG terminology: 2021 version. J Clin Neurophysiol 38:1–29
Derbyshire AJ, Rempel B, Forbes A, Lambert EF (1936) The effects of anesthetics on action potentials in the cerebral cortex of the cat. Am J Physiol Leg Content 116:577–596
Swank RL, Watson CW (1949) Effects of barbiturates and ether on spontaneous electrical activity of dog brain. J Neurophysiol 12:137–160
Raurale SA, Boylan GB, Lightbody G, O’Toole JM (2020) Identifying tracé alternant activity in neonatal EEG using an inter-burst detection approach. In: 2020 42nd Annu Int Conf IEEE Eng Med Biol Soc EMBC. 2020, pp 5984–5987
Aicardi J (2005) Aicardi syndrome. Brain Dev 27:164–171
Ohtahara S, Yamatogi Y (2006) Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res 70(Suppl 1):S58–S67
Ching S, Purdon PL, Vijayan S, Kopell NJ, Brown EN (2012) A neurophysiological-metabolic model for burst suppression. Proc Natl Acad Sci U S A 109:3095–3100
Sutter R, Pang T, Kaplan PW (2017) EEG in metabolic disorders, intoxications, and epileptic encephalopathies [Internet]. Schomer DL, lopes da silva FH, editors. Oxford university press. http://www.oxfordmedicine.com/view/10.1093/med/9780190228484.001.0001/med-9780190228484-chapter-17. Accessed 1 May 2022
Westover BM, Ching S, Kumaraswamy VM, Akeju O, Pierce E, Cash SS et al (2015) The human burst suppression electroencephalogram of deep hypothermia. Clin Neurophysiol 126:1901–1914
Brown EN, Lydic R, Schiff ND (2010) General anesthesia, sleep, and coma. N Engl J Med 363:2638–2650
Akrawi WP, Drummond JC, Kalkman CJ, Patel PM (1996) A comparison of the electrophysiologic characteristics of EEG burst-suppression as produced by isoflurane, thiopental, etomidate, and propofol. J Neurosurg Anesthesiol 8:40–46
Cobb W, Hill D (1950) Electroencephalogram in subacute progressive encephalitis. Brain J Neurol 73:392–404
Echlin FA, Arnett V, Zoll J, Peck D (1952) Paroxysmal high voltage discharges from isolated and partially isolated human and animal cerebral cortex. Trans Am Neurol Assoc 56:54–57
Henry CE, Scoville WB (1952) Suppression-burst activity from isolated cerebral cortex in man. Electroencephalogr Clin Neurophysiol 4:1–22
Lewis LD, Ching S, Weiner VS, Peterfreund RA, Eskandar EN, Cash SS et al (2013) Local cortical dynamics of burst suppression in the anaesthetized brain. Brain 136:2727–2737
Ming Q, Liou J‑Y, Yang F, Li J, Chu C, Zhou Q et al (2022) Isoflurane-induced burst suppression is a thalamus-modulated, focal-onset rhythm with persistent local asynchrony and variable propagation patterns in rats. Front Syst Neurosci. https://doi.org/10.3389/fnsys.2020.599781
Kroeger D, Florea B, Amzica F (2013) Human brain activity patterns beyond the Isoelectric line of extreme deep coma. PLoS ONE 8:e75257 (Dickson CT, editor)
Kroeger D, Amzica F (2007) Hypersensitivity of the anesthesia-induced comatose brain. J Neurosci 27:10597–10607
Ferron J‑F, Kroeger D, Chever O, Amzica F (2009) Cortical inhibition during burst suppression induced with isoflurane anesthesia. J Neurosci 29:9850–9860
Mazarati AM, Baldwin RA, Sankar R, Wasterlain CG (1998) Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res 814:179–185
Kapur J, Coulter DA (1995) Experimental status epilepticus alters gamma-aminobutyric acid type A receptor function in CA1 pyramidal neurons. Ann Neurol 38:893–900
Naylor DE, Liu H, Wasterlain CG (2005) Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci 25:7724–7733
Cho Y‑J, Kim H, Kim W‑J, Chung S, Kim Y‑H, Cho I et al (2017) Trafficking patterns of NMDA and GABAA receptors in a Mg2+-free cultured hippocampal neuron model of status epilepticus. Epilepsy Res 136:143–148
González MI, Del Angel CY, Brooks-Kayal A (2013) Down-regulation of gephyrin and GABAA receptor subunits during epileptogenesis in the CA1 region of hippocampus. Epilepsia 54:616–624
Adotevi N, Lewczuk E, Sun H, Joshi S, Dabrowska N, Shan S et al (2020) α‑amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor plasticity sustains severe, fatal status epilepticus. Ann Neurol 87:84–96
Burman RJ, Selfe JS, Lee JH, van den Berg M, Calin A, Codadu NK et al (2019) Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain J Neurol 142:3482–3501
Krishnamurthy KB, Drislane FW (1999) Depth of EEG suppression and outcome in barbiturate anesthetic treatment for refractory status epilepticus. Epilepsia 40:759–762
Rossetti AO, Logroscino G, Bromfield EB (2005) Refractory status epilepticus: effect of treatment aggressiveness on prognosis. Arch Neurol 62:1698
Hocker SE, Britton JW, Mandrekar JN, Wijdicks EFM, Rabinstein AA (2013) Predictors of outcome in refractory status epilepticus. JAMA Neurol 70:72
Kang BS, Jung K‑H, Shin J‑W, Moon JS, Byun J‑I, Lim J‑A et al (2015) Induction of burst suppression or coma using intravenous anesthetics in refractory status epilepticus. J Clin Neurosci 22:854–858
Phabphal K, Chisurajinda S, Somboon T, Unwongse K, Geater A (2018) Does burst-suppression achieve seizure control in refractory status epilepticus? BMC Neurol 18:46
Das AS, Lee JW, Izzy S, Vaitkevicius H (2019) Ultra-short burst suppression as a “reset switch” for refractory status epilepticus. Seizure 64:41–44
Peedicail J, Mehdiratta N, Zhu S, Nedjadrasul P, Ng MC (2021) Quantitative burst suppression on serial intermittent EEG in refractory status epilepticus. Clin Neurophysiol Pract 6:275–280
Hofmeijer J, Tjepkema-Cloostermans MC, van Putten MJAM (2014) Burst-suppression with identical bursts: a distinct EEG pattern with poor outcome in postanoxic coma. Clin Neurophysiol 125:947–954
Johnson EL, Martinez NC, Ritzl EK (2016) EEG characteristics of successful burst suppression for refractory status epilepticus. Neurocrit Care 25:407–414
Thompson SA, Hantus S (2016) Highly epileptiform bursts are associated with seizure recurrence. J Clin Neurophysiol 33:66–71
Rubin DB, Angelini B, Shoukat M, Chu CJ, Zafar SF, Westover MB et al (2020) Electrographic predictors of successful weaning from anaesthetics in refractory status epilepticus. Brain 143:1143–1157
An J, Jonnalagadda D, Moura V, Purdon PL, Brown EN, Westover MB (2018) Variability in pharmacologically-induced coma for treatment of refractory status epilepticus. PLoS ONE 13:e205789 (Hahn CD, editor)
Funding
Open access funding provided by University of Basel
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
U. Fisch, A.L. Jünger, L. Hert, S. Rüegg and R. Sutter declare that they have no competing interests.
The patients’ data presented are part of the STatus EPilepticus Unicenter Population (STEP UP) study (ClinicalTrials.gov ID: NCT04204863) and the REfractory Status Epilepticus Treatment: Quality and Efficacy of Coma Induction (RESET) study (NCT04333082) performed at University Hospital of Basel. Patients’ consent was waived after approval by the local ethics committee (Ethikkommission Nordwest- und Zentralschweiz).
Additional information

Scan QR code & read article online
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fisch, U., Jünger, A.L., Hert, L. et al. Therapeutically induced EEG burst-suppression pattern to treat refractory status epilepticus—what is the evidence?. Z. Epileptol. 35, 303–309 (2022). https://doi.org/10.1007/s10309-022-00539-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10309-022-00539-z