
Abstract
A major barrier to both metabolic engineering and fundamental biological studies is the lack of genetic tools in most microorganisms. One example is Clostridium thermocellum ATCC 27405T, where genetic tools are not available to help validate decades of hypotheses. A significant barrier to DNA transformation is restriction–modification systems, which defend against foreign DNA methylated differently than the host. To determine the active restriction–modification systems in this strain, we performed complete methylome analysis via single-molecule, real-time sequencing to detect 6-methyladenine and 4-methylcytosine and the rarely used whole-genome bisulfite sequencing to detect 5-methylcytosine. Multiple active systems were identified, and corresponding DNA methyltransferases were expressed from the Escherichia coli chromosome to mimic the C. thermocellum methylome. Plasmid methylation was experimentally validated and successfully electroporated into C. thermocellum ATCC 27405. This combined approach enabled genetic modification of the C. thermocellum-type strain and acts as a blueprint for transformation of other non-model microorganisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most microbial metabolic engineering for biotechnology research is performed in model organisms, because they are well studied and have a large toolbox to enable genetic modifications [7]. Non-model organisms, on the other hand, are an attractive alternative as potential industrial platforms for bioconversion, because they often possess complex phenotypes that are not currently feasible to engineer into model organisms, such as the ability to grow at extreme pH, extreme robustness/toxicity tolerance, or the ability to catabolize less common substrates such as syngas, methane, cellulose, or lignin [49, 52]. However, work with non-model organisms is limited due to a lack tools and knowledge of the organism. A major barrier to genetic tools development is the inability to efficiently transform DNA, and to routinely allow for the use of non-model organisms; a systematic process for developing transformation is needed.
One of the major barriers to successful transformation of bacteria is native DNA restriction–modification (RM) systems, which act as a bacterial immune system to cut DNA that is methylated differently than the host [2]. RM systems are classified into four types. In Types I, II, and III, a restriction enzyme typically cuts DNA that is unmethylated at a specific recognition sequence, and a corresponding DNA methyltransferase adds a methyl group to the host’s DNA to protect these same sequences from restriction enzyme activity [2]. Type I systems are comprised of three subunits: a DNA methyltransferase, a DNA recognition subunit, and a restriction enzyme. This type of system recognizes two motifs of 3–4 bases separated by any 5–8 bases [25], such as the EcoKI system that targets AACNNNNNNGTGC (N is any base), and motifs are methylated at the N-6 position of one adenine per DNA strand to form N6-methyladenine (m6A). Type II and III systems are typically comprised of a methyltransferase and a restriction enzyme subunit. Type II systems are largely studied and commonly used as tools in molecular biology. Their recognition systems are often palindromic, and they can methylate bases to form m6A, N4-methylcytosine (m4C), or 5-methylcytosine (m5C) [16, 31]. Type III systems recognize non-palindromic motifs and typically methylate to form m6A [38]. The last group, Type IV, is only comprised of a restriction enzyme, which recognizes motifs that are methylated differentially than the host [19, 38].
To successfully evade most native RM systems, DNA needs to be methylated in the same way as the target organism, and many studies have shown that overcoming these systems is important for efficient DNA transformation [4, 23, 33, 41, 50]. To rationally evade RM systems, the targeted motifs first need to be identified. Microbes protect their chromosomal DNA from restriction via DNA methylation; therefore, methylome analysis can be used to identify these motifs. While not all DNA methyltransferases are associated with restriction enzymes [3], methylome analysis does reveal all potentially active Type I, II, and III RM systems present in an organism. A common approach to microbial methylome analysis uses single-molecule real-time (SMRT) sequencing on the PacBio platform [9, 17, 24, 28], which identifies m6A and m4C motifs based on kinetic delays in nucleotide incorporation when a base is methylated [37]. This approach has been specifically used to help overcome restriction barriers to genetic transformation in a number of organisms [4, 34, 41]. SMRT sequencing can also identify m5C motifs, but the signal is less strong, so it requires immense coverage of the genome or modification of the methylated base via Tet1 oxidation [5], approaches that are not common practice. An alternate approach to detect m5C and m4C is methyl-C sequencing for whole-genome bisulfite sequencing (WGBS), in which the cytosine (but not m5C) is deaminated to uracil, followed by polymerase chain reaction (PCR) to convert the resulting uracils to thymines. The resulting PCR-amplified DNA can then be sequenced, and remaining cytosines in the sequence were previously methylated. This approach has not been routinely used in bacteria, and we have only identified a few studies that utilized WGBS for bacterial methylome analysis for characterization of RM systems [13, 44, 51].
Clostridium thermocellum is an anaerobic, thermophilic bacterium that efficiently deconstructs lignocellulosic biomass via large cell surface-associated enzyme complexes called cellulosomes. The resulting soluble sugars are fermented into products such as organic acids and ethanol [1]. There is particular interest in C. thermocellum due to its potential for biofuel production from lignocellulose via a process called consolidated bioprocessing (CBP), in which cellulolytic enzyme production and fermentation occur in a single reactor [26, 27]. The cellulosome was first discovered in the Clostiridium thermocellum-type strain, ATCC 27405 [15], and a substantial amount of work has been done on this strain to understand carbon metabolism and the genes related to cellulosome production [8, 32, 35, 43, 48]. All of these studies examined the wild-type strain using omics tools such as transcriptomics and proteomics, but to date, this strain has not been genetically modified. This is in stark contrast to C. thermocellum DSM 1313, where transformation and genetics are readily available [29] and extensive metabolic engineering has been achieved [18, 30, 40, 45, 46].
The lack of genetic tools in strain ATCC 27405 has hindered both fundamental studies and development of this strain for bioengineering. Thus far, one publication has demonstrated transformation of this strain using a custom-made electroporator [47], and additional attempts to transform C. thermocellum ATCC 27405 using commercially available equipment have been unsuccessful. One other publication demonstrated a low transformation rate of 2.5 ± 1.5 colonies per microgram of DNA with a single plasmid using large cell and DNA volumes, which makes the process hard to utilize in the future [20].
One possible reason for the difference in success of transformation between strains DSM 1313 and ATCC 27405 is differences in RM systems, which are understudied in C. thermocellum ATCC 27405. New England Biolabs Restriction Enzyme Database (REBASE) [39] predicts the RM systems encoded in both strains of C. thermocellum, where ATCC 27405 encodes eight restriction systems and DSM 1313 only encodes five. Of the eight potential RM systems in ATCC 27405, one is a putative Type I system, genes Cthe_1144–1145, though it seems to be missing the restriction enzyme subunit. There are six putative Type II systems encoded: Cthe_1511–1513, Cthe_1638–1639, Cthe_1748–1749, Cthe_2470–2471, Cthe_2319–2320, and Cthe_1748–1749. Cthe_2740 and Cthe_1728–1729 do not have annotated associated restriction enzymes, but the other four encode putative restriction enzymes do. The genome also encodes one Type III RM system, Cthe_0518-0519, with Cthe_0518 being a predicted restriction enzyme. No apparent Type IV systems are encoded in the genome. Previously, one of these RM systems was shown to be active in cell extracts of strain ATCC 27405, and extracts exhibited endonuclease activity targeting the motif GATC, similar to the MboI restriction enzyme [14]. Here, we report the methylome of C. thermocellum ATCC 27405, identify and express the active methyltransferases, validate the expression for in vivo methylation, and successfully transform C. thermocellum ATCC 27405.
Materials and methods
Strains, plasmids, and growth conditions
Escherichia coli Top 10 Δdcm::frt was created by deleting dcm in Top10 (Invitrogen) with the lambda Red recombinase system as previously described [6]. E. coli strains were grown aerobically in LB at 37 °C, and chloramphenicol was added for plasmid selection at a final concentration of 15 µg/mL. C. thermocellum ATCC 27405 was grown in CTFUD medium [40], which is comprised of (per L) 3 g sodium citrate tribasic dehydrate, 1.3 g ammonium sulfate, 1.43 g potassium phosphate monobasic, 1.8 g potassium phosphate dibasic trihydrate, 0.5 g cysteine HCL, 10.5 g MOPS sodium salt, 6 g glycerol-2-phosphate disodium, 5 g cellobiose, 4.5 g yeast extract, 0.13 g calcium chloride dehydrate, 2.6 g magnesium chloride hexahydrate, 0.0001 g ferrous sulfate heptahydrate, and 0.5 ml 0.2% (w/v) resazurin. The pH is adjusted to 7.0 after addition of MOPS with 45% (w/v) potassium hydroxide. C. thermocellum was grown at 50 or 55 °C, as indicated, in a Coy anaerobic chamber. Thiamphenicol was added to the medium when appropriate at a final concentration of 5 µg/mL.
Two plasmids, pNJ020 and pAMG216, were used for transformation of C. thermocellum, each containing the pNW33N origin of replication for C. thermocellum and the cat gene for thiamphenicol selection. Plasmid pNJ020 contains the p15a origin for medium copy-number replication in E. coli. Plasmid pAMG216 is derived from pAMG205 with the yeast machinery deleted, and it has a high copy-number pUC origin of replication for E. coli [11]. Methyltransferases were codon optimized for E. coli and synthesized with the T5Lac promoter by GenScript Biotech Corp (New Jersey, USA), amplified by PCR, and cloned into CRIM integration vectors [12] using Gibson assembly (New England Biolabs, NEB). The native C. thermocellum gene Cthe_0519 was cloned into pAH55 [12]. The bifunctional methyltransferase phi3TI, from Bacillus phage phi3T, was cloned into pAH144. Each plasmid was integrated, using the CRIM system [12], into the λ and HK022 phage attB sites of E. coli Top 10 Δdcm::frt, resulting in strain AG2006 (genotype: Top10 Δdcm::frt λ::Cthe_0519 HK022::phi3TI). Complete, annotated plasmid sequences are available in Supplemental File 1.
SMRT sequencing methylome analysis
Genomic DNA from C. thermocellum ATCC 27405 was isolated using the Genomic Tip kit (Qiagen according to the manufacturer’s instructions and sent to Expression Analysis (Durham, NC, USA) for sequencing on a Pacific Biosciences (PacBio) instrument. Single-molecule real-time (SMRT) sequencing was performed using the PacBio RS technology with four SMRT cells. Methylated sequences were determined by Expression Analysis using the SMRT Analysis software [10].
Whole-genome bisulfite sequencing analysis
MethylC-seq libraries were prepared and Illumina sequencing was performed (Genomics & Bioinformatics Core, University of Georgia) using an Illumina NextSeq 500 instrument. For data processing, raw reads were trimmed for adapters and preprocessed to remove low-quality reads using cutadapt 1.9.dev1 [21]. Qualified reads were aligned to the C. thermocellum ATCC 27405 reference genome [42]. Fully unmethylated lambda DNA was used as a control to calculate the sodium bisulfite reaction non-conversion rate of unmodified cytosines. Only cytosine sites with a minimum coverage (set as 3) were selected for subsequent analysis. Binomial test coupled with Benjamini–Hochberg correction was adopted to determine the methylation status of each cytosine. The m5C motifs were identified as previously described in [51].
Determining functionality of the methyltransferases
Plasmids pNJ020 and pAMG216 were transformed into E. coli strains AG2006 and Top10 Δdcm::frt and grown in liquid culture with 0.1 mM IPTG to induce methyltransferase expression. Plasmid pNJ020 was isolated and digested in three separate reactions with NlaIII, TseI and HindIII, and HaeIII and HindIII (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions. To determine the amount of DNA methylated, digested plasmid was separated via agarose gel electrophoresis and quantified using BioRad Gel Imager software.
Transformation of C. thermocellum ATCC 27405
Three 5 mL cultures were inoculated with C. thermocellum ATCC 27405 and grown overnight. Three 500 mL cultures were inoculated with 1% of the overnight cultures and grown at 55 °C. Cultures were harvested at an optical density (O.D.) of ~ 1.0, transferred to a 500 mL centrifuge bottle, placed on ice for 30 min, and then centrifuged at 4 °C at 5000×g for 15 min. The supernatant was decanted and cells were washed with 250 mL cold electroporation buffer (10% glycerol, 250 mM sucrose), which was added without disrupting/resuspending the cell pellet. Cells were spun again, and the wash was repeated two more times. After the last wash, the electroporation buffer was completely removed, and the cell pellets were resuspended in 200 µL electroporation buffer and transferred to a microcentrifuge tube. Using fresh electrocompetent cells from each batch, 20 µL of cells were transformed with 1 µg of DNA. Square wave electroporation was performed in a 1 mM electroporation cuvette at 1200 v with a 1.5 ms pulse. Cells were resuspended in 1 mL of CTFUD liquid medium and mixed with molten CTFUD supplemented with 1.5% agar and thiamphenicol, allowed to solidified, and incubated at 50 °C for 3–5 days, when colonies were counted.
Results
Complete methylome analysis of C. thermocellum ATCC 27405
To determine which of the eight RM systems, predicted by REBASE, are active, methylome analysis was performed. All methylated motifs in C. thermocellum ATCC 27405 were determined through two sequencing techniques, PacBio SMRT sequencing and MethylC-sequencing for WGBS. PacBio SMRT sequencing detected three m6A motifs and WGBS detected two m5C motifs (Table 1). Based on the type of motif and homology to known enzymes, each motif was tentatively assigned to a DNA methyltransferase (Table 1). Cthe_2470 and Cthe_1511 are both annotated as Dam methyltransferases, and Cthe_1511 is encoded in a putative operon with the MboI-type restriction enzyme, suggesting that these enzymes target GATC. The CNCANNNNNNTTC motif is consistent with Type I RM system motifs (two 3–4 base-specific sites separated by 5–8 Ns), and so this is presumably targeted by the only Type I enzyme encoded in the genome—Cthe1144–1145. The non-palindromic sequence GTCAT is consistent with a Type III system, and so is likely targeted by the only encoded Type III enzyme, Cthe_0519, which is experimentally validated below. Cthe_2320 is annotated as a HaeIII family restriction endonuclease, which targets GGCC, suggesting that Cthe_2321 methylates GGCC. The only remaining m5C methyltransferase encoded in the genome is Cthe_1749, suggesting that it targets the remaining m5C site—GCWGC. These results are consistent with the predictions from REBASE.
DNA methyltransferases were functionally expressed in E. coli
To methylate plasmid DNA in the same way as C. thermocellum ATCC 27405, we engineered E. coli to mimic the C. thermocellum ATCC 27405 methylome. One of the motifs, GATC, is natively methylated by the Dam methyltransferase in E. coli. Two additional methyltransferases were expressed from the E. coli chromosome. First, Cthe_0519 was integrated the chromosome to target GTCAT. Next, a previously characterized bifunctional Bacillus phage methyltransferase, phi3TI [22], was added to the chromosome. The Phi3TI methyltransferase is known to methylate both GCWGC and GGCC, the same sites methylated in C. thermocellum by Cthe_1749 and 2321, so expressing this gene enabled methylation of both sites via the expression of a single gene. Cthe_0519 and phi3TI were added to the chromosome of an E. coli dam+dcm− strain, so that Dam natively methylates GATC, a methylated motif observed in C. thermocellum, but Dcm does not methylate CCWGG, which was not seen in the C. thermocellum methylome analysis. The last methylated motif, CNCANNNNNNTTC, does not occur on pNJ020, and therefore, the corresponding methyltransferase was not expressed from the E. coli chromosome.
The in vivo activity of Cthe_0519 and Phi3TI methyltransferases was determined by restriction enzyme digestion to examine the extent of methylation by our heterologously expressed methyltransferases. Many commercially available restriction enzymes are blocked by overlapping DNA methylation. Therefore, a restriction enzyme was chosen for each motif that either partially overlaps the motif of interest or, when possible, targets the exact same motif. If the plasmid is properly methylated, then the enzyme will be blocked by the methylation and no cutting will occur. For instance, restriction enzyme NlaIII cuts unmethylated CATG, and a fraction of these NlaIII sites will overlap with GTCAT (when the sequence GTCATG occurs) and will not be cut (Fig. 1a). For pNJ020, methylation of GTCAT by Cthe_0519 blocks NlaIII-cutting between a 393 and 62 bp band, resulting in the generation of 455 bp band instead (Fig. 1a). Complete blockage of this cut site, and therefore compete methylation, was observed for Cthe_0519 (Fig. 1b, Lane 1, 2), where complete disappearance of the 393 bp band and generation of the new 455 bp band are observed. For the GGCC motif, restriction enzyme HaeIII targets the same sequence, so we would expect complete blockage of restriction activity if methylation of this sequence occurs in E. coli. We linearized the plasmid with HindIII and digested with HaeIII (Fig. 1). Unmethylated DNA (Lane 3) was completely digested, while Phi3TI methylation mostly blocked HaeIII digestion (Lane 4), suggesting nearly complete methylation of GGCC by Phi3TI. For GCWGC methylation, enzyme TseI targets the same motif, so the plasmid was linearized with HindIII and digested with TseI. Unmethylated DNA (Lane 5) was completely digested, while Phi3TI methylation fully blocked TseI digestion (Lane 6), suggesting complete methylation of GCWGW by Phi3TI.

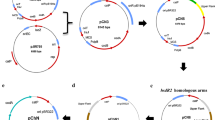
Confirmation of methyltransferase functionality in E. coli. a pNJ020 plasmid map with NlaIII restriction sites, including NlaIII* that overlaps with the GTCAT methylation site. When blocked by methylation, NlaIII* is not cut by NlaIII, resulting in a 455 bp band rather than 393 and 62 bp bands. b Agarose gel of restriction digests of plasmid pNJ020 isolated from E. coli Top10 (lanes 1, 3, and 5) and from methylating strain AG2006 (lanes 2, 4, and 6). Methylation at GTCAT is indicated by a band shift from 393 to 455 bp (lane 1 vs 2) or complete blockage of digestion by GGCC (lane 3 vs 4) and GCWGC (lane 5 vs 6)
Methylated DNA allows transformation of C. thermocellum ATCC 27405
To determine if targeted DNA methylation would allow transformation of C. thermocellum ATCC 27405, we tested transformation efficiency using pNJ020. This plasmid contains 8 GTCAT, 7 GCWGC, 5 GGCC, 5 GATC, and no CNCANNNNNNTTC sites. Plasmid DNA was methylated by the control strain Top10 Δdcm, which only methylates GATC, and pNJ020 methylated with Cthe_0519 and Phi3TI, which methylates GATC, GTCAT, GGCC (partially), and GCWGC, where all putative restriction systems should be evaded. The control transformation in which pNJ020 was only methylated with native E. coli Dam yielded no colonies, while the methylated plasmid yielded an average of 80 colony-forming units (CFU)/μg of plasmid DNA. Transformation of plasmid pAMG216 was also tested, which contains one Type I motif, and an average of 40 CFU/µg was observed (Table 2).
Discussion
Development of genetic systems in non-model microbes is a grand challenge for the study of both fundamental and applied microbiology. Here, we show a rational, systematic process for developing transformation methods in C. thermocellum ATCC 27405 by overcoming the native RM systems. This was done by first identifying the methylated motifs using high-throughput sequencing techniques, including the rarely used WGBS technique for identifying m5C motifs. Methylome analysis was followed by heterologous expression of targeted methyltransferases from the E. coli chromosome to mimic the C. thermocellum ATCC 27405 methylome. Plasmid DNA was passaged through this E. coli strain to methylate it, and functionality of the methylases was confirmed through restriction enzyme digest of the plasmid DNA. Properly methylated plasmid avoids the native RM systems and allowed transformation using commercially available equipment, which opens the door to metabolic engineering and the development of more advanced genetic tools. By enabling reliable transformation, the type strain of C. thermocellum can now be studied through genetic manipulation.
The first step to understanding and overcoming the native RM systems in C. thermocellum ATCC 27405 and other organisms is identifying the methylated motifs and the cognate methyltransferases. PacBio SMRT sequencing is the most commonly utilized method for microbial methylome analysis and can accurately identify m6A and m4C motifs. SMRT sequencing revealed three methylated motifs, and the corresponding RM systems were identified using REBASE. While SMRT sequencing is also able to detect m5C motifs, it does not always identify these motifs [37]. For example, the two m5C motifs in C. thermocellum ATCC 27405 were not discovered in the SMRT methylome analysis. Therefore, WGBS is a vital step to reliably determine the full methylome of a bacterial strain. Using WGBS on an Illumina platform, we were readily able to detect m5C motifs with less sequencing coverage. Recently, Oxford Nanopore sequencing has also been shown to detect DNA methylation [36]. As technologies advance, the simplicity of methylome analysis will likely increase.
Previous studies have expressed methyltransferases to methylate DNA prior to transformation, but functionality of these enzymes beyond examining the impact on transformation efficiency is rarely tested. Here, we tested functionality of the expressed methyltransferases through a restriction enzyme digestion assay in which restriction activity is blocked when the plasmid is methylated. While the Cthe_0519 methyltransferase fully methylated the plasmid DNA, the Phi3TI methyltransferase only partially methylated the DNA. By digesting plasmid DNA isolated from the E. coli methylation strain with an enzyme that overlaps with the methylated motif of interest, functionality can be easily determined by the percentage of DNA cut/uncut. This approach also unambiguously confirmed that Cthe_0519 methylates GTCAT.
An alternative approach to evading RM systems is to use plasmids that lack the targeted sequence. We used this approach for the putative Type I RM system (Cthe_1144–1145), where the motif was avoided using plasmid DNA (pNJ020) that does not contain the motif CNCANNNNNNTTC. Thus, isolation of pNJ020 out of the E. coli methylation strain results in plasmid DNA that fully mimicked the ATCC 27405 methylome. While this approach can be helpful, it is not always possible to avoid or remove the target motifs. For instance, when the motif is short, such as a four-base recognition sequence, it may not be feasible to remove them all. Some motifs may also happen to be in critical parts of the sequence, such as when they are in the origin of replication or in a sequence being used for homologous recombination. Therefore, expression of methyltransferases in E. coli will likely continue to be an attractive approach for developing transformation tools in new organisms.
Interestingly, Cthe_1144–1145 seems to not be part of an active restriction system. It lacks a predicted restriction enzyme, and when transformation of a plasmid containing the corresponding Type I motif (pAMG216) was tested, there was not a substantial difference in transformation efficiency relative to pNJ020, suggesting that this system, indeed, lacks a restriction enzyme. The twofold difference in efficiency can likely be explained by the difference in plasmid size (pNJ020 is 3561 bp, while pAMG216 is 4700 bp), where the smaller plasmid has a greater number of plasmid molecules per microgram of DNA and may enter the cell more easily due to its smaller physical size.
We have demonstrated reproducible transformation of C. thermocellum strain ATCC 27405 and are now poised to improve these methods to increase frequency. Improved methylation by the Phi3TI methyltransferase will likely increase transformation efficiency. In addition, for reasons unknown, strain ATCC 27405 does not form a cell pellet during centrifugation as well as strain DSM 1313. Therefore, identifying growth conditions under which ATCC 27405 forms tight cell pellets would make competent cell preparation simpler and presumably increase competent cell concentration, potentially leading to increased efficiencies. However, all approaches for genetic manipulation in C. thermocellum to date have relied on use of replicating plasmids, even for gene deletions [29], so the full suite of C. thermocellum genetic tools is now available in strain ATCC 27405, even at the current transformation efficiency.
In conclusion, the combination of complete methylome analysis via both PacBio SMRT sequencing and WGBS and validated DNA methyltransferase expression allowed the rational development of transformation methods for C. thermocellum ATCC 27405. We anticipate that the approach for obtaining transformation demonstrated in this work may be applied to many other bacterial strains, especially in new, non-model organisms.
References
Akinosho H, Yee K, Close D, Ragauskas A (2014) The emergence of Clostridium thermocellum as a high utility candidate for consolidated bioprocessing applications. Front Chem. https://doi.org/10.3389/fchem.2014.00066
Arber W, Linn S (1969) DNA modification and restriction. Annu Rev Biochem 38:467–500. https://doi.org/10.1146/annurev.bi.38.070169.002343
Blow MJ, Clark TA, Daum CG, Deutschbauer AM, Fomenkov A, Fries R, Froula J, Kang DD, Malmstrom RR, Morgan RD, Posfai J, Singh K, Visel A, Wetmore K, Zhao Z, Rubin EM, Korlach J, Pennacchio LA, Roberts RJ (2016) The epigenomic landscape of prokaryotes. PLoS Genet 12:e1005854. https://doi.org/10.1371/journal.pgen.1005854
Chung D, Farkas J, Westpheling J (2013) Overcoming restriction as a barrier to DNA transformation in Caldicellulosiruptor species results in efficient marker replacement. Biotechnol Biofuels 6:82. https://doi.org/10.1186/1754-6834-6-82
Clark TA, Lu X, Luong K, Dai Q, Boitano M, Turner SW, He C, Korlach J (2013) Enhanced 5-methylcytosine detection in single-molecule, real-time sequencing via Tet1 oxidation. BMC Biol 11:4. https://doi.org/10.1186/1741-7007-11-4
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. https://doi.org/10.1073/pnas.120163297
Davis RH (2004) The age of model organisms. Nat Rev Genet 5:69. https://doi.org/10.1038/nrg1250
Ellis LD, Holwerda EK, Hogsett D, Rogers S, Shao X, Tschaplinski T, Thorne P, Lynd LR (2012) Closing the carbon balance for fermentation by Clostridium thermocellum (ATCC 27405). Bioresour Technol 103:293–299. https://doi.org/10.1016/j.biortech.2011.09.128
Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC, Jabado OJ, Deikus G, Clark TA, Luong K, Murray IA, Davis BM, Keren-Paz A, Chess A, Roberts RJ, Korlach J, Turner SW, Kumar V, Waldor MK, Schadt EE (2012) Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat Biotechnol 30:1232–1239. https://doi.org/10.1038/nbt.2432
Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW (2010) Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7:461–465. https://doi.org/10.1038/nmeth.1459
Guss AM, Olson DG, Caiazza NC, Lynd LR (2012) Dcm methylation is detrimental to plasmid transformation in Clostridium thermocellum. Biotechnol Biofuels 5:30. https://doi.org/10.1186/1754-6834-5-30
Haldimann A, Wanner BL (2001) Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure–function studies of bacteria. J Bacteriol 183:6384–6393. https://doi.org/10.1128/JB.183.21.6384-6393.2001
Johnston CD, Skeete CA, Fomenkov A, Roberts RJ, Rittling SR (2017) Restriction–modification mediated barriers to exogenous DNA uptake and incorporation employed by Prevotella intermedia. PLoS One 12:e0185234. https://doi.org/10.1371/journal.pone.0185234
Klapatch TR, Demain AL, Lynd LR (1996) Restriction endonuclease activity in Clostridium thermocellum and Clostridium thermosaccharolyticum. Appl Microbiol Biotechnol 45:127–131. https://doi.org/10.1007/s002530050659
Lamed R, Setter E, Bayer EA (1983) Characterization of a cellulose-binding, cellulase-containing complex in Clostridium thermocellum. J Bacteriol 156:828–836
Lauster R, Trautner TA, Noyer-Weidner M (1989) Cytosine-specific type II DNA methyltransferases. A conserved enzyme core with variable target-recognizing domains. J Mol Biol 206:305–312
Lee WC, Anton BP, Wang S, Baybayan P, Singh S, Ashby M, Chua EG, Tay CY, Thirriot F, Loke MF, Goh KL, Marshall BJ, Roberts RJ, Vadivelu J (2015) The complete methylome of Helicobacter pylori UM032. BMC Genom 16:424. https://doi.org/10.1186/s12864-015-1585-2
Lin PP, Mi L, Morioka AH, Yoshino KM, Konishi S, Xu SC, Papanek BA, Riley LA, Guss AM, Liao JC (2015) Consolidated bioprocessing of cellulose to isobutanol using Clostridium thermocellum. Metab Eng 31:44–52. https://doi.org/10.1016/j.ymben.2015.07.001
Liu G, Ou HY, Wang T, Li L, Tan H, Zhou X, Rajakumar K, Deng Z, He X (2010) Cleavage of phosphorothioated DNA and methylated DNA by the type IV restriction endonuclease ScoMcrA. PLoS Genet 6:e1001253. https://doi.org/10.1371/journal.pgen.1001253
Maki ML, Armstrong L, Leung KT, Qin W (2013) Increased expression of β-glucosidase A in Clostridium thermocellum 27405 significantly increases cellulase activity. Bioengineered 4:15–20. https://doi.org/10.4161/bioe.21951
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12
Mermelstein LD, Papoutsakis ET (1993) In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 59:1077–1081
Mermelstein LD, Welker NE, Bennett GN, Papoutsakis ET (1992) Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Bio/Technology 10:190–195. https://doi.org/10.1038/nbt0292-190
Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ (2012) The methylomes of six bacteria. Nucleic Acids Res 40:11450–11462. https://doi.org/10.1093/nar/gks891
Murray NE (2000) Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol Mol Biol Rev 64:412–434
Ng TK, Weimer TK, Zeikus JG (1977) Cellulolytic and physiological properties of Clostridium thermocellum. Arch Microbiol 114:1–7
Ng TK, Zeikus JG (1981) Comparison of extracellular cellulase activities of Clostridium thermocellum LQRI and Trichoderma reesei QM9414. Appl Environ Microbiol 42:231–240
O’Loughlin JL, Eucker TP, Chavez JD, Samuelson DR, Neal-McKinney J, Gourley CR, Bruce JE, Konkel ME (2015) Analysis of the Campylobacter jejuni genome by SMRT DNA sequencing identifies restriction–modification motifs. PLoS One 10:e0118533. https://doi.org/10.1371/journal.pone.0118533
Olson DG, Lynd LR (2012) Transformation of Clostridium thermocellum by electroporation. Methods Enzymol 510:317–330. https://doi.org/10.1016/b978-0-12-415931-0.00017-3
Papanek B, Biswas R, Rydzak T, Guss AM (2015) Elimination of metabolic pathways to all traditional fermentation products increases ethanol yields in Clostridium thermocellum. Metab Eng 32:49–54. https://doi.org/10.1016/j.ymben.2015.09.002
Pingoud A, Jeltsch A (1997) Recognition and cleavage of DNA by type-II restriction endonucleases. Eur J Biochem 246:1–22
Poudel S, Giannone RJ, Rodriguez M, Raman B, Martin MZ, Engle NL, Mielenz JR, Nookaew I, Brown SD, Tschaplinski TJ, Ussery D, Hettich RL (2017) Integrated omics analyses reveal the details of metabolic adaptation of Clostridium thermocellum to lignocellulose-derived growth inhibitors released during the deconstruction of switchgrass. Biotechnol Biofuels 10:14. https://doi.org/10.1186/s13068-016-0697-5
Purdy D, O’Keeffe TA, Elmore M, Herbert M, McLeod A, Bokori-Brown M, Ostrowski A, Minton NP (2002) Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol Microbiol 46:439–452
Pyne ME, Bruder M, Moo-Young M, Chung DA, Chou CP (2014) Technical guide for genetic advancement of underdeveloped and intractable Clostridium. Biotechnol Adv 32:623–641. https://doi.org/10.1016/j.biotechadv.2014.04.003
Raman B, Pan C, Hurst GB, Rodriguez M Jr, McKeown CK, Lankford PK, Samatova NF, Mielenz JR (2009) Impact of pretreated switchgrass and biomass carbohydrates on Clostridium thermocellum ATCC 27405 cellulosome composition: a quantitative proteomic analysis. PLoS One 4:e5271. https://doi.org/10.1371/journal.pone.0005271
Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, Paten B (2017) Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods 14:411–413. https://doi.org/10.1038/nmeth.4189
Rhoads A, Au KF (2015) PacBio sequencing and its applications. Genom Proteom Bioinform 13:278–289. https://doi.org/10.1016/j.gpb.2015.08.002
Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden DT, Dybvig K, Firman K, Gromova ES, Gumport RI, Halford SE, Hattman S, Heitman J, Hornby DP, Janulaitis A, Jeltsch A, Josephsen J, Kiss A, Klaenhammer TR, Kobayashi I, Kong H, Kruger DH, Lacks S, Marinus MG, Miyahara M, Morgan RD, Murray NE, Nagaraja V, Piekarowicz A, Pingoud A, Raleigh E, Rao DN, Reich N, Repin VE, Selker EU, Shaw PC, Stein DC, Stoddard BL, Szybalski W, Trautner TA, Van Etten JL, Vitor JM, Wilson GG, Xu SY (2003) A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31:1805–1812
Roberts RJ, Vincze T, Posfai J, Macelis D (2015) REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43:D298–D299. https://doi.org/10.1093/nar/gku1046
Rydzak T, Lynd LR, Guss AM (2015) Elimination of formate production in Clostridium thermocellum. J Ind Microbiol Biotechnol 42:1263–1272. https://doi.org/10.1007/s10295-015-1644-3
Sandoval NR, Venkataramanan KP, Groth TS, Papoutsakis ET (2015) Whole-genome sequence of an evolved Clostridium pasteurianum strain reveals Spo0A deficiency responsible for increased butanol production and superior growth. Biotechnol Biofuels 8:227. https://doi.org/10.1186/s13068-015-0408-7
Schmitz RJ, He Y, Valdes-Lopez O, Khan SM, Joshi T, Urich MA, Nery JR, Diers B, Xu D, Stacey G, Ecker JR (2013) Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res 23:1663–1674. https://doi.org/10.1101/gr.152538.112
Stevenson DM, Weimer PJ (2005) Expression of 17 genes in Clostridium thermocellum ATCC 27405 during fermentation of cellulose or cellobiose in continuous culture. Appl Environ Microbiol 71:4672–4678. https://doi.org/10.1128/aem.71.8.4672-4678.2005
Suzuki H, Takahashi S, Osada H, Yoshida K (2011) Improvement of transformation efficiency by strategic circumvention of restriction barriers in Streptomyces griseus. J Microbiol Biotechnol 21:675–678
Tian L, Papanek B, Olson DG, Rydzak T, Holwerda EK, Zheng T, Zhou J, Maloney M, Jiang N, Giannone RJ, Hettich RL, Guss AM, Lynd LR (2016) Simultaneous achievement of high ethanol yield and titer in Clostridium thermocellum. Biotechnol Biofuels 9:116. https://doi.org/10.1186/s13068-016-0528-8
Tripathi SA, Olson DG, Argyros DA, Miller BB, Barrett TF, Murphy DM, McCool JD, Warner AK, Rajgarhia VB, Lynd LR, Hogsett DA, Caiazza NC (2010) Development of pyrF-based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl Environ Microbiol 76:6591–6599. https://doi.org/10.1128/AEM.01484-10
Tyurin MV, Desai SG, Lynd LR (2004) Electrotransformation of Clostridium thermocellum. Appl Environ Microbiol 70:883–890. https://doi.org/10.1128/aem.70.2.883-890.2004
Wilson CM, Rodriguez M, Johnson CM, Martin SL, Chu TM, Wolfinger RD, Hauser LJ, Land ML, Klingeman DM, Syed MH, Ragauskas AJ, Tschaplinski TJ, Mielenz JR, Brown SD (2013) Global transcriptome analysis of Clostridium thermocellum ATCC 27405 during growth on dilute acid pretreated Populus and switchgrass. Biotechnol Biofuels 6:179. https://doi.org/10.1186/1754-6834-6-179
Yan Q, Fong SS (2017) Challenges and advances for genetic engineering of non-model bacteria and uses in consolidated bioprocessing. Front Microbiol. https://doi.org/10.3389/fmicb.2017.02060
Yasui K, Kano Y, Tanaka K, Watanabe K, Shimizu-Kadota M, Yoshikawa H, Suzuki T (2009) Improvement of bacterial transformation efficiency using plasmid artificial modification. Nucleic Acids Res 37:e3. https://doi.org/10.1093/nar/gkn884
Yu M, Ji L, Neumann DA, Chung D-h, Groom J, Westpheling J, He C, Schmitz RJ (2015) Base-resolution detection of N 4-methylcytosine in genomic DNA using 4mC-Tet-assisted-bisulfite-sequencing. Nucleic Acids Res 43:e148
Zeldes BM, Keller MW, Loder AJ, Straub CT, Adams MW, Kelly RM (2015) Extremely thermophilic microorganisms as metabolic engineering platforms for production of fuels and industrial chemicals. Front Microbiol 6:1209. https://doi.org/10.3389/fmicb.2015.01209
Acknowledgements
Funding was provided by The BioEnergy Science (BESC) and The Center for Bioenergy Innovation (CBI), U.S. Department of Energy Bioenergy Research Centers supported by the Office of Biological and Environmental Research in the DOE Office of Science. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U.S. DOE under contract DE-AC05-00OR22725. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This manuscript has been authored by UT-Battelle, LLC, under Contract No. DE-AC0500OR22725 with the U. S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for the United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Riley, L.A., Ji, L., Schmitz, R.J. et al. Rational development of transformation in Clostridium thermocellum ATCC 27405 via complete methylome analysis and evasion of native restriction–modification systems. J Ind Microbiol Biotechnol 46, 1435–1443 (2019). https://doi.org/10.1007/s10295-019-02218-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-019-02218-x