
Abstract
Purpose
This review aimed to provide a complete overview of the current stance and recent developments in antiarrhythmic neuromodulatory interventions, focusing on lifethreatening vetricular arrhythmias.
Methods
Both preclinical studies and clinical studies were assessed to highlight the gaps in knowledge that remain to be answered and the necessary steps required to properly translate these strategies to the clinical setting.
Results
Cardiac autonomic imbalance, characterized by chronic sympathoexcitation and parasympathetic withdrawal, destabilizes cardiac electrophysiology and promotes ventricular arrhythmogenesis. Therefore, neuromodulatory interventions that target the sympatho-vagal imbalance have emerged as promising antiarrhythmic strategies. These strategies are aimed at different parts of the cardiac neuraxis and directly or indirectly restore cardiac autonomic tone. These interventions include pharmacological blockade of sympathetic neurotransmitters and neuropeptides, cardiac sympathetic denervation, thoracic epidural anesthesia, and spinal cord and vagal nerve stimulation.
Conclusion
Neuromodulatory strategies have repeatedly been demonstrated to be highly effective and very promising anti-arrhythmic therapies. Nevertheless, there is still much room to gain in our understanding of neurocardiac physiology, refining the current neuromodulatory strategic options and elucidating the chronic effects of many of these strategic options.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
With every beat, cardiovascular afferent nerves convey sensory information to various centers of the cardiac autonomic nervous system. The subsequent multi-tiered integration of this information results in the meticulous titration of sympathetic (excitatory) and parasympathetic (inhibitory) efferent outflow, modulating cardiac physiology on a beat-to-beat basis [1,2,3]. While this system has evolved to primarily protect and preserve cardiovascular homeostasis, derangement of this system is an elemental characteristic of a plethora of cardiac diseases [2, 4,5,6]. The derangement-induced autonomic imbalance, which is commonly characterized by sympathetic overactivity and parasympathetic withdrawal, induces electrical instability and thereby greatly enhances the risk of tachycardic ventricular arrhythmias (VAs), both monomorphic and polymorphic, and thus sudden cardiac death [1]. Correspondingly, β-blockers that impede the sympathetic effects on the myocardium are one of the cornerstones of current antiarrhythmic treatments [7]. However, not all patients benefit from treatment with β-blockers, and their systemic effects cause side effects [8, 9]. It is therefore unsurprising that much research has aimed to decipher the complex interplay between the autonomic nervous system and ventricular arrhythmogenesis, in order to develop innovative, effective and more targeted antiarrhythmic therapies [10,11,12,13]. Whereas much of this research remains in the preclinical phase, some neuromodulatory strategies are already implemented in the clinical setting. For that purpose, this review assesses both preclinical studies and clinical studies, thereby providing an overview of recent developments in the field of sympathetic modulation of ventricular electrical activity.
Animal models in preclinical studies
As this review assesses both preclinical studies and clinical studies, it is crucial to recognize the main differences in anatomy, neurophysiology and electrophysiology between different animal models and humans and to realize that no animal model is a precise replica of human cardiac anatomy and physiology. In order to describe and interpret preclinical data in the right context and to appreciate the limitations that are inherent to translating preclinical results to clinical practice, this review will briefly discuss the limitations of murine, canine and porcine models.
Murine models are widely used in preclinical studies, as they are easy to handle, a relatively cheap experimental model, and genetically manipulable, with many of such strains commercially available [14]. However, the main limitation to using mice in arrhythmia research is that their electrophysiological properties are substantially different from those of humans. For example, their resting heart rate is much higher (±600 beats/min) [15] and ionic currents contribute differently to the action potential (e.g. the calcium current is less dominant, and murine repolarization relies heavily on the transient outward current [Ito]), which also causes their action potential to look greatly different from those of humans [14, 16]. Moreover, with regard to the cardiac autonomic nervous system, autonomic tone is dominated by the sympathetic nervous system [17], in contrast to the parasympathetic predominance in humans (and also dogs and pigs). Hence, these fundamental differences in cardiac physiology, in combination with the smaller size of the murine heart, make it less susceptible to spontaneous ventricular arrhythmias [18], indicating that these differences significantly affect the process of ventricular arrhythmogenesis. Therefore, one should consider these limitations of murine arrhythmia models when interpreting results on neural remodeling and the effects of cardiac autonomic innervation on ventricular electrophysiology and be aware that most results cannot be directly extrapolated to humans.
Secondly, canine models have been of great value to the field of ventricular arrhythmogenesis, a significant advantage being that all major currents that are present in human myocytes are also found in dogs, resulting in a very similar action potential morphology [19, 20]. Moreover, the social nature of dogs allows for awake experiments, which might be especially beneficial when studying the autonomic nervous system, as anesthesia can pose a confounding factor in such experiments [21]. However, canine models also come with various disadvantages and limitations. For example, dogs have a shorter action potential duration and a higher resting heart rate and respiratory rate [20]. Moreover, though parasympathetic tone is dominant in resting humans, the resting autonomic tone in dogs is even more heavily influenced by the parasympathetic nervous system, which is also reflected in their more pronounced respiratory sinus arrhythmia and increased heart rate variability [22]. This difference in autonomic tone must thus be carefully considered in the setting of preclinical studies in neurocardiology. Lastly, dogs are a costly, time-consuming and labor-intensive animal model, which greatly limits their use [20].
Pigs are another well-established animal model in the research areas of ventricular arrhythmias and neural remodeling. A great advantage of the pig model is that its coronary circulation is highly similar to that of humans [23]. As a result, the pathophysiology of coronary ligation closely mimics the effects a myocardial infarction has in humans [23]. Moreover, the pig's heart size, heart-to-body weight and inflammatory response are also all very similar to those of humans [23]. However, in comparison to humans, there are some differences in electrophysiology. For example, Purkinje fibers are richly distributed throughout the ventricular walls of pigs [23]. Consequently, the activation wave spreads faster, but the Purkinje fibers might also facilitate the development of sustained ventricular tachycardias [23]. The latter corresponds to the observation that pigs are much more susceptible to ventricular fibrillation then humans, which presents a limitation when it comes to translating results on arrhythmic inducibility and sudden cardiac death to humans. Moreover, one must keep in mind that many experimental groups use pigs that are less than a year old (corresponding to 18 human years) [24]. Therefore, the relatively young age of these animals should be kept in mind when extrapolating the effects of neural and/or cardiac remodeling to the human setting, as aging affects the ability and extent to which organs can adapt to a diseased state.
Cardiac neuraxis
Anatomically, the cardiac neuraxis can be divided into three main levels (Fig. 1). On the surface of the heart, the local intrinsic cardiac nervous system (ICNS) represents level 1 of cardiac–neural interplay. This “little brain on the heart” comprises multiple clusters of ganglia, called ganglionated plexi, that are located in epicardial fat pads on the myocardium [2]. The second anatomical level is established by the intrathoracic extracardiac components, which amongst other structures comprise the cervicothoracic ganglia. Thirdly, the extrathoracic extracardiac components include nodose ganglia, dorsal root ganglia and various areas within the spinal cord and brainstem that are involved in cardiovascular regulation (Fig. 1) [2, 25].

Simplified, schematic overview of the cardiac neuraxis. Neural control is effectuated through reflex loops at different levels of the cardiac neuraxis. ICNS intrinsic cardiac nervous system. Blue arrows: afferent nerves. Green arrows: parasympathetic nerves. Red arrows: sympathetic nerves. Purple arrows: ICNS reflex loop
Functionally, there are multiple characteristics based on which the cardiac neuraxis can be further organized. For example, neurons can be classified as (1) afferent, (2) efferent or (3) local circuit neurons (Fig. 1). Collectively, these neurons work in concert to establish feedback loops that convey afferent (sensory) information to integration centers from where efferent (motor) signals are transmitted to the myocardium [2]. Of note, efferent and afferent pathways often comprise series of multiple neurons. In the case of efferent signals, transmission between two such neurons generally occurs within a ganglion, such as the stellate ganglion. Here, preganglionic neurons synapse upon postganglionic neurons, and additional (afferent) information can be integrated, which might modulate the efferent signal. In this manner, multiple feedback loops between the heart and different levels of the cardiac neuraxis are established (Fig. 1) [2, 4].
Another, parallel categorization method allocates neural pathways to the sympathetic or parasympathetic branch of the autonomic nervous system. In the case of the sympathetic nervous system, efferents originate in the intermediolateral cell column of the cervical and thoracic spinal cord (C7-T6). These postganglionic neurons project to the superior, middle, cervicothoracic and stellate ganglia, where they synapse upon postganglionic nerves that travel towards the myocardium [26,27,28]. Similarly, parasympathetic efferents originate in the dorsal motor nucleus and nucleus ambiguus of the medulla [29]. These pre-ganglionic nerves mainly run through the vagus nerve and its dorsal root ganglion. However, parasympathetic pre- to postganglionic transmission occurs at the level of the heart, as its post-ganglionic nerves constitute the majority of the ICNS [30, 31].
Nevertheless, even though anatomical structures can be roughly assigned to the sympathetic or parasympathetic division, it is important to keep in mind that many of these structures comprise a combination of both divisions; for example, the ICNS also contains sympathetic nerves [32]. In contrast, separate neurons are either sympathetic or parasympathetic and exert their antagonizing effects through different neurotransmitters: whereas norepinephrine (NE), binding to β-adrenergic receptors on myocytes, is the main neurotransmitter of sympathetic nerves, the parasympathetic nerves release acetylcholine (ACh), which interacts with muscarinic receptors on the heart. Of note, ACh, binding onto nicotinic receptors is the primary neurotransmitter in pre- to postganglionic neural transmission for both divisions.
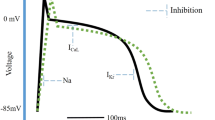
Collectively, this complex yet elegant network allows for continuous feedback between the myocardium and the different levels of the cardiac neuraxis, which results in the beat-to-beat regulation and modulation of all facets of cardiac function, including cardiac electrophysiology [4]. The release of NE and other co-transmitters, such as neuropeptide Y (NPY), from sympathetic cardiac nerve terminals and secretion of epinephrine from adrenal glands initiates various signaling pathways that collectively establish the sympathetic effects on cardiac electrophysiology [33]. For example, both NE and epinephrine can bind to β-adrenergic receptors on myocytes, which initiates a phosphorylation cascade through cAMP-dependent activation of protein kinase A (PKA) [34]. As such, PKA-mediated phosphorylation of the L-type calcium channels increases the systolic intracellular calcium concentration and thereby enhances contractional force [34]. Simultaneously, cAMP-dependent PKA phosphorylates ryanodine receptors on the sarcoplasmic reticulum, thereby enhancing the calcium-induced calcium release. This sarcoplasmic release of calcium is especially important for increasing systolic calcium concentrations. However, PKA also phosphorylates phospholamban, which stimulates the sarcoplasmic re-uptake of calcium and thereby speeds relaxation [34]. Moreover, the slowly activating potassium channels (IK,s) also become phosphorylated and thereby increased in activity [35, 36], which shortens repolarization to counterbalance calcium-induced prolongation of action potential duration and facilitates an increase in the heart rate by shortening the effective refractory period [37, 38]. Lastly, phosphorylation of INa increases ventricular conduction velocities, which also accommodates the increased heart rate (Fig. 2) [39,40,41].

Schematic representation of the electrical effects of sympathetic stimulation under health conditions and in the setting of sympathoexcitation. Healthy: The binding of norepinephrine (NE) or epinephrine to the β-receptor results in cAMP-mediated activation of protein kinase A (PKA). Next, PKA phosphorylates the L-type calcium channel (LTCC), promoting the influx of calcium. Simultaneously, the calcium-induced calcium release from the sarcoplasmic reticulum (SR) is also enhanced by the phosphorylation of the ryanodine receptor (RYR). Simultaneously, phospholamban is also phosphorylated, which enhances the calcium re-uptake through the sarco/endoplasmic reticulum calcium ATPase (SERCA). Lastly, PKA also phosphorylates INa and IK,s, which accommodate the faster heart rate by increasing conduction velocity and shortening repolarization, respectively. Sympathoexcitation: At the cellular level, sympathetic excitation results in the same effects as under healthy conditions. However, the increased calcium influx through the LTCC and the increased SR calcium release (due to increased calcium loading) improve contractile force, but also predispose to the development of early (EAD) or delayed afterdepolarization (DAD; an EAD is shown, a normal action potential is depicted under “healthy” for reference). These afterdepolarizations are caused by the reactivation of the LTCC during the prolonged plateau phase of the action potential or by sodium-calcium exchanger (NCX)-medicated depolarization due to spontaneous calcium release. This predisposition to the development of an early afterdepolarization is further enhanced by the stimulation of INa, and by the slower effect of sympathetic stimulation on IK,s activation, both of which cause a prolongation in action potential duration
Parasympathetic effects, on the other hand, caused by the interaction between ACh and muscarinic receptors, generally counterbalance these sympathetic effects and, amongst other effects, result in a slower heart rate, decrease the L-type calcium channel current and activate the acetylcholine-activated potassium current (IK,Ach) [42].
Neural remodeling
In the setting of cardiac injury and impaired cardiac function, the autonomic nervous system remodels to preserve adequate systemic circulation. Consequently, a condition with chronic sympathetic overexcitation and parasympathetic withdrawal is established.
Moreover, these functional changes are mirrored by structural adaptations in the ICNS and stellate and nodose ganglia. For example, in the stellate ganglia, neuronal and glial hypertrophy and increased inflammatory processes have been reported [43], and in post-infarct murine models, sympathetic nerves in stellate ganglia have been demonstrated to undergo cholinergic transdifferentiation [44, 45]. Similarly, neurons in nodose ganglia and ICNS also enlarge, but show an increased sympathetic or decreased cholinergic phenotype, respectively [46,47,48]. Lastly, parasympathetic withdrawal is also characterized by abnormal activity of vagal neurons [49] and structural and functional changes in the dorsal motor nucleus of the vagus nerve [48].
At the level of the heart, cardiac injury, especially a myocardial infarction, can result in degeneration of nerve fibers in both the scar and surrounding viable tissue [50,51,52,53]. Even though these nerves can regenerate, the extent to which this process develops occurs in a spatially heterogeneous manner, thereby inducing a condition wherein sympathetic hyperinnervation and denervation co-exist [50,51,52,53].
In animal and human studies, myocardial infarction results in structural and phenotypic changes in ICNS characteristics [46, 47]. Moreover, these phenotypic changes most likely reflect functional adaptations, as also observed in a porcine model of myocardial infarction [47]. In this model, myocardial infarction changed the activity pattern of afferent, processing and efferent neurons of the ICNS. Heterogeneous activation of the ICNS has also been shown to be pro-arrhythmic [54, 55], whereas blunting of this effect was antiarrhythmic [56,57,58] Additionally, in the presence of extracardiac remodeling, heterogeneous nerve sprouting and local autonomic denervation [51], heterogenous activity of the ICNS is most likely only further amplified. Hence, ICNS remodeling seems to significantly contribute to the establishment of an arrhythmia-susceptible condition.
Sympathetic modulation of myocardial electrical activity in the diseased heart
As mentioned, the autonomic nervous system modulates cardiac electrophysiology. It is thus unsurprising that neural remodeling, especially in the presence of diseased or injured myocardium, can adversely impact the electrical stability of the heart. In this way, overt sympathetic innervation can promote the development of arrhythmic triggers and/or increase the arrhythmic susceptibility of the myocardial substrate [59].
With regard to modulation of the arrhythmic trigger, early or delayed afterdepolarizations are promoted by sympathetic-induced changes in ion handling, mainly calcium. As mentioned above, sympathetic stimulation increases the systolic calcium concentration and simultaneously augments calcium loading in the sarcoplasmic reticulum [34]. The former can induce early afterdepolarizations through the reactivation of the L-type calcium channels during the prolonged phase 2 of the action potential [60]. The latter can predispose the myocyte to spontaneous calcium release from the sarcoplasmic reticulum and subsequent activation of the sodium-calcium exchanger, thereby triggering an early or delayed afterdepolarization (Fig. 2) [61]. In addition, stimulation of INa can prolong action potential duration and thereby predispose to the development of afterdepolarizations [62]. This risk for afterdepolarizations is especially increased during the initial phase of stimulation, as the slower increase in IK,s cannot completely counterbalance the faster effects on calcium handling. Hence, a biphasic effect of sympathetic stimulation can be observed, with an initial and temporary prolongation of action potential duration, which promotes the development of early afterdepolarizations [63, 64].
Second, sympathetic stimulation also promotes the development of re-entry. This effect becomes more pronounced during prolonged stimulation, when increases in IK,s cause the effective refractory period to become shortened. Moreover, it is not just the cellular electrical instability that promotes re-entry. At the tissue level, nerve sprouting and its heterogeneous nature might increase (transmural) dispersion of repolarization [65]. Especially in regions of hyperinnervation, overt release of NE can regionally exacerbate the aforementioned sympathetic-induced cellular instability [59, 66]. Moreover, in these regions of hyperinnervation, chronic overt sympathetic innervation can result in the downregulation of potassium channels and prolong action potential duration [67], which results in increased spatial dispersion of repolarization. On the contrary, local sympathetic hypo-innervation can promote spatial dispersion of repolarization and also result in super-sensitivity of the local β-adrenergic receptors, causing pro-arrhythmic conditions when exposed to catecholamines [68]. Correspondingly, following a myocardial infarction and associated neural remodeling, the myocardium has been reported to become more sensitive to circulating catecholamines, whereas the effects of direct nerve stimulation were decreased [69]. This was in contrast to healthy situations wherein direct nerve stimulation, rather than circulating catecholamines, increased spatial dispersion of repolarization [70].
Hence, sympathetic stimulation destabilizes cardiac electrophysiology at the cellular level and thereby predisposes the heart to the development of arrhythmic triggers, whilst simultaneously increasing the arrhythmic susceptibility of the myocardial substrate [59]. As such, cardiac autonomic imbalance strongly promotes the initiation and perpetuation of ventricular arrhythmias.
Interventions
As autonomic disbalance drives electrical instability of the myocardium and thereby predisposes to ventricular arrhythmias, antiarrhythmic neuromodulatory therapies aim to restore this balance. These interventions are primarily designed to decrease sympathetic efferent signaling and/or increase parasympathetic activity. This review has divided such interventions into three groups, based on their primary modulatory target. Therefore, neuromodulatory strategies are classified as modulating (i) the ICNS or myocytes, (ii) cardiac efferents or (iii) cardiac afferent signaling (and thus also affecting the integration of autonomic stimuli) (Fig. 3; Table 1).

Schematic overview of neuromodulatory interventions that are currently being developed or are already clinically implemented. Interventions are divided into three groups based on their respective primary target for modulation. The first group of interventions modulates the intrinsic cardiac nervous system (ICNS) or the myocytes and includes pharmacological inhibition of sympathetic overdrive, ICNS disruption or glial modulation. Glial modulation has been studied in the ICNS and in the stellate ganglia and is therefore connected to both. The second group (consisting of cardiac sympathetic denervation, stellate ganglia block, thoracic epidural anesthesia and (auricular) vagal nerve stimulation) directly modulate cardiac efferent and thereby either decrease sympathetic outflow or increase parasympathetic tone. The arrows next to the (auricular branch of the) vagal nerve depict the presence of afferent and/or efferent nerves. Thoracic epidural anesthesia affects cardiac efferents and afferents to a comparable extent and is therefore placed at the border of the second and the third group. Lastly, the third group (including spinal cord stimulation, carotid sinus stimulation and renal denervation) primarily modulates cardiac autonomic balance indirectly by changing cardiac afferent activity and thereby influencing the efferent outflow that is established in integration centers across the cardiac neuraxis
Modulation of cardiac neurons and myocytes
Pharmacological inhibition of the sympathetic nervous system
Ever since James Black introduced β-blockers as an antianginal drug in the 1950s, this group of sympathetic blocking drugs have revolutionized the pharmacological treatment of a plethora of cardiovascular diseases. Moreover, in the setting of ventricular arrhythmias, β-blockers are the only drugs that improve survival when used for either primary or secondary prevention of SCD [9], and are thus not surprisingly the cornerstone of antiarrhythmic treatment [7]. In addition, various inherited channelopathies (e.g. long QT syndrome, catecholaminergic polymorphic ventricular tachycardia) also rely on β-blockers as the first line of therapy in SCD prevention.
β-blockers are generally classified as either nonselective (with both β-1- and β-2-blocking properties, e.g. propranolol), or β-1-selective (i.e. bisoprolol) [7]. Even though β-2 and β-3 receptors can also be selectively targeted, such drugs are not currently used as an adjuvant therapy [9]. Nevertheless, it is important to note that β-blocking properties are not solely limited to the category of β-blockers (class II of the Vaughan-Williams classification), but are also (to a lesser extent) shared by class I and class III antiarrhythmic drugs, such as propafenone and amiodarone, respectively.
However, even at maximally tolerated doses, β-blocker therapy can be insufficient to suppress recurrent VAs [8]. As such, novel pharmacological strategies have been developed that impede the sympathetic effects on the heart by targeting neurotransmitters or neuropeptides other than NE. NPY in particular has emerged as a promising target for next-generation neuromodulatory pharmacological therapies. This sympathetic co-transmitter is released during high-level sympathetic stimulation and has various (cardiovascular) effects including vasoconstriction [71], parasympathetic inhibition [72] and cardiac electrical modulation [73] by increasing myocyte calcium loading [74]. Ajijola et al. [75] highlighted the possibly detrimental role of NPY in congestive heart failure, as they showed that elevated coronary sinus NPY levels (NPY ≥ 130 pg/mL) were significantly associated with a significantly increased risk in the composite endpoint of death, heart transplantation or implantation of a ventricular assist device [75]. Correspondingly, Kalla et al. [76] studied NPY (serum) levels in patients who underwent percutaneous coronary intervention. Their study showed that patients who suffered from recurrent VAs following the intervention had significantly higher levels of NPY [76]. Hence, even in the presence of β-blockade, NPY appears to destabilize cardiac electrical properties. These findings were corroborated by the observation that in isolated Langendorff perfused rat hearts, high-level stellate stimulation resulted in NPY release and calcium mishandling. The selective β1-blocker metoprolol could not reverse these effects, but adjuvant therapy with BIBO 3304, a NPY Y1-receptor antagonist, did effectively nullify these pro-arrhythmic conditions [76]. Similarly, Hoang et al. [73] used their in vivo porcine model to decipher the differing effects of the nonselective β-blocker propranolol and BIBO 3304. They showed that under conditions of high-frequency stimulation, propranolol could not reverse stimulation-induced changes in electrophysiological parameters. However, adjuvant therapy with BIBO 3304 resulted in stabilization of cardiac electrophysiology and positively augmented cardiac inotropy. Hence, blocking the effects of NPY on the heart and cardiac nervous system represents an interesting and novel (adjuvant) therapeutic target. Similarly, studies have looked into the possibility of blocking other sympathetic nervous system co-transmitters, including galanin [77] and dopamine [78]; however, these avenues await further studies.
ICNS disruption
As the ICNS represents both a relay station of higher centers within the cardiac neuraxis and an independent nervous system located on the heart (Fig. 1), it has a complex interaction with cardiac electrophysiology. Consequently, much of the relation between the ICNS, cardiac electrophysiology and arrhythmogenesis remain incompletely understood. Thus far, interventions directed to the ICNS have mainly been studied in the setting of atrial fibrillation (AF). For example, impeding ganglionated plexi activity through ablation [79] or botulinum toxin A injection [80, 81] was demonstrated to suppress or prevent AF occurrence, respectively. However, the opposite has also been reported [82]. Moreover, the involvement of the ICNS in the development and maintenance of ventricular arrhythmias has been even less explored. Interestingly, He et al. [83] subjected canines in the setting of an acute myocardial infarction to either ganglionated plexus ablation, or combined ganglionated plexus and stellate ganglion ablation. They showed that the incidence of ventricular fibrillation was significantly higher in animals with isolated ganglionated plexus ablation, suggesting a protective role of ganglionated plexi [83]. Hence, although ganglionated plexi affect cardiac electrophysiology and could theoretically potentiate ventricular arrhythmogenesis, their exact role of this process and their potential as a therapeutic target remain to be elucidated.
Glial cell modulation
Glia are non-neuronal cells that are found throughout the central and peripheral nervous system. These cells fulfill a plethora of functions to both support and modulate neuronal function. However, it has become increasingly clear that derangement of the reciprocal interaction between glia and neurons often contributes to the initiation, promotion and maintenance of (neurological) diseases [84].
Satellite glial cells
Satellite glial cells (SGCs), the peripheral counterparts of astrocytes, are increasingly of interest in the field of cardiac autonomics. They are found throughout the cardiac neuraxis where they envelope neurons, [85, 86] and most likely, dynamically influence synaptic activity [87, 88] Moreover, in patients with recurrent ventricular arrhythmia, SGCs have been demonstrated to be significantly enlarged and to have upregulated Gfap, a molecular marker of satellite glial cell reactivity. Hence, SGCs could be involved in the maintenance, or potentially even progression, of sympathetic overdrive. Correspondingly, Agulhon et al. (2013) [89] expressed a designer receptor exclusively activated by designer drug (DREADD) on SGCs, which allowed for artificial and selective SGC activation upon administration of clozapine-N-oxide (CNO). They demonstrated that SGC activation increased heart rate and blood pressure, which was later demonstrated to specifically result from SGC activation in the stellate ganglia [90]. Hence, SGC activation in stellate ganglia modulates local neuronal activity which translates to an increased sympathetic outflow to the heart. Therefore, impeding pathological glial activity might represent a promising future target of sympathetic modulation. Such targeted approaches appear feasible as they are already implemented in the central nervous system. Riluzole for example, a drug that inhibits glutamate neurotransmission by decreasing its presynaptic release and facilitating its uptake by glial cells, is already being used in the treatment of amyotrophic lateral sclerosis [91] and has been demonstrated to be effective in the treatment of Alzheimer's disease [92] and various psychiatric disorders in which glia (including astrocytes) are fundamental players in the pathophysiology [93]. Hence, translating such glial modulatory treatments to the peripheral nervous system might expand the boundaries of current cardiovascular treatment options.
In addition, satellite glial cells are also abundantly present within the intrinsic cardiac nervous system, [94] and have been demonstrated to become injured upon catheter ablation of atrial fibrillation [95]. Interestingly, higher serum concentrations of S100B after catheter ablation of atrial fibrillation were associated with a lower recurrence rate [95]. As S100B can induce dendrite outgrowth of myocardial nerves and can also decrease neuronal activity, [96] ablation might induce glial-modulated neural remodeling [95]. It remains unknown if and how ablation of ventricular substrates affects glia. Hence, more research is warranted into the mechanism by which glial cells of the ICNS influence neural remodeling and how their role in neural modulation might translate to anti-arrhythmic therapies.
Microglia
Lastly, microglia are also emerging as possible participants in various cardiovascular diseases. Also in the setting of recurrent ventricular arrhythmias, inflammatory processes were active in stellate ganglia, which might imply that microglia and other inflammatory cells contribute to this pathological phenotype. Though the role of microglia in ventricular arrhythmogenesis has been scarcely studied, Wang et al. 187 [97] used a post-infarct murine model to demonstrate their involvement in ventricular arrhythmogenesis. In their study, microglial activation was inhibited through light-emitting diode (LED) illumination, a fairly new non-thermal method that modulates cellular activity through photons that are absorbed by mitochondrial chromophores [98]. In their study, they compared the arrhythmic outcome between control mice and mice in which microglia had been kept inactivated following myocardial infarction. They demonstrated that LED illumination resulted in significant decreases in neural activity in the left stellate ganglion and associated this effect with the observed reduction in microglial activity and reduced expression of pro-inflammatory cytokines [97]. Moreover, these effects decreased incidence of VAs significantly, which further highlighted the potential role and therapeutic potential of microglia in ventricular arrhythmias [97].
Modulation of cardiac efferents
Cardiac sympathetic denervation
Even though increasing antiarrhythmic drugs to their maximally tolerated dose and catheter substrate ablation comprise the central steps in treatment of ventricular arrhythmias, a subset of patients suffer from recurrent ventricular tachycardia despite these treatment modalities [99, 100]. Cardiac sympathetic denervation (CSD), the surgical disruption of neural transmission within the left stellate ganglion or bilateral stellate ganglia, has emerged as an effective procedure for this patient population. Even though this strategy is still highly restricted to a few “expertise centers,” the number and size of clinical studies is ever expanding. The success of this treatment modality was reconfirmed in a meta-analysis by Murtaza et al. [101] which combined the results of 14 retrospective clinical studies that included a total of 311 patients with recurrent ventricular tachycardia or electrical storm. Their analyses showed that CSD resulted in complete abolishment of ventricular tachycardia in approximately 60% of treated patients (average follow-up time: 15 ± 10.7 months) and a significant decrease of 3.01 in mean implantable cardioverter-defibrillator (ICD) shocks [101]. Importantly, these results of CSD efficacy are similar between patients with and without an ischemic substrate underlying their ventricular arrhythmias [102, 103]. In addition, a recent study by Assis et al. [104], which represents the longest follow-up study on bilateral CSD thus far, reported ventricular tachycardia-free survival in 54.5% of the patients at 4 years. Importantly, this study also demonstrated that early recurrence (≤ 12 weeks) of ventricular tachycardia after bilateral CSD does not correlate with the long-term antiarrhythmic outcome of this intervention.
CSD has also been demonstrated to be an appropriate and effective antiarrhythmic strategy in the setting of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia [105].
Nevertheless, not all patients treated with CSD benefit from this intervention. There are various reasons for this undesirable outcome. For example, arrhythmic episodes might not be modulated by the sympathetic nervous system, and therefore CSD will not abolish the pro-arrhythmic factor. In addition, procedural differences in CSD might yield different outcomes. For example, bilateral CSD appears more effective in patients with either an ischemic substrate [106] or a non-ischemic (including cardiac sarcoidosis) cardiomyopathy [107]. whereas isolated left-sided CSD is effective and even preferred in the setting of long QT syndrome (type 1, 2 or 3) [108, 109] or catecholaminergic polymorphic ventricular tachycardia [110]. Moreover, the extent to which the sympathetic chain needs to be resected is also under much discussion. Whereas the initial reports and CSD went as low as level T7 and T8 [111, 112], current practice commonly resects the lower half of the stellate ganglia, down to the level of T4. Though not studied in the clinical setting yet, evidence from animal studies suggests that selected CSD to the level of T2 might be sufficient to be antiarrhythmic [113]. Nevertheless, interindividual variation in origin of cardiac sympathetic innervation might cause this approach to be less effective in the clinical setting [114].
Hence, even though CSD is a great alternative to current antiarrhythmic therapies and has already been successfully used to treat many patients with recurrent VAs, there are still possibilities for improvement. For example, as CSD has only been a clinical modality for recurrent VAs for a limited period, long-term results are yet to be published. As such, it is largely unknown yet whether reinnervation of postganglionic nerves, as for example observed following heart transplantation, might induce arrhythmic or other cardiac side effects after the passage of time [115]. Moreover, all trials thus far haven been prospective in character; hence, randomized controlled trials would greatly add to the evidence of CSD's success and could further elucidate the difference between left or bilateral cardiac denervation.
Stellate ganglion blockade
Whereas CSD is permanent and rather invasive, sympathetic outflow from stellate ganglia to the heart can also be interrupted via local pharmacological stellate ganglion blockade (SGB). Through a percutaneous approach in conscious patients, sodium channel blockers (i.e. bupivacaine, lidocaine) can be injected into the stellate ganglia to temporarily suppress nerve transmission in that restricted area. This temporarily block can suppress arrhythmias and thus serve as a bridge to longer lasting therapies, such as substrate ablation, CSD or heart transplantation [116]. Injection with these drugs effectively suppress drug-refractory electrical storm in over half of the patients treated (regardless of the intervention being directed to both stellate ganglia or only the left stellate ganglion) [117]. However, in order to become a more widespread implemented strategy, SGB still has to overcome several difficulties. For one, though SGB is commonly accepted as a successful and safe procedure, SGB can cause various complications. Neck hematomas and transient hoarseness (caused by the accidental block of the recurrent laryngeal nerve or a large neck hematoma) are the most common complications of SGB [118]. However, also local anesthetic systemic toxicity, a rare but life-threatening adverse event that is caused by too high plasma levels of local anesthetics, can occur [118]. Nevertheless, these complications are rare as SGB is most often guided by ultrasound or performed under fluoroscopic guidance. Moreover, it remains difficult to determine successful SGB block. Various markers of successful blockade have been proposed, such as a temperature rise in the ipsilateral arm, presence of Horner's syndrome (ipsilateral ptosis, myosis and anhidrosis resulting from impeded sympathetic innervation to the eye and ocular adnexae) and the perfusion index [118,119,120,121]. Although out of these three parameters the perfusion index has been established to be best, none of these three modalities is sufficiently sensitive in reflecting successful SGB [118,119,120,121]. Hence, for SGB to become a more customary strategy in the acute treatment of refractory VAs, more reliable and robust technical parameters will have to be developed that adequately confirm SGB.
Thoracic epidural anesthesia (TEA)
The injection of anesthetics in the high thoracic epidural space causes temporal and local nerve block, which has been repeatedly demonstrated to be an effective approach in acutely reducing the incidence of ventricular tachycardia and bridging to more permanent antiarrhythmic interventions [116, 117, 122, 123]. The efficacy of this approach has been attributed to multiple factors, including complete and bilateral blockade of all cardiac afferents and sympathetic efferents, as it blocks all nerves in segments C8–T4. With regard to ventricular electrical activity, this complete nerve block impedes arrhythmogenesis by lengthening repolarization duration and prolonging the effective refractory period [124, 125]. Moreover, since SGB impedes efferent sympathetic signaling more proximal than β-blockers, this strategy might be especially beneficial in patients with structural heart disease in which the myocardial substrate can be less receptive to pharmacological modulation [116].
Even though TEA can robustly suppress VA in the acute setting, this technique still requires some refinement. For one, reliable parameters indicative of effective TEA are needed. Moreover, many clinicians remain reluctant to perform TEA, as high thoracic anesthesia could theoretically impede cardiac pump function. Nevertheless, the opposite was demonstrated by Wink et al. [126], who subjected patients with right and/or left ventricular dysfunction to ergometric testing before and after TEA, and showed that TEA did not significantly affect or diminish their exercise-induced increases in pump function [126]. Similarly, clinical case series reported minimal effects of TEA on hemodynamic function [116, 122, 123]. However, Wink et al. [127] also demonstrated that high thoracic anesthesia diminished right ventricular systolic function. Hence, additional studies into the mechanical and hemodynamic consequences of TEA are warranted, as this technique might represent a highly effective antiarrhythmic strategy that is currently undervalued.
Vagal nerve stimulation
Vagal nerve stimulation (VNS) was primarily developed as a treatment modality for heart failure, but can also be applied in other conditions that are similarly characterized by sympathetic overdrive. Through the continuous stimulation of the parasympathetic nervous system, sympathetic overdrive is mitigated. This strategy has also been effective in the setting of preclinical models of ventricular arrhythmias, as it stabilizes cardiac electrophysiology. For instance, recruitment of IK,Ach and the subsequent shortening of repolarization duration might impede the development of early afterdepolarizations, and VNS has been demonstrated to increase the ventricular fibrillation threshold [128, 129]. Moreover, preclinical studies have highlighted the efficacy of VNS in reducing arrhythmic episodes and SCD in the setting of acute and chronic ischemia [130,131,132] and in mitigating pro-arrhythmic effects of left stellate ganglion stimulation [133].
Hence, even though VNS exerts multiple beneficial effects on the myocardium in preclinical studies, it has proven to be rather difficult to corroborate these findings in clinical studies. Especially since clinical studies for VNS were primarily designed to study its effects on heart failure progression, few clinical data are available on the effects of VNS on ventricular arrhythmias. Moreover, conflicting results of these clinical trials on the efficacy of VNS in heart failure patients [134] has constrained efforts towards further studies exploring the clinical applicability of VNS for other cardiovascular diseases. Nevertheless, these conflicting and confusing outcomes of the clinical studies were most likely not a result of VNS inefficacy, but rather a methodological problem caused by inappropriate stimulation protocols. In particular, the unsolicited stimulation of afferent vagal nerves and subsequent activation of sympathetic efferents could have induced the disappointing results. The differences in stimulation parameters and their possible effects on clinical outcome have been extensively reviewed elsewhere [135].
Nevertheless, the ANTHEM II trial, which reported a beneficial effect of VNS on heart failure, also studied the electrical effects of VNS during a 3-year follow-up and observed a reduction in the incidence of arrhythmic episodes in their cohort of patients [136, 137]. Hence, though rudimentary and still insufficiently refined, VNS remains a promising candidate for future heart failure and antiarrhythmic purposes.
In addition to the inexperience with VNS, its invasive character is also a drawback of this therapy. Therefore, the possibility of transcutaneous VNS through stimulation of the auricular branch of the vagus nerve is currently being explored. Stimulation of this branch results in centrally mediated increases in vagal efferent activity [138, 139]. This modality has already proven to be effective in suppressing ventricular arrhythmias in canine models of myocardial infarction [140, 141] and has exerted antiarrhythmic effects in the setting of acute myocardial infarctions in humans [142]. In addition, auricular VNS could possibly be more effective than conventional VNS, as the aforementioned adverse activation of vagal afferent is circumvented by this approach.
Future preclinical and clinical studies should therefore aim to optimize the stimulation parameters, to identify markers reflective of optimal stimulation and gear their therapeutic approach towards less invasive modalities.
Modulation of cardiac afferents and integration centers of the cardiac neuraxis
Spinal cord stimulation
As with many other neuromodulatory therapies, spinal cord stimulation (SCS) was initially developed to treat angina pectoris [143, 144]. Though not completely understood, local stimulation of the spinal cord affects various central and local neural circuits, which collectively impede the process of arrhythmogenesis [145,146,147,148,149,150,151,152,153]. For instance, SCS impedes the ability of afferent cardiac nerves in transferring their signal to second-order spinal cord neurons, thereby hampering the initiation of a sympathetic reflex and the development of sympathetically driven remodeling processes that are induced by myocardial ischemia [146, 147]. In addition, SCS stabilizes the ICNS and decreases left stellate ganglion activity, all of which could contribute to its antiarrhythmic effects [148, 149, 154].
Another benefit of SCS is that it is a relatively fast-acting antiarrhythmic therapy, as it has been demonstrated to be effective within 1 hour in animal models of acute myocardial ischemia [152, 155]. However, even though the preclinical success of SCS in animal studies was corroborated by an initial case series, which reported a significant reduction in arrhythmic burden in the two included patients [156], two consecutive larger clinical trials observed contrasting effects on reduction of arrhythmic episodes in patients treated with SCS [157, 158]. This inconsistency in results could be attributable to a variety of causes, including differences in SCS parameters (either “continuous and optimally programmed as determined by the physician” or with a frequency of 50 Hz, a pulse width of 200 µs and continuously paced at 90% of maximum tolerated voltage) and anatomical location of pacing (C6 vs. T2-T4) [157, 158]. Hence, SCS shows promise as an antiarrhythmic strategy in the setting of both acute and chronic cardiac injury, but insufficient knowledge on the physiology and interplay between neural centers and optimal location and stimulation parameters of SCS are impeding its advances into clinical applicability.
Carotid sinus stimulation
Carotid sinus stimulation aims to modulate the baroreflex. Normally, afferent nerves in the carotid sinus and aortic arch transmit mechanical information on arterial blood pressure to the central nervous system, from where negative feedback is exerted by adapting the relative activity of the cardiac sympathetic and parasympathetic branch. However, chronically increased sympathetic tone causes this reflex to become depressed, resulting in inadequate coordination of sympatho-vagal balance. Therefore, carotid sinus stimulation aims to restore baroreflex sensitivity through artificial activation of baroreceptor afferents, which should cause central stimulation of parasympathetic efferents and inhibition of sympathetic efferents. This elegant tactic to indirectly restore cardiac autonomic tone has been performed in multiple preclinical studies and has been demonstrated to effectively reduce ventricular arrhythmias [159,160,161]. However, no clinical trials have studied the antiarrhythmic efficacy of this approach in humans. Nevertheless, case series have demonstrated that carotid sinus stimulation can effectively decrease blood pressure in patients suffering from hypertension, an outcome that was attributed to its mitigating effect on the sympathetic nervous system [162,163,164]. Hence, even though this approach seems effective, feasible and safe, more studies are warranted to expand (clinical) experience on its applicability in the setting of (recurrent) ventricular arrhythmias and to promote its clinical implementation.
Renal denervation
Disrupting renal nerves impedes sympathetic overactivation on a more systemic level. Even though it was first performed in 1953 as a treatment for primary hypertension [165], sympathetic overdrive contributes to various other cardiovascular diseases and could thus be an adequate approach for a wider range of indications, including ventricular arrhythmias. Multiple preclinical studies have demonstrated the antiarrhythmic effects of renal denervation. For example, in pigs subjected to 20 min of left anterior descending coronary artery (LAD) occlusion, renal denervation exerted acute antiarrhythmic effects [166], and in animal models of both ischemic and non-ischemic cardiac injury, disruption of renal autonomic nerves prevented pro-arrhythmic neurohumoral cardiac remodeling [167,168,169]. Unfortunately, the disappointing results of the SYMPLICITY HTN-3 trial [170], the first sham-controlled clinical trial on the efficacy of renal nerve denervation for hypertension, greatly diminished clinical interest in this strategy. The additional critiques on the trial's experimental design and incomplete execution of renal denervation have further discredited this strategy and greatly impeded its progression [171,172,173]. Consequently, clinical data on antiarrhythmic effects of renal denervation are also scarce. Nevertheless, the clinical results that have been published thus far seem promising [174,175,176,177,178,179,180]. For example, Remo et al. [175] showed that renal denervation effectively decreased ventricular tachycardia episodes in patients with ischemic and non-ischemic cardiomyopathies who were suffering from refractory ventricular tachycardia. Similarly, Ukena et al. [177] combined 13 cases of patients suffering from chronic heart failure who had undergone renal denervation and showed that this intervention was associated with a significant reduction in arrhythmic burden. Correspondingly, Tsioufis et al. [176] showed the beneficial effect of renal denervation on reducing the occurrence of premature ventricular contractions in patients with drug-resistant and uncontrolled hypertension. Hence, renal denervation positively affects cardiac electrophysiology and exerts an antiarrhythmic effect in a wide range of cardiovascular patients. Future preclinical studies should aim to expand our understanding of the physiology underlying the antiarrhythmic efficacy of renal denervation and to establish the long-term outcome of this approach. Moreover, the ability of renal denervation to reverse pro-arrhythmic remodeling remains to be elucidated, and further investigations validating and strengthening the applicability of this treatment are warranted.
Emerging modalities of neural modulation
Thus far, this review has discussed neuromodulatory interventions that are already clinically implemented or currently progressing towards clinical application. However, novel methods of neuromodulation are continuously being developed as knowledge on the neural–cardiac interplay rapidly grows in parallel with technological advances. Therefore, this last section will briefly elaborate on interesting, novel avenues of neuromodulatory interventions that modify neural activity through the employment of magnetic or ultrasound stimulation.
Transcranial magnetic stimulation is a neuromodulatory technique that is already clinically used for pain and depression, and has much clinical potential for various other (neurological) disorders [181]. Moreover, this intervention has been observed to affect cardiac rhythm [182] and heart rate variability [183], which indicates its ability to modulate cardiac autonomic tone. Though therapeutically appealing, this technique has thus far not been studied as an antiarrhythmic therapeutic option. Nevertheless, such refinement is made difficult by various confounding factors in clinical neuromodulatory research. For example, aging, comorbidities and interindividual variation in nervous system anatomy and/or functionality result in considerable variations in neuromodulatory response. Although these confounding variables are challenging, current research on modulatory therapies is progressing rapidly.
Electromagnetic stimulation of the ICNS or vagosympathetic nerve trunks were previously demonstrated to effectively suppress atrial fibrillation through modulation of cardiac autonomic balance [184, 185]. Wang et al. [186] extrapolated this strategy to suppressing ventricular arrhythmias through low-frequency electromagnetic stimulation of the left stellate ganglia in canines with acute myocardial infarction. They showed that this strategy significantly decreased left stellate ganglion activity and reduced myocardial infarction-induced ventricular arrhythmia burden. Markman et al. [187] published a case series on five patients suffering from ventricular tachycardia storm (≥ 3 episodes of sustained ventricular tachycardia in 24 h), which were all treated with transcutaneous magnetic stimulation of the left stellate ganglion. This strategy successfully lowered arrhythmia burden and was not associated with any adverse events, further highlighting the potential of this modality to serve as a bridge to more permanent interventions. However, further (pre)clinical studies are warranted to further optimize the understanding of this approach.
Lastly, Wang et al. [188] recently reported on the possibility of using low-level ultrasound stimulation of the left stellate ganglion to reduce arrhythmic burden. In their study, dogs were exposed to 10 min of low-level ultrasound stimulation prior to the establishment of a myocardial infarction. This pretreatment was shown to possibly inhibit neural activity in the left stellate ganglion and to thereby reduce myocardial infarction-induced ventricular arrhythmias [188]. Though promising, this abstract is the first publication on the efficacy and applicability of this strategy.
It is clear that much more research is needed to advance these modalities to the clinical setting. Nevertheless, with the current pace of device development and progression, the implementation of these stimulatory techniques comparable to that of pacemakers is conceivable. Therefore, the subcutaneous implantation of a stimulatory device could be an excellent treatment option in the outpatient setting.
Conclusion
Much research has elucidated the elegance of the cardiac neuraxis and its intricate involvement in cardiac physiology. Moreover, its detrimental role in cardiovascular diseases and ventricular arrhythmogenesis is becoming increasingly clear, which has already resulted in targeted therapies for most levels of the cardiac neuraxis. Even though all these strategies appear very promising, there is still much room to gain in the treatment of these possibly fatal VAs. For example, there is much to learn about how central integration centers and the ICNS control sympatho-vagal balance and how antiarrhythmic therapies can target these structures more specifically. Additionally, even though many neuromodulatory interventions have been developed, most of these interventions would greatly benefit from additional refinement (e.g. identification of robust and reliable parameters of successful neuromodulation and/or optimal stimulatory parameters). Nevertheless, such refinement is made difficult by various confounding factors in clinical neuromodulatory research. For example, aging, comorbidities and interindividual variation in nervous system anatomy and/or functionality result in much variation in neuromodulatory response. Although these confounding variables are challenging, current research on modulatory therapies is progressing at a fast pace.
Lastly, the relatively young age of this research field has not allowed for long-term follow-up studies. Therefore, chronic effects of these therapies are yet to be elucidated. Nevertheless, current research is well on its way to fill these gaps and to further install neuromodulation as a widely implemented antiarrhythmic strategy.
References
Goldberger JJ, Arora R, Buckley U, Shivkumar K (2019) Autonomic nervous system dysfunction: JACC focus seminar. J Am Coll Cardiol 73:1189–1206
Ardell JL, Armour JA (2016) Neurocardiology: structure-based function. Comprehensive physiology 6. John Wiley & Sons Inc., London, pp 1635–1653
Jänig W (2014) Sympathetic nervous system and inflammation: a conceptual view. Auton Neurosci Basic Clin 182:4–14
Fukuda K, Kanazawa H, Aizawa Y, Ardell JL, Shivkumar K (2015) Cardiac innervation and sudden cardiac death. Circ Res 116:2005–2019
Ardell JL et al (2016) Translational neurocardiology: preclinical models and cardioneural integrative aspects. J Physiol 594:3877–3909
Shivkumar K, Ardell JL (2016) Cardiac autonomic control in health and disease. J Physiol 594:3851–3852
Grandi E, Ripplinger CM (2019) Antiarrhythmic mechanisms of beta blocker therapy. Pharmacol Res 146:104274
Chatzidou S et al (2018) Propranolol versus metoprolol for treatment of electrical storm in patients with implantable cardioverter-defibrillator. J Am Coll Cardiol 71:1897–1906
Al-Khatib SM et al (2018) 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American college of cardiology/American heart association task force on clinical practice guidelines and heart rhythm society. J Am Coll Cardiol 138:e210–e271
Shivkumar K et al (2016) Clinical neurocardiology defining the value of neuroscience-based cardiovascular therapeutics. J Physiol 594:3911–3954
Kapa S, DeSimone C, Asirvatham SJ (2016) Innervation of the heart: an invisible grid within a black box. Trends Cardiovasc Med 26:245
Chatterjee NA, Singh JP (2015) Novel interventional therapies to modulate the autonomic tone in heart failure. JACC Hear Fail 3:786–802
Waldron NH, Fudim M, Mathew JP, Piccini JP (2019) Neuromodulation for the treatment of heart rhythm disorders. JACC Basic Transl Sci 4:546
Choy L, Yeo JM, Tse V, Chan SP, Tse G (2016) Cardiac disease and arrhythmogenesis: mechanistic insights from mouse models. IJC Heart Vasc 12:1–10
Mitchell GF, Jeron A, Koren G (1998) Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol-Hear Circ Physiol 274:747–751
Boukens BJ, Rivaud MR, Rentschler S, Coronel R (2014) Misinterpretation of the mouse ECG: ‘musing the waves of Mus musculus.’ J Physiol 592:4613–4626
Janssen BJA, Smits JFM (2002) Autonomic control of blood pressure in mice: basic physiology and effects of genetic modification. Am J Physiol-Regul Integr Comp Physiol. https://doi.org/10.1152/ajpregu.00714.2001
London B (2001) Cardiac arrhythmias: from (transgenic) mice to men. J Cardiovasc Electrophysiol 12:1089–1091
Kaese S et al (2013) The ECG in cardiovascular-relevant animal models of electrophysiology. Herzschrittmacherther Elektrophysiol 24:84–91
Loen V, Vos MA, Heyden MAG (2021) The canine chronic atrio-ventricular block model in cardiovascular preclinical drug research. Br J Pharmacol. https://doi.org/10.1111/bph.15436
Billman GE (2006) A comprehensive review and analysis of 25 years of data from an in vivo canine model of sudden cardiac death: implications for future anti-arrhythmic drug development. Pharmacol Ther 111:808–835
Moïse NS, Flanders WH, Pariaut R (2020) Beat-to-beat patterning of sinus rhythm reveals non-linear rhythm in the dog compared to the human. Front Physiol. https://doi.org/10.3389/fphys.2019.01548
Hamlin RL (2007) Animal models of ventricular arrhythmias. Pharmacol Ther 113:276–295
Tohyama S, Kobayashi E (2019) Age-appropriateness of porcine models used for cell transplantation. Cell Transplant 28:224–228
Hadaya J, Ardell JL (2020) Autonomic Modulation for Cardiovascular Disease. Front Physiol 11:617459
Randall WC (1994) Efferent sympathetic innervation of the heart” in neurocardiology. Oxford University Press, New York
Armour JA, Ardell JL (2004) Basic and clinical neurocardiology. Oxford University Press
Jänig W (2006) The integrative action of the autonomic nervous system: neurobiology of homeostasis. The integrative action of the autonomic nervous system: neurobiology of homeostasis. Cambridge University Press
Hopkins DA, Bieger D, De Vente J, Steinbusch HWM (1996) Vagal efferent projections: viscerotopy, neurochemistry and effects of vagotomy. Prog Brain Res 107:79–96
Pauza DH et al (2014) A combined acetylcholinesterase and immunohistochemical method for precise anatomical analysis of intrinsic cardiac neural structures. Ann Anat 196:430–440
Armour JA, Collier K, Kember G, Ardell JL (1998) Differential selectivity of cardiac neurons in separate intrathoracic autonomic ganglia. Am J Physiol Integr Comp Physiol 274:R939–R949
Wink J et al (2020) Human adult cardiac autonomic innervation: Controversies in anatomical knowledge and relevance for cardiac neuromodulation. Auton Neurosci Basic Clin 227:102674
Herring N (2015) Autonomic control of the heart: going beyond the classical neurotransmitters. Exp Physiol 100:354–358
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415:198–205
Walsh KB, Kass RS (1991) Distinct voltage-dependent regulation of a heart-delayed I(K) by protein kinases A and C. Am J Physiol-Cell Physiol. https://doi.org/10.1152/ajpcell.1991.261.6.C1081
Walsh KB, Kass RS (1988) Regulation of a heart potassium channel by protein kinase A and C. Science 242:67–69
Bartos DC, Grandi E, Ripplinger CM (2015) Ion channels in the heart. Compr Physiol 5:1423–1464
Grandi E et al (2017) Potassium channels in the heart: structure, function and regulation. J Physiol 595:2209–2228
Herren AW, Bers DM, Grandi E (2013) Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol-Heart Circ Physiol. https://doi.org/10.1152/ajpheart.00306.2013
Ng GA, Brack KE, Patel VH, Coote JH (2007) Autonomic modulation of electrical restitution, alternans and ventricular fibrillation initiation in the isolated heart. Cardiovasc Res 73:750–760
Ajijola OA et al (2017) Sympathetic modulation of electrical activation in normal and infracted myocardium: Implications for arrhythmogenesis. Am J Physiol-Hear Circ Physiol. 312:H608–H621
Tomson TT, Arora R (2016) Modulation of cardiac potassium current by neural tone and ischemia. Card Electrophysiol Clin 8:349–360
Ajijola OA et al (2017) Inflammation, oxidative stress, and glial cell activation characterize stellate ganglia from humans with electrical storm. JCI Insight 2:e94715
Olivas A et al (2016) Myocardial infarction causes transient cholinergic transdifferentiation of cardiac sympathetic nerves via gp130. J Neurosci 36:479–488
Wang L et al (2020) Cardiac sympathetic nerve transdifferentiation reduces action potential heterogeneity after myocardial infarction. Am J Physiol Heart Circ Physiol 318:H558–H565
Hopkins DA, Macdonald SE, Murphy DA, Armour JA (2000) Pathology of intrinsic cardiac neurons from ischemic human hearts. Anat Rec 259:424–436
Rajendran PS et al (2016) Myocardial infarction induces structural and functional remodelling of the intrinsic cardiac nervous system. J Physiol 594:321–341
Salavatian S et al (2017) Myocardial infarction causes both structural and functional remodeling in cardiac neurons of the inferior vagal (nodose) ganglia: implications for mechanisms behind parasympathetic withdrawal in heart disease. Circulation 136:A17355
Vaseghi M et al (2017) Parasympathetic dysfunction and antiarrhythmic effect of vagal nerve stimulation following myocardial infarction. JCI Insight 2:e86715
Yokoyama T et al (2017) Quantification of sympathetic hyperinnervation and denervation after myocardial infarction by three-dimensional assessment of the cardiac sympathetic network in cleared transparent murine hearts. PLoS One. https://doi.org/10.1371/journal.pone.0182072
Cao JM et al (2000) Nerve sprouting and sudden cardiac death. Circ Res 86:816–821
Li W, Knowlton D, Van Winkle DM, Habecker BA (2004) Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Circ Physiol 286:H2229–H2236
Lorentz CU et al (2013) Sympathetic denervation of peri-infarct myocardium requires the p75 neurotrophin receptor. Exp Neurol 249:111–119
Armour JA et al (2005) Origin and pharmacological response of atrial tachyarrhythmias induced by activation of mediastinal nerves in canines. Auton Neurosci 118:68–78
Beaumont E et al (2013) Network interactions within the canine intrinsic cardiac nervous system: Implications for reflex control of regional cardiac function. J Physiol 591:4515–4533
Richer L-P et al (2008) α-Adrenoceptor blockade modifies neurally induced atrial arrhythmias. Am J Physiol Integr Comp Physiol 295:R1175–R1180
Gibbons DD et al (2012) Neuromodulation targets intrinsic cardiac neurons to attenuate neuronally mediated atrial arrhythmias. Am J Physiol Integr Comp Physiol 302:R357–R364
Ardell JL et al (2014) Chronic spinal cord stimulation modifies intrinsic cardiac synaptic efficacy in the suppression of atrial fibrillation. Auton Neurosci Basic Clin 186:38–44
Shen MJ, Zipes DP (2014) Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res 114:1004–1021
Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z (2010) Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm 7:1891–1899
Bers DM (2008) Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70:23–49
Clancy CE, Rudy Y (1999) Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature 400:566–569
Meijborg VMF et al (2020) Stellate ganglion stimulation causes spatiotemporal changes in ventricular repolarization in pig. Heart Rhythm 17:795–803
Xie Y, Grandi E, Puglisi JL, Sato D, Bers DM (2013) β-adrenergic stimulation activates early afterdepolarizations transiently via kinetic mismatch of PKA targets. J Mol Cell Cardiol 58:153–161
Jiang H et al (2007) Relationship between sympathetic nerve sprouting and repolarization dispersion at peri-infarct zone after myocardial infarction. Auton Neurosci Basic Clin 134:18–25
Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM (2012) Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res 110:1454–1464
Aflaki M et al (2014) Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained β-adrenergic activation in guinea pig hearts. Circ Res 114:993–1003
Gardner RT et al (2015) Targeting protein tyrosine phosphatase σ after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nat Commun. https://doi.org/10.1038/ncomms7235
Tapa S et al (2020) Adrenergic supersensitivity and impaired neural control of cardiac electrophysiology following regional cardiac sympathetic nerve loss. Sci Rep. https://doi.org/10.1038/s41598-020-75903-y
Yagishita D et al (2015) Sympathetic nerve stimulation, not circulating norepinephrine, modulates T-peak to T-end interval by increasing global dispersion of repolarization. Circ Arrhythmia Electrophysiol 8:174–185
Malmström RE (2002) Pharmacology of neuropeptide Y receptor antagonists: focus on cardiovascular functions. Eur J Pharmacol 447:11–30
Herring N, Lokale MN, Danson EJ, Heaton DA, Paterson DJ (2008) Neuropeptide Y reduces acetylcholine release and vagal bradycardia via a Y2 receptor-mediated, protein kinase C-dependent pathway. J Mol Cell Cardiol 44:477–485
Hoang JD, Salavatian S, Yamaguchi N, Swid MA, Vaseghi M (2020) Cardiac sympathetic activation circumvents high-dose beta blocker therapy in part through release of neuropeptide Y. JCI Insight. https://doi.org/10.1172/jci.insight.135519
Heredia MDP et al (2005) Neuropeptide Y rapidly enhances [Ca 2+] i transients and Ca 2+ sparks in adult rat ventricular myocytes through Y 1 receptor and PLC activation. J Mol Cell Cardiol 38:205–212
Ajijola OA et al (2020) Coronary sinus neuropeptide y levels and adverse outcomes in patients with stable chronic heart failure. JAMA Cardiol 5:318–325
Kalla M et al (2020) The cardiac sympathetic co-transmitter neuropeptide Y is pro-arrhythmic following ST-elevation myocardial infarction despite beta-blockade. Eur Heart J 41:2168–2179
Herring N et al (2012) The cardiac sympathetic co-transmitter galanin reduces acetylcholine release and vagal bradycardia: Implications for neural control of cardiac excitability. J Mol Cell Cardiol 52:667–676
Yamaguchi T et al (2020) Cardiac dopamine D1 receptor triggers ventricular arrhythmia in chronic heart failure. Nat Commun 11:1–8
Katritsis DG et al (2013) Autonomic denervation added to pulmonary vein isolation for paroxysmal atrial fibrillation: a randomized clinical trial. J Am Coll Cardiol 62:2318–2325
Romanov A et al (2019) Long-term suppression of atrial fibrillation by botulinum toxin injection into epicardial fat pads in patients undergoing cardiac surgery: three-year follow-up of a randomized study. Heart Rhythm 16:172–177
Waldron NH et al (2019) Temporary autonomic modulation with botulinum toxin type A to reduce atrial fibrillation after cardiac surgery. Heart Rhythm 16:178–184
Lo LW et al (2013) Paradoxical long-term proarrhythmic effects after ablating the head station ganglionated plexi of the vagal innervation to the heart. Heart Rhythm 10:751–757
He B et al (2013) Effects of ganglionated plexi ablation on ventricular electrophysiological properties in normal hearts and after acute myocardial ischemia. Int J Cardiol 168:86–93
Valles SL et al (2019) Function of glia in aging and the brain diseases. Int J Med Sci 16:1473–1479
Hanani M (2010) Satellite glial cells: more than just rings around the neuron. Neuron Glia Biol 6:1–2
Hanani M, Spray DC (2020) Emerging importance of satellite glia in nervous system function and dysfunction. Nat Rev Neurosci 21:485–498
Feldman-Goriachnik R, Wu B, Hanani M (2018) Cholinergic responses of satellite glial cells in the superior cervical ganglia. Neurosci Lett 671:19–24
Enes J et al (2020) Satellite glial cells modulate cholinergic transmission between sympathetic neurons. PLoS One 15:e0218643
Agulhon C et al (2013) Modulation of the autonomic nervous system and behaviour by acute glial cell G q protein-coupled receptor activation in vivo. J Physiol 591:5599–5609
Xie AX, Lee JJ, McCarthy KD (2017) Ganglionic GFAP+ glial Gq-GPCR signaling enhances heart functions in vivo. JCI Insight 2:e90565
Bensimon G, Lacomblez L, Meininger V (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med 330:585–591
Vallée A, Vallée JN, Guillevin R, Lecarpentier Y (2020) Riluzole: a therapeutic strategy in Alzheimer’s disease by targeting the WNT/β-catenin pathway. Aging (Albany NY) 12:3095–3113
Pittenger C et al (2008) Riluzole in the treatment of mood and anxiety disorders. CNS Drugs 22:761–786
Hanna P et al (2020) Innervation and neuronal control of the mammalian sinoatrial node: a comprehensive atlas. bioRxiv. https://doi.org/10.1101/2020.10.28.359109
Scherschel K et al (2019) Cardiac glial cells release neurotrophic S100B upon catheter-based treatment of atrial fibrillation. Sci Transl Med 11:7770
Sahoo N, Tröger J, Heinemann SH, Schönherr R (2010) Current inhibition of human EAG1 potassium channels by the Ca2+ binding protein S100B. FEBS Lett 584:3896–3900
Wang S et al (2019) Light-emitting diode therapy protects against ventricular arrhythmias by neuro-immune modulation in myocardial ischemia and reperfusion rat model. J Neuroinflammation 16:1–10
Li N et al (2013) Efficient and bright organic light-emitting diodes on single-layer graphene electrodes. Nat Commun 4:1–7
Tung R et al (2015) Freedom from recurrent ventricular tachycardia after catheter ablation is associated with improved survival in patients with structural heart disease: an international VT ablation center collaborative group study. Heart Rhythm 12:1997–2007
Borne RT, Varosy PD, Masoudi FA (2013) Implantable cardioverter-defibrillator shocks: epidemiology, outcomes, and therapeutic approaches. JAMA Intern Med 173:859–865
Murtaza G et al (2020) Role of cardiac sympathetic denervation in ventricular tachycardia: a meta-analysis. Pacing Clin Electrophysiol 43:828–837
Dusi V et al (2021) Arrhythmic risk profile and outcomes of patients undergoing cardiac sympathetic denervation for recurrent monomorphic ventricular tachycardia after ablation. J Am Heart Assoc 10:e018371
Vaseghi M et al (2017) Cardiac sympathetic denervation for refractory ventricular arrhythmias. J Am Coll Cardiol 69:3070–3080
Assis FR et al (2021) Long-term outcomes of bilateral cardiac sympathetic denervation for refractory ventricular tachycardia. JACC Clin Electrophysiol. https://doi.org/10.1016/j.jacep.2021.02.003
Hong JC, Crawford T, Tandri H, Mandal K (2017) What is the role of cardiac sympathetic denervation for recurrent ventricular tachycardia? Curr Treat Options in Cardiovasc Med. https://doi.org/10.1007/s11936-017-0512-z
Vaseghi M et al (2014) Cardiac sympathetic denervation in patients with refractory ventricular arrhythmias or electrical storm: intermediate and long-term follow-up. Heart Rhythm 11:360–366
Ajijola OA et al (2012) Bilateral cardiac sympathetic denervation for the management of electrical storm. J Am Coll Cardiol 59:91–92
Schwartz PJ et al (2004) Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation 109:1826–1833
Dusi V, De Ferrari GM, Pugliese L, Schwartz PJ (2019) Cardiac sympathetic denervation in channelopathies. Front Cardiovasc Med 6:27
Wilde AAM et al (2008) Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med 358:2024–2029
Estes EH, Izlar HL (1961) Recurrent ventricular tachycardia. A case successfully treated by bilateral cardiac sympathectomy. Am J Med 31:493–497
Zipes D et al (1968) Treatment of ventricular arrhythmia by permanent atrial pacemaker and cardiac sympathectomy. Ann Intern Med 68:591–597
Buckley U et al (2016) Targeted stellate decentralization: Implications for sympathetic control of ventricular electrophysiology. Heart Rhythm 13:282–288
Ajijola O, Vaseghi M, Mahajan A, Shivkumar K (2012) Bilateral cardiac sympathetic denervation: why, who and when? Expert Rev Cardiovasc Ther 10:947–949
Schwartz PJ (2014) Cardiac sympathetic denervation to prevent life-threatening arrhythmias. Nat Rev Cardiol 11:346–353
Bourke T et al (2010) Neuraxial modulation for refractory ventricular arrhythmias: value of thoracic epidural anesthesia and surgical left cardiac sympathetic denervation. Circulation 121:2255–2262
Tian Y et al (2019) Effective Use of Percutaneous Stellate Ganglion Blockade in Patients with Electrical Storm. Circ Arrhythmia Electrophysiol. https://doi.org/10.1161/CIRCEP.118.007118
Ganesh A et al (2020) Stellate ganglion blockade: an intervention for the management of ventricular arrhythmias. Curr Hypertens Rep 22:100
Ginosar Y et al (2009) Pulse oximeter perfusion index as an early indicator of sympathectomy after epidural anesthesia. Acta Anaesthesiol Scand 53:1018–1026
Aleanakian R, Chung BY, Feldmann RE, Benrath J (2020) Effectiveness, safety, and predictive potential in ultrasound-guided stellate ganglion blockades for the treatment of sympathetically maintained pain. Pain Pract 20:626–638
Şahin ÖF, Tarıkçı Kılıç E, Aksoy Y, Kaydu A, Gökçek E (2018) The importance of perfusion index monitoring in evaluating the efficacy of stellate ganglion blockage treatment in Raynaud’s disease. Libyan J Med. https://doi.org/10.1080/19932820.2017.1422666
Mahajan A, Moore J, Cesario DA, Shivkumar K (2005) Use of thoracic epidural anesthesia for management of electrical storm: a case report. Heart Rhythm 2:1359–1362
Do DH et al (2017) Thoracic epidural anesthesia can be effective for the short-term management of ventricular tachycardia storm. J Am Heart Assoc. https://doi.org/10.1161/JAHA.117.007080
Meissner A et al (2001) Effects of thoracic epidural anesthesia with and without autonomic nervous system blockade on cardiac monophasic action potentials and effective refractoriness in awake dogs. Anesthesiology 95:132–138
Blomberg S, Ricksten SE (1988) Thoracic epidural anaesthesia decreases the incidence of ventricular arrhythmias during acute myocardial ischaemia in the anaesthetized rat. Acta Anaesthesiol Scand 32:173–178
Wink J et al (2021) Biventricular function in exercise during autonomic (thoracic epidural) block. Eur J Appl Physiol. https://doi.org/10.1007/s00421-021-04631-6
Wink J et al (2016) Thoracic epidural anesthesia reduces right ventricular systolic function with maintained ventricular-pulmonary coupling. Circulation 134:1163–1175
Ng GA (2014) Vagal modulation of cardiac ventricular arrhythmia. Exp Physiol 99:295–299
Brack KE, Winter J, Ng GA (2013) Mechanisms underlying the autonomic modulation of ventricular fibrillation initiation-tentative prophylactic properties of vagus nerve stimulation on malignant arrhythmias in heart failure. Heart Fail Rev 18:389–408
Shinlapawittayatorn K et al (2013) Low-amplitude, left vagus nerve stimulation significantly attenuates ventricular dysfunction and infarct size through prevention of mitochondrial dysfunction during acute ischemia-reperfusion injury. Heart Rhythm 10:1700–1707
Vanoli E et al (1991) Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ Res 68:1471–1481
Vaseghi M et al (2017) Parasympathetic dysfunction and antiarrhythmic effect of vagal nerve stimulation following myocardial infarction. JCI Insight 2:e86715
Huang J et al (2015) Vagus nerve stimulation reverses ventricular electrophysiological changes induced by hypersympathetic nerve activity. Exp Physiol 100:239–248
Schwartz PJ et al (2008) Long term vagal stimulation in patients with advanced heart failure. First experience in man. Eur J Heart Fail 10:884–891
Anand IS et al (2020) Comparison of symptomatic and functional responses to vagus nerve stimulation in ANTHEM-HF, INOVATE-HF, and NECTAR-HF. ESC Hear Fail 7:75–83
Libbus I, Nearing BD, Amurthur B, KenKnight BH, Verrier RL (2016) Autonomic regulation therapy suppresses quantitative T-wave alternans and improves baroreflex sensitivity in patients with heart failure enrolled in the ANTHEM-HF study. Heart Rhythm 13:721–728
Nearing BD et al (2021) Vagus nerve stimulation provides multiyear improvements in autonomic function and cardiac electrical stability in the ANTHEM-HF study. J Card Fail 27:208–216
Kaniusas E et al (2019) Current directions in the auricular vagus nerve stimulation II - an engineering perspective. Front Neurosci. https://doi.org/10.3389/fnins.2019.00772
Kaniusas E et al (2019) Current directions in the auricular vagus nerve stimulation I-A physiological perspective. Front Neurosci. https://doi.org/10.3389/fnins.2019.00854
Buajieer-guli N et al (2018) Vagus nerve stimulation reduces ventricular arrhythmias and increases ventricular electrical stability. Pacing Clin Electrophysiol. https://doi.org/10.1111/pace.13585
Yu L et al (2016) Chronic intermittent low-level stimulation of tragus reduces cardiac autonomic remodeling and ventricular arrhythmia inducibility in a post-infarction canine model. JACC Clin Electrophysiol 2:330–339
Yu L et al (2017) Low-level tragus stimulation for the treatment of ischemia and reperfusion injury in patients with st-segment elevation myocardial infarction: a proof-of-concept study. JACC Cardiovasc Interv 10:1511–1520
Mannheimer C et al (2002) The problem of chronic refractory angina: report from the ESC joint study group on the treatment of refractory angina. Eur Heart J 23:355–370
Zhang TC, Janik JJ, Grill WM (2014) Mechanisms and models of spinal cord stimulation for the treatment of neuropathic pain. Brain Res 1569:19–31
Ardell JL (2016) Heart failure: mechanisms of spinal cord neuromodulation for heart disease. Nat Rev Cardiol 13:122–123
Ardell JL, Cardinal R, Vermeulen M, Armour JA (2009) Dorsal spinal cord stimulation obtunds the capacity of intrathoracic extracardiac neurons to transduce myocardial ischemia. Am J Physiol Integr Comp Physiol 297:R470–R477
Salavatian S et al (2019) Thoracic spinal cord neuromodulation obtunds dorsal root ganglion afferent neuronal transduction of the ischemic ventricle. Am J Physiol Circ Physiol 317:H1134–H1141
Cardinal R et al (2004) Spinal cord activation differentially modulates ischaemic electrical responses to different stressors in canine ventricles. Auton Neurosci Basic Clin 111:37–47
Cardinal R et al (2006) Spinal cord stimulation suppresses bradycardias and atrial tachyarrhythmias induced by mediastinal nerve stimulation in dogs. Am J Physiol-Regul Integr Comp Physiol. https://doi.org/10.1152/ajpregu.00056.2006
Issa ZF et al (2005) Thoracic spinal cord stimulation reduces the risk of ischemic ventricular arrhythmias in a postinfarction heart failure canine model. Circulation 111:3217–3220
Foreman RD et al (2000) Modulation of intrinsic cardiac neurons by spinal cord stimulation: implications for its therapeutic use in angina pectoris. Cardiovasc Res 47:367–375
Wang S et al (2015) Spinal cord stimulation protects against ventricular arrhythmias by suppressing left stellate ganglion neural activity in an acute myocardial infarction canine model. Heart Rhythm 12:1628–1635
Howard-Quijano K et al (2017) Spinal cord stimulation reduces ventricular arrhythmias during acute ischemia by attenuation of regional myocardial excitability. Am J Physiol Circ Physiol 313:H421–H431
Foreman RD et al (2000) Modulation of intrinsic cardiac neurons by spinal cord stimulation: implications for its therapeutic use in angina pectoris. Cardiovasc Res 47:367–375
Odenstedt J et al (2011) Spinal cord stimulation effects on myocardial ischemia, infarct size, ventricular arrhythmia, and noninvasive electrophysiology in a porcine ischemia-reperfusion model. Heart Rhythm 8:892–898
Grimaldi R, De Luca A, Kornet L, Castagno D, Gaita F (2012) Can spinal cord stimulation reduce ventricular arrhythmias? Heart Rhythm 9:1884–1887
Tse HF et al (2015) Thoracic spinal cord stimulation for heart failure as a restorative treatment (SCS heart study): first-in-man experience. Heart Rhythm 12:588–595
Zipes DP et al (2016) Determining the feasibility of spinal cord neuromodulation for the treatment of chronic systolic heart failure: the DEFEAT-HF study. JACC Hear Fail 4:129–136
Liao K et al (2014) Low-level carotid baroreceptor stimulation suppresses ventricular arrhythmias during acute ischemia. PLoS One 9:e109313
Liao K et al (2014) Carotid baroreceptor stimulation prevents arrhythmias induced by acute myocardial infarction through autonomic modulation. J Cardiovasc Pharmacol 64:431–437
Zhou X et al (2015) Low-level carotid baroreceptor stimulation: A promising feasible modulator for ventricular and atrial arrhythmias. Int J Cardiol 199:430–431
Scheffers IJM et al (2010) Novel baroreflex activation therapy in resistant hypertension: results of a European multi-center feasibility study. J Am Coll Cardiol 56:1254–1258
Bisognano JD et al (2011) Baroreflex activation therapy lowers blood pressure in patients with resistant hypertension: results from the double-blind, randomized, placebo-controlled rheos pivotal trial. J Am Coll Cardiol 58:765–773
Hoppe UC et al (2012) Minimally invasive system for baroreflex activation therapy chronically lowers blood pressure with pacemaker-like safety profile: results from the Barostim neo trial. J Am Soc Hypertens 6:270–276
Smithwick RH, Thompson JE (1953) Splanchnicectomy for essential hypertension: results in 1266 cases. J Am Med Assoc 152:1501–1504
Linz D et al (2013) Renal denervation suppresses ventricular arrhythmias during acute ventricular ischemia in pigs. Heart Rhythm 10:1525–1530
Dai Z et al (2014) Renal sympathetic denervation suppresses ventricular substrate remodelling in a canine high-rate pacing model. Eurointervention 10:392–399
Zhang WH et al (2018) Renal denervation reduced ventricular arrhythmia after myocardial infarction by inhibiting sympathetic activity and remodeling. J Am Heart Assoc. https://doi.org/10.1161/JAHA.118.009938
Yamada S et al (2017) Beneficial effect of renal denervation on ventricular premature complex induced cardiomyopathy. J Am Heart Assoc. https://doi.org/10.1161/JAHA.116.004479
Bhatt DL et al (2014) A controlled trial of renal denervation for resistant hypertension. N Engl J Med 370:1393–1401
Kandzari DE et al (2015) Predictors of blood pressure response in the SYMPLICITY HTN-3 trial. Eur Heart J 36:219–227
Papademetriou V, Tsioufis C, Doumas M (2014) Renal denervation and symplicity HTN-3: ‘dubium sapientiae initium’ (doubt is the beginning of wisdom). Circ Res 115:211–214
Epstein M, De Marchena E (2015) Is the failure of SYMPLICITY HTN-3 trial to meet its efficacy endpoint the ‘end of the road’ for renal denervation? J Am Soc Hypertens 9:140–149
Ukena C et al (2012) Renal sympathetic denervation for treatment of electrical storm: first-in-man experience. Clin Res Cardiol 101:63–67
Remo BF et al (2014) Safety and efficacy of renal denervation as a novel treatment of ventricular tachycardia storm in patients with cardiomyopathy. Heart Rhythm 11:541–546
Tsioufis C et al (2014) Drug-resistant hypertensive patients responding to multielectrode renal denervation exhibit improved heart rate dynamics and reduced arrhythmia burden. J Hum Hypertens 28:587–593
Ukena C et al (2016) Renal denervation for treatment of ventricular arrhythmias: data from an international multicenter registry. Clin Res Cardiol 105:873–879
Scholz EP et al (2015) Rescue renal sympathetic denervation in a patient with ventricular electrical storm refractory to endo- and epicardial catheter ablation. Clin Res Cardiol 104:79–84
Hoffmann BA, Steven D, Willems S, Sydow K (2013) Renal sympathetic denervation as an adjunct to catheter ablation for the treatment of ventricular electrical storm in the setting of acute myocardial infarction. J Cardiovasc Electrophysiol. https://doi.org/10.1111/jce.12282
Pokushalov E et al (2012) A randomized comparison of pulmonary vein isolation with versus without concomitant renal artery denervation in patients with refractory symptomatic atrial fibrillation and resistant hypertension. J Am Coll Cardiol 60:1163–1170
Lefaucheur JP et al (2014) Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS). Clin Neurophysiol 125:2150–2206
Cabrerizo M et al (2014) Induced effects of transcranial magnetic stimulation on the autonomic nervous system and the cardiac rhythm. Sci World J 2014:12
Yoshida T et al (2001) Effects of slow repetitive transcranial magnetic stimulation on heart rate variability according to power spectrum analysis. J Neurol Sci 184:77–80
Scherlag BJ et al (2004) Magnetism and cardiac arrhythmias. Cardiol Rev 12:85–96
Yu L et al (2015) The use of low-level electromagnetic fields to suppress atrial fibrillation. Heart Rhythm 12:809–817
Wang S, Zhou X, Huang, B et al (2016) Noninvasive low-frequency electromagnetic stimulation of the left stellate ganglion reduces myocardial infarction-induced ventricular arrhythmia. Sci Rep 6, 30783. https://doi.org/10.1038/srep30783
Markman TM, Hamilton RH, Marchlinski FE, Nazarian S (2020) Case series of transcutaneous magnetic stimulation for ventricular tachycardia storm. JAMA - J Am Med Assoc 323:2200–2202
Wang S, Li B, Li X, Wu L, Jiang H (2019) Abstract: low intensity ultrasound stimulation might reduce ventricular arrhythmia by modulating sympathetic neural activity in myocardial infarction canine model. J Am Coll Cardiol 73:531
Funding
National Heart, Lung, and Blood Institute (DP2OD024323), NIH Office of the Director (OT2OD028201 and OT2OD023848).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van Weperen, V.Y.H., Vos, M.A. & Ajijola, O.A. Autonomic modulation of ventricular electrical activity: recent developments and clinical implications. Clin Auton Res 31, 659–676 (2021). https://doi.org/10.1007/s10286-021-00823-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10286-021-00823-4