
Abstract
Purpose
Investigate single nucleotide variants and short tandem repeats in 39 genes related to spinocerebellar ataxia in clinical and pathologically defined cohorts of multiple system atrophy.
Methods
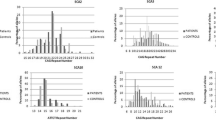
Exome sequencing was conducted in 28 clinical multiple system atrophy patients to identify single nucleotide variants in spinocerebellar ataxia-related genes. Novel variants were validated in two independent disease cohorts: 86 clinically diagnosed multiple system atrophy patients and 166 pathological multiple system atrophy cases. Expanded repeat alleles in spinocerebellar ataxia genes were evaluated in 36 clinically diagnosed multiple system atrophy patients, and CAG/CAA repeats in TATA-Box Binding Protein (TBP, causative of SCA17) were screened in 216 clinical and pathological multiple system atrophy patients and 346 controls.
Results
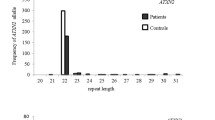
No known pathogenic spinocerebellar ataxia single nucleotide variants or pathogenic range expanded repeat alleles of ATXN1, ATXN2, ATXN3, CACNA1A, AXTN7, ATXN8OS, ATXN10, PPP2R2B, and TBP were detected in any clinical multiple system atrophy patients. However, four novel variants were identified in four spinocerebellar ataxia-related genes across three multiple system atrophy patients. Additionally, four multiple system atrophy patients (1.6%) and one control (0.3%) carried an intermediate length 41 TBP CAG/CAA repeat allele (OR = 4.11, P = 0.21). There was a significant association between the occurrence of a repeat length of longer alleles (> 38 repeats) and an increased risk of multiple system atrophy (OR = 1.64, P = 0.03).
Conclusion
Occurrence of TBP CAG/CAA repeat length of longer alleles (> 38 repeats) is significantly associated with increased multiple system atrophy risk. This discovery warrants further investigation and supports a possible genetic overlap of multiple system atrophy with SCA17.

Similar content being viewed by others

Data availability
All relevant data generated or analysed during this study are included in this published article (and its supplementary information files). Full datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T, Holton JL (2012) The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol 38:4–24
Trojanowski JQ, Revesz T (2007) M.S.A. Neuropathology Working Group on, Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 33:615–620
Bower JH, Maraganore DM, McDonnell SK, Rocca WA (1997) Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology 49:1284–1288
Bjornsdottir A, Gudmundsson G, Blondal H, Olafsson E (2013) Incidence and prevalence of multiple system atrophy: a nationwide study in Iceland. J Neurol Neurosurg Psychiatry 84:136–140
Fanciulli A, Wenning GK (2015) Multiple-system atrophy. N Engl J Med 372:249–263
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71:670–676
Koga S, Dickson DW (2018) Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J Neurol Neurosurg Psychiatry 89:175–184
Koga S, Aoki N, Uitti RJ, van Gerpen JA, Cheshire WP, Josephs KA, Wszolek ZK, Langston JW, Dickson DW (2015) When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 85:404–412
Xie T, Kang UJ, Kuo SH, Poulopoulos M, Greene P, Fahn S (2015) Comparison of clinical features in pathologically confirmed PSP and MSA patients followed at a tertiary center. NPJ Parkinsons Dis 1:15007
Doherty KM, De Pablo-Fernandez E, Houlden H, Polke JM, Lees AJ, Warner TT, Holton JL (2016) MSA-C or SCA 17? A clinicopathological case update. Mov Disord 31:1582–1584
Doherty KM, Warner TT, Lees AJ (2014) Late onset ataxia: MSA-C or SCA 17? A gene penetrance dilemma. Mov Disord 29:36–38
Smetcoren C, Weckhuysen D (2016) SCA 8 mimicking MSA-C. Acta Neurol Belg 116:221–222
Lin IS, Wu RM, Lee-Chen GJ, Shan DE, Gwinn-Hardy K (2007) The SCA17 phenotype can include features of MSA-C, PSP and cognitive impairment. Parkinsonism Relat Disord 13:246–249
Klockgether T, Mariotti C, Paulson HL (2019) Spinocerebellar ataxia. Nat Rev Dis Primers 5:24
Tai G, Corben LA, Yiu EM, Milne SC, Delatycki MB (2018) Progress in the treatment of Friedreich ataxia. Neurol Neurochir Pol 52:129–139
Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, U. (2012) Rub, Brain pathology of spinocerebellar ataxias. Acta Neuropathol 124:1–21
Bird TD (1993) Hereditary ataxia overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews ((R)). University of Washington, Seattle
Kim HJ, Jeon BS, Shin J, Lee WW, Park H, Jung YJ, Ehm G (2014) Should genetic testing for SCAs be included in the diagnostic workup for MSA? Neurology 83:1733–1738
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10:1441–1448
Munhoz RP, Teive HA, Raskin S, Werneck LC (2009) CTA/CTG expansions at the SCA 8 locus in multiple system atrophy. Clin Neurol Neurosurg 111:208–210
Mongelli A, Sarro L, Rizzo E, Nanetti L, Meucci N, Pezzoli G, Goldwurm S, Taroni F, Mariotti C, Gellera C (2018) Multiple system atrophy and CAG repeat length: a genetic screening of polyglutamine disease genes in Italian patients. Neurosci Lett 678:37–42
Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, Healy DG, Wood NW, Lees AJ, Holton JL, Revesz T (2004) The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain 127:2657–2671
S. Gabriel, L. Ziaugra, D. Tabbaa (2009) SNP genotyping using the Sequenom MassARRAY iPLEX platform, Curr Protoc Hum Genet, Chapter 2 Unit 2 12.
Ai SX, Xu Q, Hu YC, Song CY, Guo JF, Shen L, Wang CR, Yu RL, Yan XX, Tang BS (2014) Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. J Neurol Sci 337:123–128
Al-Chalabi A, Durr A, Wood NW, Parkinson MH, Camuzat A, Hulot JS, Morrison KE, Renton A, Sussmuth SD, Landwehrmeyer BG, Ludolph A, Agid Y, Brice A, Leigh PN, Bensimon G, N.G.S. Group (2009) Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS ONE 4:e7114
Guo XY, Chen YP, Song W, Zhao B, Cao B, Wei QQ, Ou RW, Yang Y, Yuan LX, Shang HF (2014) SNCA variants rs2736990 and rs356220 as risk factors for Parkinson’s disease but not for amyotrophic lateral sclerosis and multiple system atrophy in a Chinese population. Neurobiol Aging 35:2882e2881-2882e2886
Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wullner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Del Sorbo F, Schneider S, Bhatia KP, Gasser T (2009) SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 65:610–614
Sklerov M, Kang UJ, Liong C, Clark L, Marder K, Pauciulo M, Nichols WC, Chung WK, Honig LS, Cortes E, Vonsattel JP, Alcalay RN (2017) Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov Disord Clin Pract 4:574–581
Whaley NR, Fujioka S, Wszolek ZK (2011) Autosomal dominant cerebellar ataxia type I: a review of the phenotypic and genotypic characteristics. Orphanet J Rare Dis 6:33
Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, Yamada M, Takahashi H, Tsuji S (1999) A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet 8:2047–2053
Shin JH, Park H, Ehm GH, Lee WW, Yun JY, Kim YE, Lee JY, Kim HJ, Kim JM, Jeon BS, Park SS (2015) The pathogenic role of low range repeats in SCA17. PLoS ONE 10:e0135275
Nanda A, Jackson SA, Schwankhaus JD, Metzer WS (2007) Case of spinocerebellar ataxia type 17 (SCA17) associated with only 41 repeats of the TATA-binding protein (TBP) gene. Mov Disord 22:436
Origone P, Gotta F, Lamp M, Trevisan L, Geroldi A, Massucco D, Grazzini M, Massa F, Ticconi F, Bauckneht M, Marchese R, Abbruzzese G, Bellone E, Mandich P (2018) Spinocerebellar ataxia 17: full phenotype in a 41 CAG/CAA repeats carrier. Cerebellum Ataxias 5:7
Gill G, Tjian R (1992) Eukaryotic coactivators associated with the TATA box binding protein. Curr Opin Genet Dev 2:236–242
Martianov I, Viville S, Davidson I (2002) RNA polymerase II transcription in murine cells lacking the TATA binding protein. Science 298:1036–1039
Friedman MJ, Shah AG, Fang ZH, Ward EG, Warren ST, Li S, Li XJ (2007) Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci 10:1519–1528
Yang S, Li XJ, Li S (2016) Molecular mechanisms underlying Spinocerebellar Ataxia 17 (SCA17) pathogenesis. Rare Dis 4:e1223580
Gardiner SL, van Belzen MJ, Boogaard MW, van Roon-Mom WMC, Rozing MP, van Hemert AM, Smit JH, Beekman ATF, van Grootheest G, Schoevers RA, Oude Voshaar RC, Comijs HC, Penninx B, van der Mast RC, Roos RAC, Aziz NA (2017) Large normal-range TBP and ATXN7 CAG repeat lengths are associated with increased lifetime risk of depression. Transl Psychiatry 7:e1143
Gardiner SL, Harder AVE, Campman YJM, Trompet S, Gussekloo J, van Belzen MJ, Boogaard MW, Roos RAC, Jansen IE, Pijnenburg YAL, Scheltens P, van der Flier WM, Aziz NA (2019) Repeat length variations in ATXN1 and AR modify disease expression in Alzheimer’s disease. Neurobiol Aging 73:230.e9-230.e17
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rub U, Auburger G, Trojanowski JQ, Lee VM, Van Deerlin VM, Bonini NM, Gitler AD (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069–1075
Acknowledgements
We would like to thank all those who have contributed to our research, particularly the patients and families who donated brain and blood samples for this work. We are grateful to all patients, family members, and caregivers who agreed to brain donation. Without their donation these studies would not have been possible. We also acknowledge expert technical assistance of Virginia Phillips for histology and Monica Castanedes-Casey for immunohistochemistry and Audrey Strongosky for her assistance in blood sample collection and brain donation arrangements. Mayo Clinic is an American Parkinson Disease Association (APDA) Mayo Clinic Information and Referral Center, an APDA Center for Advanced Research, and the Mayo Clinic Lewy Body Dementia Association (LBDA) Research Center of Excellence and a LBD Center WithOut Walls (U54 NS110435). Samples included in this study were clinical controls or brain donors to the Mayo Clinic Brain Bank in Jacksonville, which is supported by CurePSP and the Tau Consortium. SK is supported by a Jaye F. and Betty F. Dyer Foundation Fellowship in Progressive supranuclear palsy research and CorticoBasal Degeneration Solutions Research Grant. ZKW is partially supported by the Mayo Clinic Center for Regenerative Medicine, the gifts from The Sol Goldman Charitable Trust, and the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, and The Albertson Parkinson's Research Foundation. The funding organizations and sponsors had no role in any of the following: design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Funding
Mayo Clinic is an American Parkinson Disease Association (APDA) Mayo Clinic Information and Referral Center, an APDA Center for Advanced Research, and the Mayo Clinic Lewy Body Dementia Association (LBDA) Research Center of Excellence and a LBD Center WithOut Walls (U54 NS110435). SK is supported by a Jaye F. and Betty F. Dyer Foundation Fellowship in Progressive supranuclear palsy research and CorticoBasal Degeneration Solutions Research Grant.
Author information
Authors and Affiliations
Contributions
AIW: study concept and design; analysis or interpretation of data; sample preparations or genotyping; drafting/revising the manuscript; RLW: sample preparations and genotyping; drafting/revising the manuscript; AIS: sample preparation and genotyping; drafting/revising the manuscript; SK: sample preparation; drafting/revising the manuscript; MGH: analysis and interpretation of data, drafting/revising the manuscript; LMM: contribution of patients; drafting/revising the manuscript; RRV: drafting/revising the manuscript; DHZ: contribution of patients; drafting/revising the manuscript; DK: contribution of patients; drafting/revising the manuscript; AH: contribution of patients; drafting/revising the manuscript; RJU: contribution of patients; drafting/revising the manuscript; WPC: contribution of patients; drafting/revising the manuscript; WS: contribution of patients; drafting/revising the manuscript; ZKW: contribution of patients; drafting/revising the manuscript; DWD: study concept and design; diagnosis and provision of samples; drafting/revising the manuscript; PAL: contribution of patients; drafting/revising the manuscript; OAR: study concept and design; analysis or interpretation of data; sample preparations or genotyping; drafting/revising the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Anna I. Wernick, Ronald L. Walton, Alexandra I. Soto-Beasley, Shunsuke Koga, and Rebecca R. Valentino report no disclosures. Michael G. Heckman is a Statistical Editor of Parkinsonism & Related Disorders, and is on the editorial board of Molecular Neurodegeneration. Lukasz M. Milanowski is supported by the Polish National Agency for Academic Exchange Iwanowska’s Programme PPN/IWA/2018/1/00006/U/00001/01. Dorota Hoffman-Zacharska report no disclosures. Dariusz Koziorowski report no disclosures. Anhar Hassan is on the editorial board of Parkinsonism and Related Disorders, and receives research support from Intrabio. Ryan J. Uitti is an Associate Editor of Neurology. William P. Cheshire is an Associate Editor of Clinical Autonomic Research and is on the editorial boards of Autonomic Neuroscience and Parkinsonism and Related Disorders. Wolfgang Singer received research support from NIH (R01NS092625, U54NS065736), FDA (R01FD004789), Sturm Foundation, and Mayo Funds. He is associate editor of Clinical Autonomic Research and serves on the editorial board of Autonomic Neuroscience. He has advisory board and consulting agreements with Lundbeck, Catalyst, and Astellas. Zbigniew K. Wszolek receives research support from P50-NS072187, NIH/NIA (primary) and NIH/NINDS (secondary) 1U01AG045390-01A1, Mayo Clinic Center for Regenerative Medicine, the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch, The Sol Goldman Charitable Trust, and Donald G. and Jodi P. Heeringa, the Haworth Family Professorship in Neurodegenerative Diseases fund, and by the Albertson Parkinson's Research Foundation. He serves as Co-Editor-in-Chief of Neurologia i Neurochirurgia Polska and is on the editorial boards of European Journal of Clinical and Experimental Medicine, Clinical and Experimental Medical Letters, and Wiadomosci Lekarskie; holds and has contractual rights for receipt of future royalty payments from patents for “A Novel Polynucleotide Involved in Heritable Parkinson’s Disease”; and received royalties from publishing Parkinsonism and Related Disorders (Elsevier, 2016, 2017) and the European Journal of Neurology (Wiley Blackwell, 2016, 2017). ZKW serves as PI or Co-PI on Abbvie, Inc. (M15-562 and M15-563), Biogen, Inc. (228PD201) grant, and Biohaven Pharmaceuticals, Inc. (BHV4157-206 and BHV3241-301). He serves as PI of the Mayo Clinic American Parkinson Disease Association (APDA) Information and Referral Center, and as Co-PI of the Mayo Clinic APDA Center for Advanced Research. Dennis W. Dickson (DWD) receives support from P50-AG016574, P50-NS072187, P01-AG003949, and CurePSP: Foundation for PSP | CBD and Related Disorders. DWD is an editorial board member of Acta Neuropathologica, Annals of Neurology, Brain, Brain Pathology, and Neuropathology and is Editor in Chief of American Journal of Neurodegenerative Disease and International Journal of Clinical and Experimental Pathology. Phillip A. Low receives support from Supported by NIH (P01NS44233, U54NS065736, R01 FD004789, R01 NS092625, Cure MSA Foundation, Sturm and Mayo Funds. Owen A. Ross (OAR) receives support from R01-NS078086, P50-NS072187, and U54 NS100693, The Little Family Foundation, and the Michael J. Fox Foundation. OAR is an editorial board member of American Journal of Neurodegenerative Disease and Molecular Neurodegeneration.
Ethical approval
Written consent from each subject or next-of-kin was collected for all patients included in this study and IRB approval was obtained from the Mayo Clinic institutional review board for ethical conduct of research.
Supplementary Information
Below is the link to the electronic supplementary material.
10286_2020_759_MOESM1_ESM.docx
Supplementary Table 1 aGenes likely responsible for SCA but require further confirmation. 1: Gene was screened for known pathogenic point mutations and novel point mutations via exome sequencing (N = 28). 2: Gene was screened for repeats by duplication analysis; however, duplication analysis was not possible for BEAN and DAB1 due to the absence of a positive control (DOCX 19 KB)
Rights and permissions
About this article

Cite this article
Wernick, A.I., Walton, R.L., Soto-Beasley, A.I. et al. Frequency of spinocerebellar ataxia mutations in patients with multiple system atrophy. Clin Auton Res 31, 117–125 (2021). https://doi.org/10.1007/s10286-020-00759-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10286-020-00759-1