
Abstract
That the brain may be involved in cardiovascular regulation has been acknowledged for over a century. That cardiac arrhythmias may result from cortical derangement has been less well recognized. That cortical cardiac representation may be lateralized is even more controversial. Recent evidence implicates several cortical structures, especially the insula, in cardiac rate and rhythm control. Experimental models indicate that insular lesions may be arrhythmogenic. Accumulating data show similar lesion effects in humans. In the rat, monkey and man sympathetic cardiovascular control is generally represented in the right insula, although pronounced insulo-insular connectivity has been demonstrated. Proarrhythmic shifts in cardiac sympathovagal balance occur after human stroke, including left insular lesions. This evidence implicates the cortex in the promotion and even generation of cardiovascular dysfunction under appropriate circumstances.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Perspective
Evidence for sympathetic upregulation as a mechanism for cerebral influences on cardiac structure and function
In 1913 Goodman Levy [24, 25] showed that chloroform-induced ventricular tachyarrhythmias were abolished by cardiac sympathetic denervation. Additionally, sympathetic activation induced identical ventricular tachyarrhythmias, indicating a direct neurological rather than cardiac effect of chloroform. Subsequently, Beattie [3] showed that these tachyarrhythmias were abolished by mid-collicular, but not higher, diencephalic sectioning. Histological examination indicated degeneration of a pathway originating in the hypothalamus and terminating in the intermediolateral column (close to the cells of origin of sympathetic preganglionic fibers).
Another decade passed. Aschenbrenner [1] reported seven young patients with cortical and diencephalic tumors who developed ventricular ectopy or nodal rhythm. These patients lacked cardiac history or post mortem ischemic cardiac pathology to explain their death. Subendocardial hemorrhages adjacent to the ventricular cavity were noted as an unusual feature. These resembled the changes seen after sudden death in epilepsy reported earlier by Neuberger [30].
Much of this was forgotten with the advent of World War II. In the aftermath, single case reports of intriguing ECG repolarization changes after subarachnoid hemorrhage (SAH) were reported: inverted T waves (often tall, symmetrical downward peaking) and prolonged QTc interval with ST elevation or depression. Cropp [9] initially thought these changes indicated coronary artery disease and delayed surgery, with fatal consequences. Clinical and pathological findings were reviewed. No patient had a cardiac history, most were young and no autopsied case showed coronary artery pathology. Aneurysm position led to implication of inferior frontal structures and possible vagal mechanisms in the etiology of these cardiac changes.
Recently, Kono [19] compared 7 SAH patients with repolarization changes and 5 in whom no ECG abnormalities were detected. Reduced myocardial contractility occurred only in association with ECG changes. All patients underwent catheterization without evidence of coronary artery disease.
Greenhoot [15] extended the range of cardiac pathology observed after subarachnoid hemorrhage. Additional to subendocardial hemorrhages, scattered myocyte necrosis was observed surrounded by infiltrating monocytes and interstitial hemorrhages (myocytolysis). Electron microscopy showed clustering of cardiac pathology around nerves and indicated a neural rather than vascular origin.
Others investigated stress as a pointer to cerebral-cardiac interactions. In a Holter study, stress induced ventricular ectopy when healthy individuals addressed an audience [47]. In those with documented cardiac disease, frequency and severity of ventricular ectopy was markedly increased. All arrhythmias were abolished by β-blockade. Toivonen [49] investigated arousal effects on the ambulatory ECG of 30 “healthy physicians” without known heart disease. Recordings were compared just before and during emergency calls. The T wave inverted in 67 % subjects and asymptomatic ST depression was noted in 33%. As RR interval shortened, the QT interval failed to show the expected decrease. Thus, arousal can significantly affect ventricular repolarization even in putatively normal hearts, presumably through sympathetic mechanisms.
Coupled with the resemblance of myocytolysis to phaeochromocytoma cardiomyopathy, the cardiac pathology accompanying catecholamine infusion and prolonged stress, these observations strongly indicate that sympathetic neural activation can destabilize cardiac structure and function. Much recent evidence indicates the arrhythmogenic potential of catecholamines. Mid-myocardial cells have been identified in the dog and human ventricle [27,48] sensitive to catecholamines which may underlie the pro-arrhythmic effects of cardiac sympathetic activation. In this regard, Cao [5] compared cardiac innervation density in patients with or without history of ventricular arrhythmias. Increased sympathetic nerve density was found around blood vessels and areas of myocardial damage only in those with an arrhythmia history. They then infused nerve growth factor (NGF) into canine left stellate ganglia [7]. When combined with AV block and experimental MI, VT and sudden cardiac death occurred only in the NGF-treated animals. This suggested that sympathetic neural remodeling might facilitate ventricular arrhythmias and contribute to sudden cardiac death. Conversely, Manitius-Robeck [28] reported ictal sinus arrest during temporal lobe seizures. In one patient, cortical dysplasia was associated with a near-complete absence of MIBG-SPECT determined cardiac sympathetic innervation. The bradyarrhythmias were abolished by surgery indicating a permissive interaction and developmental link between cortical dysplasia and reduced cardiac sympathetic innervation.
Stroke is associated with an adverse long-term cardiac outlook [41], possibly relating to altered sympathovagal balance. Barron [2] reported that compared with age/gender matched controls, total spectral power of RR intervals was reduced after stroke; power associated with cardiac parasympathetic neural activity was particularly affected indicating a shift in sympathovagal balance. Strittmatter [45] reported that cardiac sympathetic tone was increased within 5 days of stroke and similar findings were reported by Giubilei [13],who also showed persistence for at least 3 weeks. Additionally, Korpelainen et al. [20] demonstrated that diurnal cardiac autonomic variability was disturbed after acute stroke with a striking loss of nocturnal vagal dominance. This normalized by 6 months while sympathovagal balance was still abnormal [21]. Sander [42, 44] reported that absence of circadian cardiovascular variation was particularly striking after insular infarction. These patients had higher plasma no-radrenaline levels, increased nocturnal blood pressure, a higher incidence of QTc interval prolongation and ventricular arrhythmias than patients with stroke in other locations. However, in these, and many other such studies the contribution of concomitant coronary artery disease and medications were not assessed.
Collectively these data strongly indicate that the brain has a major influence on cardiac structure and function and that this is likely mediated through alterations in patterning of sympathovagal relationships.
Clinical implications of cerebrocardiac interactions
Frequency and consequences of cerebrally induced cardiac dysfunction
While evidence for cerebral involvement in cardiac dysfunction is persuasive, frequency, predisposing circumstances and relevance for outcome are unclear. Assessment is confounded by coincident (often asymptomatic) cardiac disease. In this regard, early traces of (non-ischemic) myocytolysis were found in 26% of patients dying of non-ischemic causes (pneumonia, sepsis) and in 89% of SAH, 71% of intracerebral hemorrhage, and 52% of ischemic stroke deaths [18]. While indicative, these data come from a select group of patients for whom post mortem data were available. Relevance to the overall stroke population is unclear.
Stroke may provide a good model to investigate neurocardiac influences: lesion location may be identified with precision, and onset categorized with some accuracy. However few studies detail arrhythmia incidence after stroke, and even fewer have controlled for concomitant coronary artery disease. Comparing ECGs before and after stroke, Lavy [23] indicated a 39 % incidence of new onset cardiac arrhythmias in patients without cardiac history. Goldstein [14] reviewed 53 acute stroke patients’ (including SAH) ECGs obtained within 24 hours of admission and recordings taken an average of 4 months earlier. A control group comprised age and sex matched patients. QT prolongation of new onset was seen in 32% of strokes (against 2% of controls); T wave inversion and U waves were present in 15% of stroke patients and in none of the controls. New onset cardiac arrhythmias (of which atrial fibrillation was the most frequent) were present in 25% of all stroke patients, and 3% of controls. Curiously, there was no difference in ventricular arrhythmia incidence between the two groups. However, this may not be too surprising since the ECG underestimates arrhythmia incidence. In other studies, where Holter recording has been used, ventricular arrhythmia incidence after stroke was higher, varying from 25–75% [31,38,39] without control for co-existing cardiac disorders or for whether these were new or pre-existing. In Yamour’s uncontrolled study of intracerebral hemorrhage patients [52], comparison with pre-event ECG status indicated a new onset ventricular arrhythmia in 10% of patients and interestingly correlated with a temporoparietal location.
In our recent unpublished observations, ventricular tachyarrhythmias were not observed in ECG recordings of 189 acute stroke patients. Ventricular premature beats occurred in 2.4% of strokes, and in 2% of 37 patients admitted with transient ischemic attacks (TIA). TIA patients served as controls with similar demographics, incidence of coronary artery disease (70%), atherosclerotic risk factors and medication experience. However, Holter monitoring revealed a ventricular tachyarrhythmia incidence of 12% in stroke, and 3% in TIA patients, bigeminy in 21% and 5% respectively, ventricular ectopy in 71% and 73% respectively, and atrial fibrillation in 9% and 3% respectively. These figures confirm differences in arrhythmia ascertainment between ECG and continuous monitoring.
Atrial fibrillation is commonly found in ECG studies of stroke [14, 23]. However, when pre-event ECGs are used for comparison, it is unclear whether paroxysmal arrhythmia preceded and in some cases, caused stroke because of embolic potential. Additionally, most reports do not control for cardiac concomitants (indeed, 40% of acute stroke patients without cardiac symptoms are reported to have > 70% stenosis on coronary angiography [17]).
Cardiac arrhythmia incidence is reportedly highest after SAH (98 %), of which multifocal ventricular beats occur on cardiac monitoring in 54%, couplets in 40% and unsustained ventricular tachycardia in 29 % [10]. This correlates with QTc prolongation, but not with cardiac history.
Cardiac arrhythmias may be associated with adverse outcome: 80% mortality in acute stroke patients with ventricular tachyarrhythmias, compared to 23% in those without [14]. Similarly, an association was documented between poor outcome (including death) in patients with arrhythmias or ECG changes after SAH [10].
Wong [51] reported that after adjusting for overt cardiac disease and traditional ischemic risk factors, stroke patients with QTc prolongation in lead V6 had a 2.8 relative risk of cardiac death. If the QTc exceeded 480ms then the specificity was 94% for cardiac death prediction within five years. Eckhardt [12] showed that insular strokes were particularly associated with QT dispersion lengthening compared with those in other locations.
Collectively, these data indicate that significant cardiac arrhythmogenesis can occur following stroke. This is most pronounced after subarachnoid hemorrhage, but may also accompany intracerebral hemorrhage and ischemia. In many reported series, precision is reduced by failure to account for concomitant ischemic cardiac disorders as a confounding variable, and by the use of ECG rather than Holter data. Additionally, most studies are of short duration, performed in the acute phase and do not investigate the longer-term cardiovascular consequences of the lesion. It is this latter which may prove to be of importance in determining survival, and, as yet, definitive data on this point are lacking.
Cerebral lateralization of cardioregulatory function
Clinical and physiological considerations
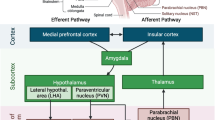
In many species the insular cortex plays a pivotal role in the integration of autonomic function. Viscerotopic organization has been demonstrated in the insula of the rat [6]. Cardiac chronotropic organization was subsequently identified in the rostral posterior insula [32]. Phasic microstimulation of the rat left insula resulted in QT prolongation, ST depression, pronounced bradycardia, complete heart block and idioventricular rhythm ending in asystole [33]. Myocytolysis was apparent on cardiac examination and plasma noradrenaline levels were elevated, without a change in adrenaline (which in the rat indicates neural rather than adrenal origin).Neither was provoked by stimulation of peri-insular sensory cortex.Thus, cerebral stimulation can reproducibly generate lethal cardiac arrhythmias and pathology resembling changes seen after stroke or sudden unexpected death in epilepsy.
Lesions confined mainly to the right posterior insula of the rat increase blood pressure and heart rate without altering baroreceptor sensitivity [54]. Conversely, left posterior insular lesions do not alter cardiovascular variables, but increase baroreceptor sensitivity. Previously, we had shown that damage to the right hemisphere in a rat middle cerebral artery occlusion model increased both QT interval and plasma norepinephrine [16]. Chemical lesions confined to the right insula also increase heart rate and blood pressure [4].
Direct recording from cardiac sympathetic nerves is difficult; however surrogate measures of sympathovagal balance may be assessed from spectral analysis of heart rate.We have shown in the rat that right posterior insular stimulation increases cardiac sympathetic tone in the absence of heart rate, blood pressure or respiration changes [36]. Interestingly, baroreceptor sensitivity decreased, a finding also linked to increased mortality after stroke [40].
These laterality issues are of interest because of previous observations that in the human, right carotid amylobarbital infusion produces bradycardia, and left carotid infusion is accompanied by tachycardia [53].Additionally, an increased incidence of supraventricular tachycardia was reported in patients with right middle cerebral artery stroke [22]. Our investigations in the human indicate that left caudal anterior insular stimulation during surgery for intractable epilepsy increases the frequency of bradycardia and depressor responses, whereas stimulation of a similar region of the right anterior insula is associated with heart rate and diastolic blood pressure elevation [34].Although both types of response were elicitable from either insula, the proportion varied, and the degree of bradycardia was greater on left insular stimulation. These data indicate that in the human at least, some lateralization of cardiovascular representation may exist with sympathetic predominance of cardiovascular regulation being a right insular function, and parasympathetic cardiac neural regulation relating to the left insula.
Recently we reported a series of patients with lesions confined to the left insula [35]. Consistent with the hypothesis of left insular cardioinhibitory representation, these patients had a higher basal heart rate than age-gender matched controls. Cardiac autonomic balance was shifted towards sympathetic predominance and mirrored the effects of rising from sitting to standing in the control group. ISE and approximate entropy data indicated a decrease in heart rate complexity. Interestingly 40% of these patients developed ECG changes not seen on premorbid traces, comprising tachycardia, T wave inversion, prominent U waves and QTc prolongation. None reported a cardiac history. No similar changes were seen in controls.The abnormalities resolved within 2 months of stroke. A similar case has also been reported of an arrhythmia resolving after evacuation of a left insular hematoma [46].We have also encountered a patient who developed transient ST depression 3 days after left insular infarction in the absence of cardiac history, coronary artery disease, or echocardiographic abnormalities [8].
Others have indicated a significant role for the right insular cortex in cardiovascular decompensation after stroke. Tokgozoglu [50] showed that compared to age-gender matched controls total spectral energy was reduced after ischemic stroke as noted previously by others. However, this was particularly marked in patients with stroke involving the right middle cerebral artery, or the insular cortex. Sudden death was observed in 11%, but occurred more often after right MCA lesions. Naver [29] indicated that heart rate variability entrained to deep breathing (a parasympathetic relationship) was reduced with right sided stroke, whereas peripheral sympathetic influences were equally distributed between the two sides. Sympathetic skin response and pulse rate variation (parasympathetic measure) were suppressed in patients with both right or left hemisphere lesions compared with controls [11]. This effect was more marked with right hemisphere lesions, although the authors failed to show a statistical difference when right and left sides were compared. On the other hand, Li [26] indicated that supraventricular arrhythmias were more frequently encountered after right insular infarction compared with strokes in other locations. Interestingly, ST abnormalities were more frequent after left insular involvement in comparison with controls or strokes in other locations. Sander [43] showed that right hemisphere infarction reduced circadian blood pressure variability and increased nocturnal blood pressure compared to left hemisphere infarcts. Additionally, higher serum noradrenaline levels, longer QTc prolongation and more cardiac arrhythmias were observed after right hemisphere infarction. In general, changes were greatest when there was insular involvement, under which circumstances no laterality effects were observed.
Whereas these changes indicate that left insular lesions may disrupt interactions of central oscillators regulating cardiac rhythmicity, there is some inconsistency in the data. Certainly, right rostral posterior insular stimulation in the rat increases cardiac sympathetic tone; however, lesions involving this region also increase heart rate and blood pressure. These seemingly incompatible effects may relate to anesthetic effects on descending inhibitory and excitatory pathways. Under baseline anesthesia conditions inhibitory pathways (probably insular efferents to the lateral hypothalamic area [37]) may be maximally operational, and this inhibition is released by insular lesioning. On the other hand, stimulation recruits excitatory pathways, possibly quiescent under basal anesthetic conditions. Human investigations were conducted in minimally anesthetized individuals and so the relationships may differ. In the rat, prolonged left insular stimulation results in both cardiac parasympathetic and sympathetic activation [33]. The major cardiac effect, however, appears related to parasympathetic upregulation (bradyarrhythmias and complete heart block).
Conclusions
Considerable evidence for the role of the cortex in the regulation of cardiac rate and rhythm exists from historical sources, contemporary clinical observation and animal experimentation. There is a large body of evidence indicating that insular involvement may adversely affect cardiac prognosis after stroke. Whereas there is little doubt now that lateralization of cardiovascular function occurs within the cortex of many species, there is some discrepancy regarding the evidence as to whether lesions of the left or right insula may be more clinically significant. In some part, this may reflect interspecies extrapolations, state dependency of the observed responses, whether or not clinical data refer to lesions confined solely to the insular cortex (as in our reports) or merely involve the insula in addition to adjacent regions (as in the case of others). For example, we have identified an inhibitory input to the insula from adjacent regions; when damaged, but sparing the insula itself, this may result in cardiovascular disinhibition, an effect differing from involvement of the insula alone. On balance however, it is most likely that in the human, lesions ablating part or all of the left anterior insula and its efferent connections and ablation of inhibitory circuminsular efferents to the right insular cortex are of consequence with regard to determining cardiovascular outcomes after neurological damage. Temporal dephasing of the relationships between the neuraxially dispersed oscillators controlling sympathovagal activity is the likely underlying mechanism which, interacting with concomitant ischemic cardiac and vascular disease if present, poses a potential threat to the integrity and function of the heart. These effects may not be manifest in the acute phase, but may become so chronically, especially with central synaptic reorganization after stroke. Environmental, and especially psychological, stressors may activate a reorganized system leading to upregulation and dysfunction of sympathetic neural input with resultant cardiac arrhythmia generation, cardiac structural damage and decompensation. Clearly, long-term follow-up studies of stroke patients could be exceptionally useful to determine the validity of this argument.
References
Aschenbrenner R, Bodechtel G (1938) Ueber EKG veranderungen bie hirntumorkranken. Klin Wochen 17:298–02
Barron SA, Rogovski Z, Hemli J (1994) Autonomic consequences of cerebral hemisphere infarction. Stroke 25:113–16
Beattie J, Brow GR, Long CNH (1930) Physiological and anatomical evidence for the existence of nerve tracts connecting the hypothalamus with spinal sympathetic centers. Proc R Soc Lond (Biol) 106:253–75
Butcher KS, Hachinski V, Cechetto DF (1995) Insular lesion evokes autonomic effects of stroke in normotensive and hypertensive rats. Stroke 26:459–65
Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS (2000) Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation 101:1960–969
Cechetto DF, Saper CB (1987) Evidence for a viscerotopic sensory representation in the cortex and thalamus in the rat. J Comp Neurol 262:27–5
Chen PS, Chen LS, Cao JM, Sharifi B, Karagueuzian HS, Fishbein MC (2001) Sympathetic nerve sprouting, electrical remodeling and the mechanisms of sudden cardiac death. Cardiovasc Res 50:409–16
Chua HC, Sen S, Cosgriff RF, Gerstenblith G, Beauchamp N, Oppenheimer S (1999) Neurogenic ST depression in stroke. Clin Neurol Neurosurg 101:44–8
Cropp GJ, Manning GW (1960) Electrocardiographic changes simulating myocardial ischemia and infarction associated with spontaneous intracranial hemorrhage. Circulation 22:25–8
DiPasquale G, Pinelli G, Andreoli A (1987) Holter detection of cardiac arrhythmias in intracranial subarachnoid hemorrhage. Am J Cardiol 59:596–00
Erciyas AH, Topalkara K, Topaktas S, Akyuz A, Dener S (1999) Suppression of cardiac parasympathetic functions in patients with right hemispheric stroke. Eur J Neurol 6:685–90
Eckardt M, Gerlach L, Welter FL (1999) Prolongation of the frequency-corrected QT dispersion following cerebral strokes with involvement of the insula of Reil. Eur Neurol 42:190–93
Giubilei F, Strano S, Lino S, Calcagnini G, Tisei P, Fiorelli M, Ferretti C, Cerutti S, Fieschi C (1998) Autonomic nervous activity during sleep in middle cerebral artery infarction. Cerebrovasc Dis 8:118–23
Goldstein DS (1979) The electrocardiogram in stroke: relationship to pathophysiological type and comparison with prior tracings. Stroke 10:253–59
Greenhoot JH, Reichenbach DD (1969) Cardiac injury and subarachnoid hemorrhage: a clinical, pathologic, and physiologic correlation. J Neurosurg 30:521–31
Hachinski VC, Cechetto DF, Guiraudon C, Wilson JX, Oppenheimer SM (1992) Asymmetry of the cardiovascular consequences of stroke. Arch Neurol 49:697–02
Hertzer NR, Young JR, Beven EG, Graor RA, O’Hara PJ, Ruschhaupt WF 3rd, deWolfe VG, Maljovec LC (1985) Coronary angiography in 506 patients with extracranial cerebrovascular disease. Arch Intern Med 145:849–52
Kolin A, Norris JW (1984) Myocardial damage from acute cerebral lesions. Stroke 15:990–93
Kono T, Morita H, Kurowa T, Onaka H, Takatsuka H, Fujiwara A (1994) Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: neurogenic stunned myocardium. J Am Coll Cardiol 24:636–40
Korpelainen JT, Sotaniemi KA, Huikuri HV, Myllyla VV (1997) Circadian rhythm of heart rate variability is reversibly abolished in ischemic stroke. Stroke 28:2150–154
Korpelainen JT, Sotaniemi KA, Makikallio A, Huikuri HV, Myllyla VV (1999) Dynamic behavior of heart rate in ischemic stroke. Stroke 30:1008–013
Lane R, Wallace J, Petrovsky P (1992) Supraventricular tachycardia in patients with right hemisphere strokes. Stroke 23:362–66
Lavy S, Yaar I, Melamed E (1974) The effect of acute stroke on cardiac functions as observed in an intensive stroke care unit. Stroke 5:775–80
Levy AG (1913) The exciting causes of ventricular fibrillation in animals under chloroform anesthesia. Heart 4:319–26
Levy AG (1919) Further remarks on ventricular extrasystoles and fibrillation under chloroform. Heart 7:105–12
Li C, Dong W (1999) Abnormal dynamic electrocardiogram in patients with acute cerebral infarction. Zhonghua Nei Ke Za Zhi 8:239–41
Li GR, Feng J, Yue L, Carrier M (1998) Transmural heterogeneity of action potentials and Ito1 in myocytes isolated from the human right ventricle. Am J Physiol 27:H369–H377
Manitius-Robeck S, Schuler P, Feistel H, Platsch G, Stefan H (1998) Ictal syncopes. Cardiac sympathetic innervation disorder as the etiology? Nervenarzt 69:712–16
Naver HK, Blomstrand C, Wallin BG (1996) Reduced heart rate variability after right-sided stroke. Stroke 27:247–51
Neuberger K (1933) Ueber die Herzmuskelveraenderungen bei Epileptikern und ihre Bezichungen zur Angina Pectoris. Frnkfurter Zietsch Patholog 46:14–1
Norris JW, Froggatt GM, Hachinski V (1978) Cardiac arrhythmias in acute stroke. Stroke 4:392–96
Oppenheimer SM, Cechetto DF (1990) Cardiac chronotropic organization of the rat insular cortex. Brain Res 533:66–2
Oppenheimer SM, Wilson JX, Guiraudon C, Cechetto DF (1991) Insular cortex stimulation produces lethal cardiac arrhythmias: a mechanism of sudden death? Brain Res 550:115–21
Oppenheimer SM, Gelb AW, Girvin JP, Hachinski VC (1992) Cardiovascular effects of human insular stimulation. Neurology 42:1727–732
Oppenheimer SM, Martin WM, Kedem G (1996) Left insular cortex lesions perturb cardiac autonomic tone. Clin Auton Res 6:131–40
Oppenheimer SM, Zhang ZH, Boekholdt M (1998) Electrical stimulation of the right posterior insular cortex increases cardiac sympathetic tone in the rat. Soc Neurosci Abstracts 24:1134
Oppenheimer SM, Zhang ZH (1999) Effects of bilateral stimulation of the posterior insula on lateral hypothalamic baroreceptor related neurons in the rat. Soc Neurosci Abstracts 25:1957
Orlandi G, Fanucchi S, Strata G, Pataleo L, Landucci Pellegrini L, Prontera C, Martini A, Murri L (2000) Transient autonomic nervous system dysfunction during hyperacute stroke. Acta Neurol Scand 102:317–21
Rem JA, Hachinski V, Boughner D (1985) Value of electrocardiographic monitoring and echocardiography in TIA and stroke patients. Stroke 16:950–56
Robinson TG, Dawson SL, Eames PJ, Panerai RB, Potter JF (2003) Cardiac baroreceptor sensitivity predicts long-term outcome after acute ischemic stroke. Stroke 34:705–12
Saito D, Shiraki T, Oka T, Kajiyama A, Takamura T (2002) Risk factors indicating recurrent myocardial infarction after recovery from acute myocardial infarction. Circ J 66:877–80
Sander D, Klingelhofer J (1994) Changes of circadian blood pressure patterns after hemodynamic and thromboembolic brain infarction. Stroke 25:1730–737
Sander D, Klingelhofer J (1995) Changes of circadian blood pressure patterns and cardiovascular parameters indicate lateralization of sympathetic activation following hemispheric brain infarction. J Neurol 242:313–18
Sander D, Klingelhofer J (1996) Extent of autonomic activation following cerebral ischemia is different in hypertensive and normotensive humans. Arch Neurol 53:890–94
Strittmatter M, Meyer S, Fischer C, Georg T, Schmitz B (2003) Location-dependent patterns in cardio-autonomic dysfunction in ischaemic stroke. Eur Neurol 50:30–8
Svigelj V, Grad A, Tekavcic I, Kiauta T (1994) Cardiac arrhythmia associated with reversible damage to insula in a patient with subarachnoid hemorrhage. Stroke 25:1053–055
Taggart P, Carruthers M, Somerville W (1973) Electrocardiogram, plasma catecholamines and lipids and their modification by oxprenolol when speaking before an audience. Lancet 18:341–46
Takei M, Sasaki Y, Yonezawa T, Lakhe M, Aruga M, Kiyosawa K (1999) The autonomic control of the transmural dispersion of ventricular repolarization in anesthetized dogs. J Cardiovasc Electrophysiol 10:981–89
Toivonen L, Helenius K, Viitasalo M (1997) Electrocardiographic repolarization during stress from awakening on alarm call. J Am Coll Cardiol 30:774–79
Tokgozoglu SL, Batur MK, Top uoglu MA, Saribas O, Kes S, Oto A (1999) Effects of stroke localization on cardiac autonomic balance and sudden death. Stroke 30:1307–311
Wong KY, Mac Walter RS, Douglas D, Fraser HW, Ogston SA, Struthers AD (2003) Long QTc predicts future cardiac death in stroke survivors. Heart 89:377–81
Yamour BJ, Sridharan MR, Rice JF (1980) Electrocardiographic changes in cerebrovascular hemorrhage. Am Heart J 99:294–00
Zamrini EY, Meador KJ, Loring DW, Nichols FT, Lee GP, Figueroa RE (1990) Unilateral cerebral inactivation produces differential right and left heart rate responses. Neurology 40:1408–411
Zhang ZH, Oppenheimer SM (1998) Insular cortex lesions alter baroreceptor sensitivity in the urethane-anesthetized rat. Brain Res 813:73–1
Acknowledgment
This review contains the basis of a lecture given as part of the Satellite Symposium on the Vagus Nerve Stimulation, Sudden Death in Epilepsy, and the Autonomic Nervous System at the 14th International Symposium on the Autonomic Nervous System. St. Thomas, US Virgin Islands, 5th–8th November 2003. This was part funded by PHS grants: PHS NS33770 and RR-00052.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Oppenheimer, S. Cerebrogenic cardiac arrhythmias:. Clin Auton Res 16, 6–11 (2006). https://doi.org/10.1007/s10286-006-0276-0
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s10286-006-0276-0