
Abstract
Calcitonin gene-related peptide (CGRP) is linked to neurogenic inflammation and to migraine. Activation of the trigeminovascular system plays a prominent role during migraine attacks with the release of CGRP. The trigeminal ganglion (TG) contains three main cell types: neurons, satellite glial cells (SGC) and Schwann cells; the first two have before been studied in vitro separately. Culture of rat TG provides a method to induce inflammation and the possibility to evaluate the different cell types in the TG simultaneously. We investigated expression levels of various inflammatory cytokines on mRNA level using RT-PCR arrays and qRT-PCR; furthermore expression at protein level was studied using immunohistochemistry. We report that (1) organ culture of the TG is possible with preserved morphology, (2) organ culture is associated with enhanced expression of cytokines and mitogen-activated protein kinases (MAPKs) primarily in neurons, (3) CGRP can induce expression of some cytokines and (4) cytokine expression is still upregulated following MAPK pathway inhibition by MEK inhibitor U0126 and pp38 inhibitor SB202192, but the cytokine expression is abolished when co-incubating with the JNK inhibitor SP600125. This method may be of value to examine local TG inflammation, putatively involved in the pathophysiology of some forms of primary headaches.
Similar content being viewed by others

Introduction
There is a growing body of evidence to suggest that certain forms of primary headache disorders and inflammation are linked together, and often the term neurogenic inflammation is used. Activation of the trigeminal system has been shown to be associated with release of vasoactive neuropeptides such as calcitonin gene-related peptide (CGRP) and substance P (SP), while acute migraine or cluster headache attacks result in CGRP release [1, 2]. Detailed study of the trigeminal ganglia (TG) revealed that there are two types of neurons: large ones without CGRP, but with CGRP receptors, and small-to-mid-sized neurons that are CGRP positive but lack CGRP receptors [3]. More than 90% of cells in the TG are satellite glial cells (SGC) and Schwann cells, both of which are CGRP negative but some SGC have CGRP receptors [3, 4]. Thalakoti et al. [5] have shown that activation of the V3 branch of the TG can result in transganglionic activation causing upregulation of both mitogen-activated protein kinase (MAPK) and inflammatory proteins, and the upregulation can even be seen in the V1 and V2 regions of the TG. This activation may hypothetically occur via intraganglionic spread of activity involving both neurons and glia. It is further hypothesized that this activity may lead to downstream activation of pro-inflammatory mediators such as interleukin 1 (IL-1) and bradykinin [6]. In support, there are studies showing an increased level of the pro-inflammatory tumour necrosis factor α (TNFα) in acute as well as chronic migraine patients when compared with tension type headache and controls [7–9]; in addition, TNFα may increase the production of CGRP [10]. Administration of nitroglycerine can result in an inflammatory response in the dura mater with upregulation of IL-1β and IL-6 [11]. The cytokine release from SGC has been shown to sensitize nociceptive neurons in the TG [12]. Such data support the possibility that in some forms of persistent primary headaches inflammatory mediators add to the pathophysiology [2].
Traditionally, glial cells have been considered merely as supportive cells keeping the neurons together, but this view is changing [13]. A recent report infers signalling between neurons and SCGs through gap junctions [5]; however, other work on CGRP translocation suggests otherwise [14, 15]. SGCs form an encapsulation of each of the neurons and this isolating boundary has been hypothesized to regulate signalling to and from neurons in the TG [16].
There is enhanced expression of CGRP following TG neuronal cell culture [17, 18], and glial cells have been implicated in localized inflammation [18]. Since some SGC have CGRP receptors [3, 4] they may be part of a local mechanism, where IL-1β located in SGCs is upregulated by CGRP released from the TG neurons, putatively in a positive feedback loop [19]. Furthermore, cytokines are known to activate different signalling pathways such as the MAP kinase signalling cascade [17]. TNFα has in vitro been shown to phosphorylate and hence activate, the inactive forms of extracellular signal-regulated kinase (ERK), p38 proteins and Jun N-terminal kinases (JNK) [20]. The transcription of CGRP is regulated by MAP kinases at least in the TG, possibly linking this signalling cascade to the onset of—or regulation in migraine [21].
Since previous studies mainly involved either culture of TG neurons or SGC [17, 18, 22–24], the aim of this study was to (1) examine if it was possible to organ culture intact TG and retain well-preserved morphology examined using IHC, (2) to study if organ culture was associated with enhanced expression of cytokines and MAPKs, (3) to evaluate if CGRP would be able to induce further expression of cytokines, and (4) if these effects could be blocked by specific MAPK pathway blockers. MAP kinases, cytokines and cytokine receptors were verified both at RNA and protein level using RT-PCR and immunohistochemistry.
Methods
Material
Sprague–Dawley rats (n = 24; male, b. w. 250 g) were used for tissue acquisition. The animals were kept at controlled temperature and humidity with free access to food and water. The experimental procedures were approved by the Danish veterinary authorities (#2006-561-1139). The animals were killed using CO2 and decapitation. Immediately after decapitation the main body of the TG was removed, but not the trigeminal nerve to the TNC or the main branches of the TG (I, II or III), and transferred to 1×Tris buffered saline (TBS) on ice.
Treatment of trigeminal ganglia
After dissection the main body of the fresh TGs they were directly transferred to either Trizol solution (Invitrogen, USA) for RNA extraction or embedded in Tissue-Tek (Sakura Finetek, USA) and snap frozen on dry ice for immunohistochemistry.
Whole TG (main body) used for tissue culture was transferred to 1 ml of Dulbeccos modified eagle medium (DMEM containing 1 g/L d-Glucose, l-Glutamine and Pyruvate) (Gibco, Invitrogen, USA) and incubated in a heat incubator at 37°C in the presence of 5% CO2 for 24 h. To avoid contamination, a mixture of penicillin (100 U/ml), streptomycin (100 μg/ml) and amphotericin B (25 μg/ml) (Invitrogen, USA) was added to the media prior to incubation. The TG tissues were either incubated alone or co-incubated with one of the following (all Sigma, USA): CGRP [10−6 M], CGRP8–37 [10−6 M], U0126 [10−6 M], SB202190 [38 × 10−9 M], SP600125 [10−5 M].
RNA extraction
Following the manufacturer’s protocols, total RNA preparations were obtained using Purelink a Trizol RNA isolation and purification kit (Invitrogen, USA). Briefly, the TG was homogenized using lysing matrix D tubes in a FastPrep fp120 machine (both MP Biomedicals, France) in the presence of 1 ml of Trizol. Centrifugation at 4°C was undertaken to remove fat and debris. The supernatant was transferred to a new tube for further processing. The samples were incubated at room temperature for 5 min and 0.2 ml of 1-Bromo-3-chloro-propane (Sigma, USA) was added. The samples were gently mixed by hand and incubated at room temperature for 3 min before centrifuging at 12,000g for 15 min at 4°C. After phase separation 400 μl of the upper phase containing RNA was transferred to a new tube and an equal amount of 70% ethanol was added. The obtained RNA was washed through a series of on column binding and washing steps before resuspending in 30 μl DEPC water. RNA concentration and purity was assessed using a NanoDrop 2000c machine (Thermo Fischer Scientific, USA). Both the 260/280 ratio and the 260/230 ratio were measured using 1 μl RNA dilute in the NanoDrop and typical values for both ratios were between 1.8 and 2.0 indicating a high degree of purity. Typical RNA concentrations were of 100 ng/μl RNA or more.
RT-PCR cytokine-assay
Reverse transcription of total RNA to single-strand cDNA was carried out using a RT2 First strand kit (SABiosciences, USA) in a GeneAmp PCR system 2400 (Perkin Elmer, USA) using 800 ng of RNA in a 20-μl reaction volume using random hexamers as primers. Real-time PCR was performed on a 7500 fast Real-Time PCR detection system (Applied Biosystems, USA), using 25 μl reactions with 1.13 μl of cDNA as template in a premade assay plate from SABioscience (PARN-021) detecting 84 different “common” cytokines and 6 controls (SABioscience, USA). For each treatment the detection was performed in triplicate and run for 40 cycles. The expressions were normalized to the internal on plate controls to account for differences in cDNA reaction efficiency.
The data were analysed using the online program RT2 profiler PCR array data analysis, provided by SABiosciences http://www.sabiosciences.com/pcr/arrayanalysis.php). The group of fresh samples was used as control group and compared with the organ-cultured treated group. Furthermore, to study effects of various treatments the group of organ cultured samples was compared with groups treated with CGRP, CGRP8–37, U0126, SB202190 and SP600125. To visualize the differences volcano plots are shown together with the accompanying fold change values [25]. Fold changes more than ±3 with a statistical significance of P < 0.05 are shown (Fig. 2).
qRT-PCR
Reverse transcription of total RNA to single-strand cDNA was carried out using a RT2 First strand kit (SABiosciences, USA) in a GeneAmp PCR system 2400 (Perkin Elmer, USA) using 800 ng of RNA in a 20-μl reaction volume using random hexamers as primers. Real-time PCR was performed on a 7500 fast Real-Time PCR detection system (Applied Biosystems, USA), using 25 μl reactions with 9 μl of cDNA as template in reaction mixes containing 10 μl Taqman fast universal PCR mastermix and 1 μl specific primer to amplify TNFα (Rn99999017) and rat GAPDH (4352338E), the latter as endogenous control (Applied Biosystems, USA).
Immunohistochemistry (IHC)
Fresh and in vitro cultured trigeminal ganglia were transferred to Tissue-Tek (Sakura Finetek, USA), snap frozen on dry ice and stored at −80°C until further processing. Longitudinal sectioning of the whole frozen TG containing proximal parts of regions V1–V3 was carried out in a CM3050s cryostat (Leica, Germany) with a section thickness of 14 μm. The TG sections were placed on super frosted tissue slides and left to dry out for no more than 2 h. Staining method in brief: samples were fixed for 20 min using Stefanini’s fixative (2% paraformaldehyde and picric acid 2%), permeabilized in 1× phosphate buffered saline (PBS) containing 0.25% triton X 100 (Sigma, USA) and blocked for 1 h to prevent unspecific primary antibody binding using 1× PBS with 0.25% triton X 100 and 1% bovine serum albumine (BSA) (dilution buffer) mixed with 5% donkey serum (Jackson Immunoresearch, USA). Samples were incubated overnight with primary antibodies (see Table 1) diluted in a buffer containing 2% donkey serum. The following day slides were incubated with secondary antibodies (Table 1) for 1 h and counterstained for DNA using the DNA-specific dye DAPI (Invitrogen, USA) 1:5,000 diluted in 1× PBS with 5 min incubation. Finally, the slides were mounted with Clearmount mounting media (Invitrogen, USA) and left at 60°C for 30 min to dry before proceeding to image capturing.
Experiments were setup as double staining where all samples were stained with Hoechst (Invitrogen, USA) to detect nuclei. All samples were primary antibody labelled with antibodies directed towards effectors and receptors known from migraine and inflammation (Table 1). All staining were conducted as doublets, and negative control omitting primary antibodies were routinely made in all staining batches.
Microscopy
Staining intensity and staining patterns in the samples were recorded using a Nikon C1 confocal microscope (Nikon, Japan). The microscopy was carried out using a 60× oil immersion lens with numerical aperture (NA) 1.4 in xy mode with an averaging of six scans in order to lower the contribution of unspecific random noise seen as background. The pictures were acquired using frame λ, meaning that the different laser channels were collected one by one instead as simultaneously. Fluorescence intensity measurements were performed as a region of interest for all samples in neuron-rich areas in the TG. The laser channels used were 405 nm excitation with filter 450/35 collecting Hoechst staining and 543 nm excitation with filter 605/75 collecting Dylight 549 (Jackson Immunoresearch, USA). Image and data analyses were conducted using NIS basic research software (Nikon, Japan).
Statistics
GraphPad Prism 5 (GraphPad, USA) was used for Statistical analysis except on the PCR-array data where the SABioscience online program was used. Analysis of data from qRT-PCR and IHC on TG material treated either as fresh, organ cultured or organ cultured in the presence of U0126 (Sigma, USA) was performed as one-way ANOVA. These data were post tested using Tukey’s multiple comparison test to test for differences between classes.
The immunohistochemistry receptor data from fresh versus organ-cultured TG tissue was tested using unpaired t test. Data are presented as ±standard error of the mean (SEM) and the significance *P < 0.05, **P < 0.01, ***P < 0.001 shown.
Results
Regulation of inflammatory cytokines after organ culture
Our goal was to investigate if culture of rat TG, in which the morphological relation between SGC and neurons was preserved, elicits an inflammatory type of response and if it is suitable for studying the effects of inflammation on neuron—SGC signalling. We examined the influence of TG 24 h incubation, a time-point based on previous studies [18], with and without various specific substances (see below), and compared with fresh TG using a RT-PCR array system, qRT-PCR and IHC.
Tissue culture
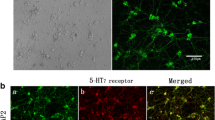
As can be seen in Fig. 1 the morphology was well preserved after 24 h of organ culture. There was a strong and significant upregulation of pro-inflammatory cytokines, which is in accordance with earlier work [26, 27]. IL-6 and leukaemia inhibiting factor (lif) were highly upregulated after organ culture (RT-PCR n = 3, IHC n = 6), and a significant down regulation was seen for a number of genes (Fig. 2a; Table 2).

IL-6 receptor localization in fresh and cultured TG tissue. An increase in fluorescence intensity was seen when going from fresh to incubated tissue, suggesting an inflammatory response. In the fresh TG IL-6R fluorescence was located in the cytoplasm (arrowheads), whereas in the cultured TG IL-6R fluorescence was located primarily close to the cell membrane in neurons and in SGC (arrows). Scale bar 50 μm (Blue colour the nuclear-specific stain DAPI, red colour the IL-6R) (colour figure online)

Volcano plots showing up/down regulation of cytokine genes as response to different treatments of the trigeminal ganglia body tissue. a Fresh versus organ culture, huge significant upregulation of the pro-inflammatory cytokines IL-6 and lif can be seen; furthermore, a number of cytokines are down regulated as response to organ culture. b Organ culture versus CGRP treatment, additional significant upregulation of a number of cytokines. c Organ culture versus CGRP8–37 treatment: significant down regulation of the pro-inflammatory cytokine IL-18; otherwise no effect of this treatment. d Organ culture versus U0126 treatment: significant upregulation of the anti-inflammatory cytokine IL-10; otherwise no effect of this treatment. e organ culture versus SB202190 treatment: little effect of this treatment. f Organ culture versus SP600125 treatment: significant down regulation of a number of cytokines indicating an anti-inflammatory effect of the JNK inhibitor. Red circles indicate upregulation and green circles indicate down regulation. Values are considered statistically significant when fold change is larger than 3 and P < 0.05 (above line) (colour figure online)
CGRP and CGRP8–37 co-incubation
To study a possible direct inflammatory link to migraine, the effect of co-incubation with CGRP (10−6 M) in the medium was investigated. Generally, we found a further upregulation of a number of cytokines, which is in agreement with results by Vause and Durham [18] (Fig. 2b; Table 2b). Having established the CGRP response, it was tested if the response could be abolished by use of the CGRP antagonist CGRP8–37 (10−6 M). This substance per se did not modify the cytokine expression much, although the pro-inflammatory cytokine IL-18 was significantly down regulated (Fig. 2c; Table 2c), but inhibited the CGRP effect (data not shown). Thus, we found CGRP to induce enhanced expression of some of the studied cytokines, whereas CGRP8–37 seems to have little effect on the expressional pattern.
qRT-PCR
After the PCR-array study we confirmed some of the results with quantitative PCR; thus, qRT-PCR of TNFα, which is one of the key cytokines in inflammation revealed regulatory effects. TNFα showed a highly significant upregulation (P < 0.001) when co-incubated with CGRP; this upregulation was somewhat counteracted by the addition of CGRP8–37 (P < 0.01), when compared with fresh tissues and other treatments (Fig. 3).

Regulation of TNFα in rat trigeminal ganglia following different in vitro culturing treatments. Up/down-regulation as compared with fresh TG. Data were tested using one-way ANOVA, and post tested with Tukeys multiple comparison test. Data are presented as ±standard error of the mean (SEM) **P < 0.01, ***P < 0.001
Protein immunohistochemistry
Following tissue culture (induced inflammation) IHC was used in order to show (1) localization of proteins, and to illustrate if (2) the mRNA upregulation corresponded to the protein expression. The IL-1 receptor was upregulated and found in the cytoplasm, whereas IL-1β had decreased fluorescence intensity and was observed in the TG neurons. The IL-6 receptor tended to be upregulated following culture while IL-6 showed a significant decrease in the cytoplasm of the TG neurons. The two toll-like receptors, TLR-2 and TLR-4, showed reduced fluorescence intensity after inflammation (Fig. 4).

Regulation of the pro-inflammatory cytokines IL1 and IL6, their receptors and toll-like receptor 2 and 4 in fresh versus cultured TG tissue. The interleukin receptors are upregulated following tissue incubation, whereas the interleukins are down-regulated after incubation. The toll-like receptors are downregulated after tissue incubation. Data were analysed using unpaired t test and results are presented as ±standard error of the mean (SEM) **P < 0.01, ***P < 0.001
The staining for most of the receptors and cytokines revealed an even cytoplasmic distribution that varied in intensity depending on treatment. The IL-6 receptor was evenly located in neuron cytoplasm of fresh TG, but when the TG tissue sections had been cultured there was a tendency towards receptor upregulation and receptor location shifted from an even cytoplasmic distribution in the neurons to being located in the outer membrane of the neurons. It could also be seen in the SGCs (Fig. 1).
Signal transduction
To investigate the regulatory means of the inflammatory response, different places in the MAP kinase signalling cascade were blocked with specific inhibitors of MAP kinases in order to obtain information on the regulatory status of the studied cytokines at the mRNA level. Vause and Durham [18] showed cytokine upregulation after 8 and 24 h of treatment with CGRP in glia-enriched cultures. In addition, they observed upregulation of several MAP kinases in the signalling cascades following treatment of CGRP.
mRNA level
We blocked the MEK1/2 step in the constitutively expressed MAP kinase pathway by co-incubating the TG tissue with U0126. Boldt and Kolch [28] have shown U0126 to specifically block the MEK1/2 step. When incubating TG in the presence of U0126 we still found an upregulation of the anti-inflammatory cytokine IL-10 with all other cytokines behaving as was seen for the organ-cultured state of TG tissue (Fig. 2; Table 2). SB202190, a p38 blocker [29], showed much the same response on gene expression regulation as found with U0126, namely almost none compared with organ culture per se (Fig. 2; Table 2). Co-incubation of TG tissue with SP600125, a JNK inhibitor [28], resulted in down regulation of a number of cytokine genes; hence, it seems this part of the MAPK signalling cascade has an impact on cytokine regulation in connection to neurogenic inflammation (Fig. 2; Table 2).
Protein level
Additionally, we assessed the influence of MAP kinase pathways on our inflammatory model at the protein level by immunohistochemistry. TG was treated as fresh, cultured or cultured in the presence of the specific MEK1/2 inhibitor U0126 [10−6 M]. When comparing organ-cultured TG with fresh tissue with IHC, there was a significant upregulation of phosphorylated ERK, p38 and JNK; thus, all three MAPK signalling pathways are activated by organ culture (Fig. 5a). Furthermore, U0126, on the other hand, tended to increase pERK1/2 and pp38 and hence to further increase in their fluoresce intensity. The fluorescence intensity measured for phosphorylated JNK returned to a value close to what was seen in fresh tissue (Fig. 5a), thus confirming the array results.

a Regulation of the pERK, pp38 and pJNK in fresh TG tissue compared with tissue incubation and tissue incubation with U0126. There is increased fluorescence intensity, interpreted as an upregulation, of all three pathways after incubating the tissue comparing with the fresh state. Furthermore, an additional fluorescence intensity increase is seen for pERK and pp38 after co-incubation with U0126. This increase is quenched in the case of pJNK. Data were tested using one-way ANOVA, and post tested with Tukey’s multiple comparison test. Data are presented as ±standard error of the mean (SEM) **P < 0.01, ***P < 0.001. b Phosphorylated ERP (pERK) localization in TG tissue as fresh, cultured and cultured in the presence of U0126. In the fresh condition most pERK was localized to the nucleus of the neuron (arrowheads). In the cultured ganglia pERK relocated and was found in the cytoplasm of large- and small-size neurons (arrows). The TG tissue co-incubated with U0126 had pERK located to the cytoplasm of large and small neurons, some SGC (arrows) and to nucleus of the neurons (arrowheads). Scale bar 20 μm (Blue colour the nuclear-specific stain DAPI, red colour pERK) (colour figure online)
Fresh tissue showed slight staining for pERK1/2 in the neuronal nucleus (Fig. 5b). After culture the pERK1/2 fluorescence signal intensified and was now apparent in the cytoplasm of the neurons (Fig. 5). When co-incubated with U0126, the fluorescence intensity further intensified and the staining was again found in the nucleus and also in the cytoplasm of the neurons (Fig. 5b). Staining for pp38 and pJNK revealed that both located to the cytoplasm of the TG neurons, and it did not change localization after the different treatments (data not shown) (Fig. 5a).
Discussion
The present study contains several novel observations: (1) organ culturing of the TG is technically possible and the morphology is well preserved as examined with immunohistochemistry, (2) organ culture is associated with enhanced expression of cytokines and MAPKs, (3) CGRP can induce further expression of some cytokines, (4) cytokine expression is not modified following MAPK pathway inhibition by MEK inhibitor U0126 and pp38 inhibitor SB202192, but the cytokine expression returns roughly to fresh tissue levels when co-incubating with the JNK inhibitor SP600125 when studied using IHC. Obviously, organ culture of the whole trigeminal ganglion or of the individual cell types, neuron or glial cells, are not models of any primary headache type. However, they provide a tool to understand at some depth how neurons and glial cells may act and interact, molecular mechanisms involved and receptors present. By further such experiments we may advance the understanding of how this tissue may react in pathophysiological situations.
Our initial aim was to assess and validate a model for the study of in vitro induced inflammation through organ/tissue culture for 24 h [6]. Since some forms of primary headaches are putatively associated with inflammation, this method could provide novel information regarding the regulation of cytokines in the TG, a key structure in this group of disorders. It was possible to provoke an inflammatory type of response in rat trigeminal ganglion by whole tissue incubation for 24 h. Furthermore, it was observed that pro-inflammatory cytokines IL-6 and lif were highly upregulated following incubation. The data strongly suggest that we can use this model to study in vitro inflammatory effects on ganglionic tissue (Fig. 2; Table 2). We have recently revealed that small- to mid-sized neurons in the human and rat TG contain CGRP; large-size neurons and SGCs, on the other hand, contain CGRP receptor elements [3]. This may suggest intraganglionic transmission.
CGRP is recognized as a major contributor involved in migraine headache [1, 30]. As such it was interesting to study if exogenously applied CGRP would modify the cytokine expression; hence we co-incubated with CGRP and its blocker. When incubating the TG with CGRP a significant upregulation was seen for some of the cytokines studied as compared with organ-cultured tissue; this is in accord with an earlier study on isolated glial cells [18]. The inflammatory effect shown for organ culture was not much affected by co-incubation with the CGRP inhibitor CGRP8–37 although there was a significant down regulation of the pro-inflammatory cytokine IL-18. Furthermore, the role of CGRP on TNFα expression was studied using qRT-PCR on the ganglia. It was revealed that co-incubation with CGRP resulted in a pronounced and significant increase in expression, which in part could be attenuated by co-incubating with the CGRP blocker CGRP8–37 (Fig. 3). In addition, CGRP was shown to be upregulated at protein level following organ culture as studied with immunohistochemistry, where a shift from unspecific cytoplasmic in the fresh TG to a more granular/vesicular distribution of CGRP was noted after organ culture [6].
To investigate if MAPK signalling cascades were involved in the inflammatory responses, measured as cytokine regulation, we co-incubated the TG tissue with specific MAPK antagonists; the MEK1/2 inhibitor U0126, the p38 blocker SB202190 and the JNK inhibitor SP600125. Little effect on the studied cytokines was seen after treatment with the MEK1/2 inhibitor U0126 and the p38 blocker SB202190 compared with organ culture, whereas the JNK inhibitor SP600125 elicited down regulation of a number of the studied cytokines (Fig. 2; Table 2).
When blocking the MEK/ERK1/2 pathway with U0126 we saw an upregulation at the protein level when studying the pERK1/2 and pp38 expression with immunohistochemistry (Fig. 5). It is fairly controversial to find a significant upregulation of activated ERK and p38 (pERK and pp38, respectively), after selectively having blocked the MEK/ERK phosphorylation step in the MAPK pathway (Fig. 5), but MAP kinases are placed downstream of the inflammatory cytokines in the signalling cascade; hence the upregulation seen may infer inactivation of a negative feedback loop. It is noteworthy though, that the staining of pERK goes from being predominantly occurring in the nucleus, with some staining in nucleus for the fresh tissue, to cytoplasmic in neurons and some SGC in the cultured tissue and last onto being localized in cytoplasm and in the nucleus of neurons and in the cytoplasm of some SGC after co-incubating the TG tissue with U0126 (Fig. 5b). There may be an underlying time relation we do not recover at the time points we study here. It would be fair to assume that initially U0126 blocks upregulation, but at 24 h it is not blocked and can be seen at high level as we observed here. In favour of this hypothesis we have data from rat arteries showing similar results [31]. After 24 h of organ culture of the rat arteries in the presence of U0126 both pERK and pp38 are significantly upregulated compared with fresh vessels. Furthermore, it was possible to show a return to control level for the expression of the endothelin B receptor after 24 h co-incubation with U0126 [31].
When phosphorylation of p38 was blocked with SB202190 the same pronounced upregulation at protein level was seen as was found when co-incubating the tissue with U0126. The pp38 staining was localized uniformly to the cytoplasm of the neurons for all three different treatments (data not shown) with a significant successive increase in pp38 fluorescence, going from fresh to incubated in the presence of SB202190 (Fig. 5a).
Finally, when incubating the TG tissue with the JNK inhibitor SP600125 down regulation of many cytokines was seen (Fig. 2; Table 2) which correlates with the results found using IHC. The pJNK immunoreactivity was located in the cytoplasm (data not shown) and there was a significant fluorescence increase after tissue incubation (Fig. 5a) which could be quenched by co-incubating with the JNK inhibitor SP600125.
The observed upregulation of activated MAP kinases (Fig. 5a) after incubation with U0126 and SB202190 is probably not attributed to cytotoxicity since both U0126 and SB202190 have low degree of in vivo toxicity [32, 33]. However, it appears that inhibiting the SAPK/JNK pathway lowers the degree of inflammatory response seen as cytokine expression in the tissue both at mRNA and protein levels. Furthermore, recent results from our laboratory on isolated blood vessels show a similar up-regulation of pERK and pp38 after 24 h organ culture, whereas the endothelin B (ETB) receptor was down-regulated on protein level after 24 h organ culture. Together, these data suggest a transient inhibition of the phosphorylation of ERK/p38 which in turn will cause a down-regulation of the ETB receptor. It could be hypothesized that the inflammatory effects of 24 h organ culture overcome the inhibitory effects of both U0126 and SB202190, and instead spin into a positive feedback loop which could enhance the upregulation of certain pro-inflammatory cytokines.
The study has revealed a complex interaction between different cell types in the TG and the activation of MAPKs and the production of cytokines. The method may be of use in dissecting the various components involved in local inflammation in the TG, the involvement of CGRP and its receptor.
References
Goadsby PJ, Edvinsson L, Ekman R (1990) Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 28:183–187, 1:STN:280:DyaK3M%2FisVSktQ%3D%3D, 10.1002/ana.410280213, 1699472
Waeber C, Moskowitz MA (2005) Migraine as an inflammatory disorder. Neurology 64:S9–S15, 15911785, 15911785
Eftekhari S, Salvatore CA, Calamari A, Kane SA, Tajti J, Edvinsson L (2010) Differential distribution of calcitonin gene-related peptide (CGRP) and CGRP receptor components (CLR and RAMP1) in the human trigeminal ganglion. Neuroscience 169:683–696, 1:CAS:528:DC%2BC3cXos1ymt7Y%3D, 10.1016/j.neuroscience.2010.05.016, 20472035
Lennerz JK, Ruhle V, Ceppa EP, Neuhuber WL, Bunnett NW, Grady EF et al (2008) Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J Comp Neurol 507:1277–1299, 1:CAS:528:DC%2BD1cXjt1Sltbc%3D, 10.1002/cne.21607, 18186028
Thalakoti S, Patil VV, Damodaram S, Vause CV, Langford LE, Freeman SE et al (2007) Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache 47:1008–1023, 10.1111/j.1526-4610.2007.00854.x, 17635592
Kristiansen KA, Edvinsson L (2010) Expressional change in calcitonin gene-related peptide, receptor activity modifying protein 1 (RAMP1) and bradykinin 1 receptor after experimen-tal inflammation in rat trigeminal ganglia. Curr Neurobiol 1:82–88, 1:CAS:528:DC%2BC3cXksVKjt70%3D
Bockowski L, Sobaniec W, Zelazowska-Rutkowska B (2009) Proinflammatory plasma cytokines in children with migraine. Pediatr Neurol 41:17–21, 10.1016/j.pediatrneurol.2009.02.001, 19520268
Perini F, D’Andrea G, Galloni E, Pignatelli F, Billo G, Alba S et al (2005) Plasma cytokine levels in migraineurs and controls. Headache 45:926–931, 10.1111/j.1526-4610.2005.05135.x, 15985111
Fidan I, Yuksel S, Ymir T, Irkec C, Aksakal FN (2006) The importance of cytokines, chemokines and nitric oxide in pathophysiology of migraine. J Neuroimmunol 171:184–188, 1:CAS:528:DC%2BD28Xkt1yr, 10.1016/j.jneuroim.2005.10.005, 16325275
Durham PL (2006) Calcitonin gene-related peptide (CGRP) and migraine. Headache 46(Suppl 1):S3–S8, 10.1111/j.1526-4610.2006.00483.x, 16927957
Reuter U, Bolay H, Jansen-Olesen I, Chiarugi A, Sanchez del RM, Letourneau R et al (2001) Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain 124:2490–2502, 1:STN:280:DC%2BD3MnovFKiuw%3D%3D, 10.1093/brain/124.12.2490, 11701602
Takeda M, Tanimoto T, Kadoi J, Nasu M, Takahashi M, Kitagawa J et al (2007) Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain 129:155–166, 1:CAS:528:DC%2BD2sXkt1eht7s%3D, 10.1016/j.pain.2006.10.007, 17127002
Allen NJ, Barres BA (2009) Neuroscience: glia—more than just brain glue. Nature 457:675–677, 1:CAS:528:DC%2BD1MXhsVCrtr4%3D, 10.1038/457675a, 19194443
Tajti J, Kuris A, Vecsei L, Xu C-B, Edvinsson L (2010) Organ culture of the trigeminal ganglion induces enhanced expression of calcitonin gene-related peptide via activation of extracellular signal-regulated protein kinase 1/2. Cephalalgia. doi:10.1177/0333102410382796
Park K-Y, Fletcher JR, Raddant AC, Russo AF (2010) Epigenetic regulation of the calcitonin/calcitonin gene-related peptide gene in trigeminal glia. Cephalalgia
Hanani M (2005) Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev 48:457–476, 1:CAS:528:DC%2BD2MXks1Cksrk%3D, 10.1016/j.brainresrev.2004.09.001, 15914252
Bowen EJ, Schmidt TW, Firm CS, Russo AF, Durham PL (2006) Tumor necrosis factor-alpha stimulation of calcitonin gene-related peptide expression and secretion from rat trigeminal ganglion neurons. J Neurochem 96:65–77, 1:CAS:528:DC%2BD28XmvF2kug%3D%3D, 10.1111/j.1471-4159.2005.03524.x, 16277606
Vause CV, Durham PL (2010) Calcitonin gene-related peptide differentially regulates gene and protein expression in trigeminal glia cells: findings from array analysis. Neurosci Lett 473:163–167, 1:CAS:528:DC%2BC3cXjvFKrurY%3D, 10.1016/j.neulet.2010.01.074, 20138125
Capuano A, De CA, Lisi L, Tringali G, Navarra P, Dello RC (2009) Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: relevance for migraine pathology. Mol Pain 5:43, 10.1186/1744-8069-5-43, 19660121
Barbin G, Roisin MP, Zalc B (2001) Tumor necrosis factor alpha activates the phosphorylation of ERK, SAPK/JNK, and P38 kinase in primary cultures of neurons. Neurochem Res 26:107–112, 1:CAS:528:DC%2BD3MXlsVKgtr4%3D, 10.1023/A:1011086426652, 11478736
Durham PL, Russo AF (2003) Stimulation of the calcitonin gene-related peptide enhancer by mitogen-activated protein kinases and repression by an antimigraine drug in trigeminal ganglia neurons. J Neurosci 23:807–815, 1:CAS:528:DC%2BD3sXhtlOrsbY%3D, 12574409
Bellamy J, Bowen EJ, Russo AF, Durham PL (2006) Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci 23:2057–2066, 10.1111/j.1460-9568.2006.04742.x, 16630053
Vause C, Bowen E, Spierings E, Durham P (2007) Effect of carbon dioxide on calcitonin gene-related peptide secretion from trigeminal neurons. Headache 47:1385–1397, 18052948, 18052948
Vause CV, Durham PL (2009) CGRP stimulation of iNOS and NO release from trigeminal ganglion glial cells involves mitogen-activated protein kinase pathways. J Neurochem 110:811–821, 1:CAS:528:DC%2BD1MXptlensrg%3D, 10.1111/j.1471-4159.2009.06154.x, 19457095
Ogawa T, Homma T, Igawa Y, Seki S, Ishizuka O, Imamura T et al (2010) CXCR3 binding chemokine and TNFSF14 over expression in bladder urothelium of patients with ulcerative interstitial cystitis. J Urol 183:1206–1212, 1:CAS:528:DC%2BC3cXjt1Gktrw%3D, 10.1016/j.juro.2009.11.007, 20096889,
Silver RF, Walrath J, Lee H, Jacobson BA, Horton H, Bowman MR et al (2009) Human alveolar macrophage gene responses to Mycobacterium tuberculosis strains H37Ra and H37Rv. Am J Respir Cell Mol Biol 40:491–504, 1:CAS:528:DC%2BD1MXktVWrtL8%3D, 10.1165/rcmb.2008-0219OC, 18787177
Myskiw C, Arsenio J, van BR, Deschambault Y, Cao J (2009) Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathways. J Virol 83:6757–6768, 1:CAS:528:DC%2BD1MXnvF2lurw%3D, 10.1128/JVI.02570-08, 19369349
Boldt S, Kolch W (2004) Targeting MAPK signalling: prometheus’ fire or pandora’s box? Curr Pharm Des 10:1885–1905, 1:CAS:528:DC%2BD2cXktV2gt7w%3D, 10.2174/1381612043384420, 15180526
Manthey CL, Wang SW, Kinney SD, Yao Z (1998) SB202190, a selective inhibitor of p38 mitogen-activated protein kinase, is a powerful regulator of LPS-induced mRNAs in monocytes. J Leukoc Biol 64:409–417, 1:CAS:528:DyaK1cXmtVOlsLo%3D, 9738669
Edvinsson L (2008) CGRP-receptor antagonism in migraine treatment. Lancet 372:2089–2090, 10.1016/S0140-6736(08)61710-9, 19036426
Ansar S, Edvinsson L (2008) Subtype activation and interaction of protein kinase C and mitogen-activated protein kinase controlling receptor expression in cerebral arteries and microvessels after subarachnoid hemorrhage. Stroke 39:185–190, 1:CAS:528:DC%2BD2sXhsVehs73I, 10.1161/STROKEAHA.107.487827, 18032736
Horstmann S, Kahle PJ, Borasio GD (1998) Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J Neurosci Res 52:483–490, 1:CAS:528:DyaK1cXjt1Gqs70%3D, 10.1002/(SICI)1097-4547(19980515)52:4<483::AID-JNR12>3.0.CO;2-4, 9589393
Martin P, Poggi MC, Chambard JC, Boulukos KE, Pognonec P (2006) Low dose cadmium poisoning results in sustained ERK phosphorylation and caspase activation. Biochem Biophys Res Commun 350:803–807, 1:CAS:528:DC%2BD28XhtVyisbbL, 10.1016/j.bbrc.2006.09.126, 17027645
Acknowledgments
This work was supported by the Lundbeck Foundation via the Lundbeck Foundation Center for Neurovascular Signalling (LUCENS).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kristiansen, K.A., Edvinsson, L. Neurogenic inflammation: a study of rat trigeminal ganglion. J Headache Pain 11, 485–495 (2010). https://doi.org/10.1007/s10194-010-0260-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10194-010-0260-x