
Abstract
Cell death is critical to normal homeostasis, although this process, when increased aberrantly, can lead to the production of pro-inflammatory mediators promoting autoimmunity. Two novel intercellular mediators of inflammation generated during cell death are high mobility group box 1 (HMGB1) protein and microparticles (MPs). HMGB1 is a nuclear protein that functions in transcription when inside the nucleus but takes on pro-inflammatory properties when released during cell death. Microparticles are small, membrane-bound structures that extrude from cells when they die and contain cell surface proteins and nuclear material from their parent cells. MPs circulate widely throughout the vasculature and mediate long-distance communication between cells. Both MPs and HMGB1 have been implicated in the pathogenesis of a broad spectrum of inflammatory diseases, including the prototypic autoimmune conditions systemic lupus erythematosus and rheumatoid arthritis. Given their range of activity and association with active disease, both structures may prove to be targets for effective therapy in these and other disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cell death is a ubiquitous and inevitable process that normally occurs without clinically evident immunologic sequelae. In the setting of inflammatory and autoimmune diseases, however, an increase in the extent of cell death or defects in the clearance of dead cell debris may contribute significantly to immune disturbances underlying autoimmunity. An increasing body of evidence suggests that two major products of cell death, extracellular high mobility group box protein 1 (HMGB1) and cellular microparticles (MPs), have important roles in inflammation and the pathogenesis of prototypic autoimmune conditions such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). These structures may also arise during cell activation, although the close linkage between immune cell activation and activation-induced cell death may complicate interpretation of their origin.
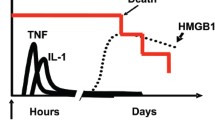
Both HMGB1 and MPs are released during the death of cells, and both induce pro-inflammatory cytokine expression, as illustrated in Fig. 1. HMGB1 is a non-histone, DNA-binding nuclear protein that has dual function. Within the nucleus, HMGB1 binds to DNA and regulates transcription and chromosome architecture. In its extracellular form, however, HMGB1 functions as a pro-inflammatory cytokine. In contrast, MPs are small, membrane-bound vesicles that display surface markers and nucleic components characteristic of the parent cells. While MPs are present in the peripheral blood of healthy individuals, marked elevations occur in many disease states characterized by high cell turnover or cell death. Furthermore, MPs can function as disease effectors, playing a role in local and long-range signaling in cellular processes that underlie inflammation and thrombosis. The association between increased blood levels of HMGB1 and MPs with active disease provides tantalizing new clues about the mechanisms of inflammation in autoimmunity, and suggests potential targets for therapeutic intervention.

This schematic depicts MP and HMBG1 release from cells and subsequent immunologic effects. Microparticles (MPs) and extracellular HMGB1 share several similar biological activities. Both MPs and HMGB1 are released from several cell types following activation, necrosis or apoptosis. MPs achieve release from cells via budding and retain cell surface markers of parent cells. HMBG1 protein becomes biologically active after release from the nucleus and extrusion into the extracellular space. Both MPs and HMBG1 exert pro-inflammatory effects, including expression of inflammatory genes and cytokines, upregulation of endothelial cell adhesion molecules, and stimulation of dendritic cells
HMGB1 and its function
HMGB1 is a 30 kDa non-histone, chromatin-binding protein ubiquitously expressed in eukaryotic cells and highly preserved across mammalian species [1]. HMGB1 contains 215 amino acids and has a tripartite structure consisting of two DNA-binding domains, the A box and the B box, and a C-terminal tail domain [2, 3]. Unlike histones, HMGB1 binds to DNA with low affinity and can move from the nucleus to the cytoplasm depending upon cell cycle phase [4].
Functionally, the role of HMGB1 depends upon its location. When inside the nucleus, HMGB1 acts as an architectural protein that binds to DNA and can impact transcription. HMBG1 recognizes particular DNA conformations (e.g., bent DNA) rather than specific nucleic acid sequences and binds in the minor groove of the DNA helix. As a result, HMGB1 can distort DNA and thereby enhance interactions with several proteins, including p53, NF-κB, progesterone receptors, estrogen receptors, and glucocorticoid receptors [5–7]. HMGB1 appears essential for survival, as suggested by its evolutionary conservation as well as the observation that HMGB1 knockout mice succumb to hypoglycemia within 24 h of birth; death in the knockout mice likely results from impaired activation of glucocorticoid receptor-responsive genes [8]. Thus, by recognizing DNA and modifying its structure, HMGB1 plays an important role in transcriptional regulation. In addition to its nuclear form, a cell-membrane form of HMGB1 (also known as amphoterin or p30) promotes neurite outgrowth, smooth muscle cell chemotaxis, and tumor cell metastasis [9–11].
Once outside the cell, HMGB1 has an entirely different role and functions as a pro-inflammatory cytokine with effects similar to TNFα. In vitro studies using purified HMGB1 demonstrate immunological activities likely implicated in inflammatory disease. Although the activity of HMGB1 preparations varies depending on the conditions of isolation and purification, both native and recombinant forms of this protein can induce expression of pro-inflammatory cytokines in vitro, including TNF-α, IL-1, IL-6 and nitric oxide (NO) in neutrophils, macrophages and pituicytes [12–14]. As a mediator of endothelial function, HMGB1 activates human umbilical venular endothelial cells (HUVEC) to upregulate adhesion molecules, and activated HUVEC cells release HMGB1 [15, 16]. Among its other functions, HMGB1 can also promote dendritic cell maturation and migration in vitro [17, 18].
The signaling systems triggered by HMGB1 are not fully understood, although receptor for advanced glycation end products (RAGE) is one receptor for HMBG-1. RAGE belongs to the IgG superfamily and is present on the surface of many cell types [19]. RAGE knockout mice have enhanced survival in sepsis models, and RAGE blockade attenuates the HMBG-1-induced inflammatory response [20]. Other in vitro evidence suggests that toll-like receptors (TLR) 2 and 4 may participate in HMGB1 signaling [21, 22].
HMGB1 release from cells
Cells release HMGB1 following cell activation or cell death, two processes central in inflammation. In activated cells including macrophages, HMGB1 translocates to the cytoplasm, enters lysosomes and, via exocytosis, reaches the extracellular milieu [23]. During the process of translocation, HMGB1 is phosphorylated [24]. As HMGB1 contains no signal sequence, the protein does not travel through the Golgi apparatus or endoplasmic reticulum [25]. In contrast, during necrosis, HMGB1 readily leaves cells, likely because its binding to DNA is weak in comparison to that of the histones, for example. As a result, when cell permeability breaks down during necrotic cell death, HMGB1 diffuses away from chromatin to enter the extracellular space. At present, Western blotting is the primary approach for measuring extracellular HMGB1.
While initial studies suggested that apoptotic cells do not release HMGB1 even when undergoing secondary necrosis, recent observations indicate that apoptotic death is also a setting for the release of this protein [25]. Following induction of apoptosis in Jurkat T cells by a variety of chemical agents, HMGB1 appears in the medium as shown by Western blotting; confocal microscopy can also demonstrate the translocation of HMGB1 during this death process [26]. Together, these findings suggest that, depending upon the cell type and the stimulus for death, HMGB1 release can follow apoptosis as well as necrosis and therefore mark death irrespective of the biochemical changes occurring in cells during these processes.
HMGB1 in inflammatory and autoimmune diseases
HMGB1 was initially described as an inflammatory protein released in vitro by LPS-stimulated macrophages and as a late mediator of murine LPS-induced sepsis [27]. Subsequent studies in animal models provided support for the role of HMGB1 in sepsis, including the attenuation of murine sepsis by anti-HMBG1 antibodies [27]. Further studies demonstrated that patients with sepsis have elevated HMGB1 levels which increase with disease severity. Beyond its role in sepsis, HMGB1 has been implicated in the pathogenesis of a broad spectrum of acute and chronic inflammatory conditions with elevated levels in the clinical settings of acute lung injury, cancer, inflammatory bowel disease, Sjogren’s syndrome, Churg–Strauss syndrome, SLE and RA [28–31]. In fact, HMGB1 may be an autoantigen recognized by perinuclear anti-neutrophil cytoplasmic antibodies (p-ANCA) [32].
Rheumatoid arthritis
In both animal models of inflammatory arthritis and in patients with RA, elevated levels of both intra- and extracellular HMGB1 are seen in synovial tissue [33]. The synovial fluid of patients with RA also shows enhanced levels of HMGB1 compared to that of osteoarthritis controls [34]. Further supporting the role of HMGB1 in inflammatory arthritis is the observation that intra-articular injection of HMGB1 into rodent knee joints induces synovitis and stimulates release of proinflammatory cytokines from synovial macrophages [35]. In animal models of collagen-induced arthritis, anti-HMGB1 antibody can produce clinical improvement in the joint count, joint pathology and cachexia similar to that seen with anti-TNFα therapy [36]. Studies investigating serum levels of HMGB1 in RA have yielded mixed results [37, 38]. Sera of ANA positive children with pauciarticular juvenile RA demonstrate antibodies to HMGB1 or HMGB-2 [39, 40]. Together, these observations suggest that HMGB1 is a mediator of inflammatory arthritis and may serve as a potential therapeutic target.
Systemic lupus erythematosus
Because it can induce proinflammatory cytokines, HMGB1 has emerged as a potential pathogenic protein in SLE. In fact, elevated levels of anti-HMGB1 antibody are present in the serum of patients with SLE and increased extracellular HMGB1 expression is seen in biopsies of cutaneous lupus lesions [37, 41]. Of particular interest is the finding that, in vitro, HMGB1 is present in DNA-containing immune complexes that stimulate TLR9-RAGE mediated dendritic cell production of cytokines, including interferon-α. These complexes also bind to and activate B cells via RAGE [42]. Additionally, anti-dsDNA sensitive, nephritogenic T cell lines from patients with active lupus nephritis proliferate in response to HMG proteins [43]. As SLE is a disease characterized by the formation of autoantibodies to cell nucleus constituents, high levels of cytokine expression (particularly interferon-α), formation of immune complexes, and autoreactive B cells, these data support the role of HMGB1 in the pathogenesis of SLE. Studies assessing the efficacy of blocking HMGB1 activity in SLE are needed to further elucidate the role of this novel inflammatory protein in SLE.
Microparticles and their function
Like HMGB1, MPs are released from activated and dying cells and promote inflammation. Originally described as inert “platelet dust,” MPs are small (0.1–1 μm) membrane-bound vesicles that circulate in the blood [44]. While platelet-derived MPs are the most abundant particle species in peripheral blood, circulating MPs can also arise from lymphocytes, monocytes, endothelial cells, and other cell types. MPs contain membrane, cytoplasmic and nuclear components characteristic of their precursor cells [45].
Despite their initial description as inert debris, MPs are potently active physiologically, and can mediate inflammation, hemostasis, thrombosis, angiogenesis, and vascular reactivity. Because of their small size, MPs widely circulate throughout the vasculature, allowing participation in both local and long-range signaling. In their interaction with cells, MPs bind via surface ligands, representing a kind of direct long-distance cell–cell communication for cells typically remote from each other. Furthermore, in a novel mechanism for the intercellular interaction, MPs can transfer surface molecules, including receptors, to other cells, as well as exchange membrane and cytoplasmic proteins [46, 47]. Receptor transfer can enlarge the responses that can be mounted by a given cell type; such transfer can also confer sensitivity to infection by an organism (e.g., HIV), requiring a particular cell surface molecule to allow entry [48].
Since platelet MPs were the first species identified, investigation of the role of these structures initially focused on the clotting system. MPs play a role in normal hemostasis and also have diverse properties that could promote the pathogenesis of thrombotic disorders. Phosphatidylserine and tissue factor are both exposed on the outer membranes of MPs and are central players in the coagulation cascade. MPs also interact with factors Va, VIII, and IXa, thereby facilitating assembly of the prothrombinase complex [49–52]. Among other activities, platelet MPs bind β-2-glycoprotein-1 antibodies, suggesting a possible role in antiphospholipid antibody syndrome (APS) [53]. As shown in in vitro experiments, MPs isolated from patients in various clinical settings (including sepsis, thrombotic thrombocytopenic purpura, sickle cell disease and cardiopulmonary bypass grafting) have procoagulant activity [54–57]. This activity may be relevant clinically since MP concentrations are elevated in prothrombotic disorders including acute coronary syndromes, venous thromboembolism, heparin-induced thrombocytopenia, vasculitis, paroxysmal nocturnal hemoglobinuria and thrombotic thrombocytopenic purpura [58–63].
In addition to their thrombotic effects, MPs have potent pro-inflammatory activities and are potentially important mediators of autoimmune and inflammatory diseases. Among these activities, platelet MPs can induce the binding of monocytes to endothelial cells and promote survival of hematopoietic cells [64]. Furthermore, platelet MPs can promote leukocyte–leukocyte aggregation, likely due to interactions between P-selectin expressed on platelet MPs and its ligand on leukocytes [65]. The mechanisms by which MPs mediate these effects may include the transfer of arachidonic acid to other cells, leading to increased adhesion of monocytes to endothelium [47, 66]. Other pro-inflammatory roles of MP include the secretion of IL-1β [67].
In contrast to actions promoting inflammation, T-cell-derived MPs can induce macrophage apoptosis. This killing action can potentially impair a key element of the immune system and in turn trigger MP release from the dying macrophage to augment other immunological and vascular events [68].
Microparticle release from cells
Similar to HMGB1, activation and cell death promote MP release, and the exact mechanisms underlying these processes remain unknown. During cell activation by a variety of stimuli, a rise in intracellular calcium is followed by remodeling of the plasma membrane. This membrane modification can cause phosphatidylserine exposure and bleb formation, leading to the extrusion of MPs to the extracellular space [69]. During apoptosis, MP release occurs in association with membrane blebbing, a characteristic feature of programmed cell death. Blebbing involves a dynamic redistribution of cellular contents, perhaps related to volume stress that occurs as cells die. Rho-associated kinase 1, ROCK-1, an effector of Rho GTPases, is essential for apoptotic membrane blebbing [68]. Not all cells bleb, however, and the blebbing process can differ during the stage of apoptosis. Like HMGB1, MP release appears to occur late in the cell death process and may occur concurrently with cell fragmentation and the formation of apoptotic bodies; apoptotic bodies represent shrunken and collapsed cells with nuclear fragmentation.
Any eukaryotic cell undergoing activation or death should theoretically release MPs, but most research has focused on hematopoietic cell types (e.g., platelets, leukocytes and erythrocytes) as well as vascular cells including endothelial cells. Other cells, including tumors, smooth muscle cell and synovial cells, can also release MPs, although these MPs may be more readily detected in the tissue or origin or at sites of inflammation (e.g., in the synovium or synovial fluid), rather than the blood.
Given the generation of MPs during exocytosis or blebbing of membranes, their origin can be tracked by cell-specific protein markers. Thus, the presence of CD4, CD3 or CD8 on the MP surface indicates lymphoid origin while platelet MPs are marked by the expression of glycoprotein IIb–IIIa, P-selectin/CD42a. Similarly, endothelial MPs display surface CD31 or CD 146 [70, 71]. MPs can express a different set of surface markers than the precursor cells, however, as has been observed with erythrocyte MPs [72]. The rules for the incorporation of different proteins into MPs are not known.
Microparticles in inflammatory and autoimmune disease
Like HMGB1 and other consequences of cell activation or death, elevated MP levels occur in the blood of patients with many different diseases. As a group, these diseases are characterized by disturbances of the immune system vasculature and include atherosclerosis, malignancy, multiple sclerosis, vasculitis RA and SLE [62, 73–75].
Rheumatoid arthritis
In the pathogenesis of RA inflammation, angiogenesis and thrombosis occur prominently, and the course of disease is marked by accelerated atherosclerosis. MPs may play a role in both articular and extraarticular manifestation, suggesting that their levels could serve as biomarkers. In patients with RA, platelet MPs levels in the blood are elevated and appear to correlate with disease activity [76]. While platelet MPs are found in the plasma of RA subjects, in the synovial fluid, MPs derived from granulocytes and monocytes dominate. Also present in synovial fluid are T cell, B cell, platelet and erythrocyte MPs. Synovial MPs from patients with RA and other inflammatory arthritides stimulate tissue factor/factor VII-dependent thrombin generation. This local hypercoaguability may promote intraarticular inflammation and the formation of fibrin clots, known as “rice bodies” [77].
In addition to promoting inflammation, MPs may contribute to the erosion of cartilage and bone via effects on synovial fibroblast activity. As observed in in vitro experiments using cells from patients with RA and other inflammatory arthritides, incubation of fibroblast-like synoviocytes with autologous MPs induces expression of MCP-1, IL-6, IL-8, VEGF, ICAM-1 and RANTES and a decrease in GMCSF [78]. Furthermore, in in vitro studies, MPs derived from T cells and monocytes can induce synovial fibroblast production of matrix metalloproteinases (MMPs), including MMP-1, MMP-3, MMP-9 and MMP-13 [78]. Taken together, these data suggest that MPs can mediate cellular interactions responsible for synovial activation and articular destruction [77].
Systemic lupus erythematosus and APS
In view of the inflammation and vascular abnormalities characteristic of SLE, the presence of MPs in peripheral blood could provide a novel marker reflecting dysregulation in the major cell populations underlying disease. Elevated levels of platelet MPs have been described in individuals with SLE [79]. In one study, patients with antiphospholipid antibodies and elevated levels of platelet MPs but not endothelial MPs predicted thrombosis [80]. In contrast, another investigation found that endothelial MPs were elevated in subjects with APS compared to controls with and without non-APS thrombosis. Additionally, in vitro, APS plasma induced a fourfold increase in the release of endothelial MPs from human umbilical endothelial cells [81]. In one study, MPs, primarily platelet-derived, were elevated in SLE and correlated with thrombin generation, although levels did not correlate with disease activity or the presence of antiphospholipid antibody [82].
Conclusion
HMGB1 and MPs are novel mediators of inflammation, liberated in the settings of cell activation and cell death. Both are potentially important players in immune-mediated diseases and can participate in local and long-range intercellular communication. In many inflammatory and autoimmune diseases, but particularly RA and SLE, MPs and HMGB1 may play important roles in pathogenesis and, as potential targets for treatment, may also provide new opportunities for therapeutic intervention.
References
Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255:332–43.
Wen L, Huang JK, Johnson BH, Reeck GR. A human placental cDNA clone that encodes nonhistone chromosomal protein HMG-1. Nucleic Acids Res. 1989;17:1197–214.
Bianchi ME, Falciola L, Ferrari S, Lilley DM. The DNA binding site of HMG1 protein is composed of two similar segments (HMG boxes), both of which have counterparts in other eukaryotic regulatory proteins. EMGO J. 1992;11:1055–63.
Falciola L, Spada F, Calogero S, Langst G, Voit R, Grummt I, et al. High mobility group 1 protein is not stably associated with the chromosomes of somatic cells. J Cell Biol. 1997;137:19–26.
Jayaraman L, Moorthy NC, Murthy KG, Manley JL, Bustin M, Prives C. High mobility group protein-1 (HMG-1) is a unique activator of p53. Genes Dev. 1998;12:462–72.
Agresti A, Lupo R, Bianchi ME, Muller S. HMGB1 interacts differently with members of the Rel family of transcription factors. Biochem Biophys Res Commun. 2003;302:421–6.
Boonyaratanakornkit V, Melvin V, Prendergast P, Altmann M, Ronfani R, Bianchi ME, et al. High-mobility group chromatin proteins 1 and 2 functionally interact with steroid hormone receptors to enhance their DNA binding in vitro and transcriptional activity in mammalian cells. Mol Cell Biol. 1998;18:4471–87.
Calogero S, Grassi F, Aguzzi A, Voigtländer T, Ferrier P, Ferrari S, et al. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet. 1999;22:276–80.
Daston MM, Ratner N. Expression of P30, a protein with adhesive properties, in Schwann cells and neurons of the developing and regenerating peripheral nerve. J Cell Biol. 1991;112:1229–39.
Debryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–206.
Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, et al. Blockade of RAGE-amphoterin signaling suppresses tumor growth. Nature. 2000;405:354–60.
Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–70.
Zimmermann K, Volkel D, Pable S, Lindner T, Kramberger F, Bahrami S, et al. Native versus recombinant high-mobility group B1 proteins: functional activity in vitro. Inflammation. 2004;28:221–9.
Wang H, Visnubhakat JM, Bloom O, Zhang M, Ombrellino M, Sama A, Tracey KJ. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery. 1999;126:389–92.
Treutiger CJ, Mullins GE, Johansson AS, Rouhiainen A, Rauvala JME, Erlandsson-Harris H, et al. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–85.
Mullins GE, Sunden-Cullberg J, Johansson AS, Rouhiainen A, Errlandsson-Harris H, et al. Activation of human umbilical vein endothelial cells leads to relocation and release of high-mobility group box chromosomal protein 1. Scand J Immunol. 2004;60:566–73.
Messmer D, Yang H, Telusma G, Knoll F, Li J, Messmer B, et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol. 2004;173:307–13.
Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol. 2007;81:59–66.
Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2004;83:876–86.
Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9.
Park JS, Svetkauskaite D, He Q, Kim HY, Strassheim D, Ishizaka A et al. Involvement of toll-like receptors 2 and 4 in the cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7.
Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim Jy, Strassheim D, et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol. 2006;290:C917–24.
Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;10:995–1001.
Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177:7889–97.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5.
Bell CW, Jiang W, Reich CF, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. J Physiol Cell Physiol. 2006;291:C1318–25.
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51.
Ueno H, Matsuda T, Hashimoto S, Amaya F, Kitamura Y, Tanaka M et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury, Am J Respir Crit Care Med. 2004;170:1306–10.
Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A et al. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. 2007;13:2836–48.
Taira T, Matsuyama W, Mitsuyama H, Kawahara KI, Higashimoto I, Maruyama I et al. Increased serum high mobility box-1 level in Churg–Strauss syndrome. Clin Exp Immunol. 2007;148:241–7.
Ek M, Popovic K, Harris HE, Naucler CS, Wahren-Herlenius M. Increased extracellular levels of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in minor salivary glands of patients with Sjogren’s syndrome. Arthritis Rheum. 2006;54:2289–94.
Sobajima J, Ozaki S, Uesugi H, Osakada F, Shirakawa H, Yoshida M et al. Prevalence and characterization of perinuclear anti-neutrophil cytoplasmic antibodies (P-ANCA) directed against HMG1 and HMG2 in ulcerative colitis (UC). Clin Exp Immunol. 1998;111:402–7.
Kokkola R, Sundberg E, Ulfgren AK, Palmblad K, Li J, Wang H et al. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum. 2002;46:2598–603.
Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48:971–81.
Pullertis R, Jonsson IM, Verdrengh M, Bokarewa M, Andersson U, Erlandsson-Harris H et al. High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthritis Rheum. 2003;48:1693–700.
Kokkola R, Li J, Sundberg E, Aveberger AC, Palmblad K, Yang H et al. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003;48:2052–8.
Goldstein RS, Bruchfeld A, Yang L, Qureshi AR, Gallowitsch-Puerta M, Patel NB et al. Cholinergic anti-inflammatory pathway activity and high mobility group box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol Med. 2007;13:210–5.
Santoro P, De Andrea M, Migliaretti G, Trapani C, Landolfo S, Gariglio M. High prevalence of autoantibodies against the nuclear high mobility group (HMG) protein SSRP1 in sera from patients with systemic lupus erythematosus, but not other rheumatic diseases. J Rheumatol. 2002;29:90–3.
Wittemann B, Neuer G, Michels H, Truckenbrodt H, Bautz FA. Autoantibodies to nonhistone chromosomal proteins HMG-1 and HMG-2 in sera of patients with juvenile rheumatoid arthritis. Arthritis Rheum. 1990;33:1378–83.
Rosenberg AM, Cordeiro DM. Relationship between sex and antibodies to high mobility group proteins 1 and 2 in juvenile idiopathic arthritis. J Rheumatol. 2000;27:2489–93.
Popovic K, Ek M, Espinosa A, Padyukov L, Harris HE, Warhren-Herlenius M et al. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheum. 2005;52:3639–45.
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–96.
Desai-Mehta A, Mao C, Rajagopalan S, Robinson T, Datta SK. Structure and specificity of T cell receptors expressed by potentially pathogenic anti-DNA autoantibody-inducing T cells in human lupus. J Clin Invest. 1995;95:531–41.
Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol. 1967;13:269–88.
Distler JHW, Pisetsky DS, Huber LC, Kalden JR, Gay S, Distler O. Microparticles as regulators of inflammation. Arthritis Rheum. 2005;52:3337–48.
Fritzsching B, Schwer B, Kartenbeck J, Pedal A, Horejsi V, Ott M et al. Release and intercellular transfer of CD81 via microparticles. J Immunol. 2002;169:5531–7.
Barry OP, Pratico D, Savani RC, FitzGerald GA. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J Clin Invest. 1998;102:136–44.
Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, Cihak J. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med. 2000;6:769–75.
Gilbert GE, Sims PJ, Wiedmer T, Furie B, Furie BC, Shattil SJ. Platelet-derived microparticles express high affinity receptors for factor VIII. J Biol Chem. 1991;266:17261–8.
Hoffman M, Monroe DM, Roberts HR. Coagulation factor IXa binding to activated platelets and platelet-derived microparticles: a flow cytometric study. Thromb Haemost. 1992;68:74–8.
Michelson AD, Rajasekhar D, Bednarek FJ, Barnard MR. Platelet and platelet-derived microparticle surface factor V/Va binding in whole blood: differences between neonates and adults. Thromb Haemost. 2000;84:689–94.
Sims PJ, Wiedmer T, Esmon CT, Weiss JH, Shattil SJ. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J Biol Chem. 1989;264:17049–57.
Vallar L, Regnault V, Latger-Cannard V, Lecompte T. Beta 2-glycoprotein I binding to platelet microparticle membrane specifically reduces immunoreactivity of glycoproteins IIb/IIIa. Thromb Haemost. 2001;85:314–219.
Nieuwland R, Berckmans RJ, McGregor S, Boing AN, Romijn FP, Westendorp RG et al. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000;95:930–5.
Jimenez JJ, Jy W, Mauro LM, Horstman LL, Yeon S, Ahn WH. Elevated endothelial microparticles in thrombotic thrombocytopenic purpura: findings from brain and microvascular cell culture and patients with active disease. Br J Haematol. 2001;112:81–90.
Shet AS, Aras O, Gupta K, Hass MJ, Rausch DJ, Saba N et al. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–83.
Nieuwland R, Berckmans RJ, Rotteveel-Eijkman RC. Cell-derived microparticles generated in patients during cardiopulmonary bypass are highly procoagulant. Circulation. 1997;96:3534–41.
Bernal-Mizrachi L, Jy W, Jimenez JJ, Pastor J, Mauro LM, Horstman LL et al. High levels of circulating endothelial microparticles in patients with acute coronary syndromes. Am Heart J. 2003;145:962–70.
Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E et al. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol. 2005;45:1467–71.
Hughes M, Hayward CP, Warkentin TE, Horsewood P, Chomeyko KA, Kelton JG. Morphological analysis of microparticle generation in heparin-induced thrombocytopenia. Blood. 2000;96:188–94.
Simak J, Holada K, Risitano AM, Zivny JH, Young NS, Vostal JG. Elevated circulating endothelial membrane microparticles in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2004;125:804–13.
Brogan PA, Shan V, Brachet C, Harnden A, Mant D, Klein N et al. Endothelial and platelet microparticles in vasculitis of the young. Arthritis Rheum. 2004;50:927–36.
Walenga JM, Jeske WP, Messmore HL. Mechanisms of venous and arterial thrombosis in heparin-induced thrombocytopenia. J Thromb Thrombolysis. 2000;10 Suppl 1:13–20.
Baj-Kryworzeka M, Majka M, Pratico D, Ratajczak J, Vilaire G, Kijowski J et al. Platelet-derived microparticles stimulate proliferation, survival, adhesion and chemotaxis of hematopoietic cells. Exp Hematol. 2002;30:450–9.
Forlow SB, McEver RP, Nollert MU. Leukocyte–leukocyte interactions mediated by platelet microparticles under flow. Blood. 2000;95:1317–23.
Barry OP, Pratico D, Lawson JA, Fitzgerald GA. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J Clin Invest. 1997;99:2118–27.
Mackenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Suprenant A. Rapid secretion of interleukin-1β by microvesicle shedding. Immunity 2001;15:825–35.
Distler JH, Huber LC, Reich CF III, Gay S, Distler O, Pisetsky DS. The release of microparticles by apoptotic cells and their effects on macrophages. Apoptosis. 2005;4:731–41.
Martinez MC, Tesse A, Zobairi F, Andrianstsitohaina R. Shed membrane microparticles from circulating and vascular cells in regulating vascular function. Am J Physiol Heart Circ Physiol. 2005;288:1004–9.
Horstman LL, Ahn YS. Platelet microparticles: a wide-angle perspective. Crit Rev Oncol Hematol. 1999;30:111–45.
Piccin A, Murphy W, Smith O. Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 2007;21:157–71.
Butikofer P, Kuypers FA, Xu CM, Chiu DT, Lubin B. Enrichment of two glycosyl-phosphatidylinositol-anchored proteins, acetylcholinesterase and decay accelerating factor, in vesicles released from human red blood cells. Blood. 1989;74:1481–5.
Mallat Z, Hugel B, Ohan J, Leseche G, Freyssinet JM, Tedgui A. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation. 1999;99:348–53.
Janowska-Wieczorek A, Marquez-Curtis LA, Wysoczynski M, Ratajczak MZ. Enhancing effect of platelet-derived microvesicles on the invasive potential of breast cancer cells. Transfusion. 2006;46:1199–209.
Minagar A, Jy W, Jimenez JJ, Sheremata WA, Maura LM, Mao WW et al. Elevated plasma endothelial microparticles in multiple sclerosis. Neurology. 2001;56:1319–24.
Knijff-Dutmer EA, Koerts J, Nieuwland R, Kalsbeek-Batenburg EM, van de Laar MA. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum. 2002;46:1498–503.
Berckmans RJ, Nieuwland R, Tak PP, Boing AN, Romijn FP, Kraan MC et al. Cell-derived microparticles in synovial fluid from inflamed arthritic joints support coagulation exclusively via a factor VII-dependent mechanism. Arthritis Rheum. 2002;46:2857–66.
Distler JH, Jungel A, Huber LC, Seemayer CA, Reich CF 3rd, Gay RE et al. The induction of matrix metalloproteinase and cytokine expression in synovial fibroblasts stimulated with immune cell microparticles. Proc Natl Acad Sci USA. 2005;102:2892–7.
Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol. 2001;115:451–9.
Jy W, Tiede M, Bidot CG, Horstman LL, Jimenez JJ, Chirinos J, et al. Platelet activation rather than endothelial injury identifies risk of thrombosis in subjects positive for antiphospholipid antibodies. Thromb Res 2007;121(3):319–25.
Dignat-George F, Camoin-Jau L, Sabatier F, Arnoux D, Anfosso F, Bardin N et al. Endothelial microparticles: a potential contribution to the thrombotic complications of the antiphospholipid antibody syndrome. Thromb Haemost. 2004;91:667–73.
Pereira J, Alfaro G, Goycoolea M, Quiroga T, Ocqueteau M, Massardo L, et al. Circulating platelet-derived microparticles in systemic lupus erythematosus: association with increased thrombin generation and procoagulant state. Thromb Haemost. 2006;95:94–9.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ardoin, S.P., Pisetsky, D.S. The role of cell death in the pathogenesis of autoimmune disease: HMGB1 and microparticles as intercellular mediators of inflammation. Mod Rheumatol 18, 319–326 (2008). https://doi.org/10.1007/s10165-008-0054-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10165-008-0054-z