
Abstract
Studies using inbred strains of mice have been invaluable for identifying alleles that adversely affect hearing. However, the efficacy of those studies is limited by the phenotypes that these strains express and the alleles that they segregate. Here, by selectively breeding phenotypically and genetically heterogeneous NIH Swiss mice, we generated two lines—the all-frequency hearing loss (AFHL) line and the high-frequency hearing loss (HFHL) line—with differential hearing loss. The AFHL line exhibited characteristics typical of severe, early-onset, sensorineural hearing impairment. In contrast, the HFHL line expressed a novel early-onset, mildly progressive, and frequency-specific sensorineural hearing loss. By quantitative trait loci (QTLs) analyses in these two lines, we identified QTLs on chromosomes 7, 8, and 10 that significantly affected hearing function. The loci on chromosomes 7 and 8 (Hfhl1 and Hfhl2, respectively) are novel and appear to adversely affect only high frequencies (≥30 kHz). Mice homozygous for NIH Swiss alleles at either Hfhl1 or Hfhl2 have 32-kHz auditory-evoked brain stem response thresholds that are 8–14 dB SPL higher than the corresponding heterozygotes. DNA sequence analyses suggest that both the Cdh23 ahl and Gipc3 ahl5 variants contribute to the chromosome 10 QTL detected in the AFHL line. The frequency-specific hearing loss indicates that the Hfhl1 and Hfhl2 alleles may affect tonotopic development. In addition, dissecting the underlying complex genetics of high-frequency hearing loss may prove relevant in identifying less severe and common forms of hearing impairment in the human population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
To maintain hearing acuity at a range of frequencies, the cochlea efficiently and accurately transmits the vibrational energy of sound to the sensory cells that traverse the length of the cochlear duct, encodes the mechanical energy of each frequency of sound as an electrochemical signal, and relays these electrochemical signals to the brain. To accomplish this, the mammalian inner ear has evolved a set of sensory hair cells—inner and outer hair cells—capable of transducing, amplifying, and tuning sound waves in the presence of an electrochemical gradient created by the stria vascularis and with the aid of the mechanical properties of the tectorial and basilar membranes (Dallos 1996; Hudspeth 1982). To further aid in sound decoding, the physical and chemical properties of the constituent parts of the cochlear coil change from the base to its apex. This creates a physiological gradient that produces an ordered range of frequency specificities (the tonotopic map) such that the base of the cochlea detects high-frequency sound while the apex detects low-frequency sound (see review by Mann and Kelley 2011). Due to the cochlea’s complexity, many proteins and isoforms are required for the development and maintenance of hearing.
Pathogenic alleles at over 100 loci have been linked to hearing deficits in humans (http://webh01.ua.ac.be/hhh/). However, the identities of many additional genes remain to be determined to account for all of the genetic variation underlying human hearing impairment. Gene–gene interactions, genetic modifiers, and probably the small additive effects of common polymorphisms complicate the identification of causal mutations and variants (Noben Trauth et al. 1997; Noguchi et al. 2006). A comprehensive understanding of most of the genes involved in hearing may also be necessary to elucidate the causes of less extreme and more common forms of hearing impairment. For a number of deafness genes, the genetic and mechanistic analysis of mouse models provided the first evidence as to its function in normal and impaired hearing (Dror and Avraham 2009).
The NIH Swiss outbred stock of mice was initially derived from a population of Swiss albino mice imported to the USA in 1926. Since the early 1960s, this stock has been maintained under strict random mating protocols to preserve genetic heterogeneity (Chia et al. 2005). In 1980, the NIH Swiss population was estimated to segregate a degree of heterozygosity that is similar to that found in natural populations (Rice and O’Brien 1980). We recently showed that the NIH Swiss stock also exhibits substantial variation in hearing thresholds (Drayton and Noben-Trauth 2006). The combination of genetic and phenotypic variation makes the NIH Swiss stock a good model system for investigating genes and alleles that affect hearing.
Methods
Population
NIH Swiss mice were obtained from Charles River Laboratories (Wilmington, MA). C3HeB/FeJ mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All studies were conducted following the guidelines approved by the Animal Care and Use Committee of the NIDCD, National Institutes of Health.
A previous study of several outbred stocks of mice indicated that the NIH Swiss stock exhibits substantial variation in hearing thresholds (Drayton and Noben-Trauth 2006). Since NIH Swiss mice are genetically heterogeneous (Rice and O’Brien 1980), we decided to investigate the genetic basis for this phenotypic variation. We began by selectively breeding members of our founding NIH Swiss population based on auditory-evoked brain stem response (ABR) thresholds for four stimuli (click, 8, 16, and 32 kHz) to produce two new lines. The first line, the all-frequency hearing loss (AFHL) line, was selected for elevated ABR thresholds at all stimuli tested. The second line, the high-frequency hearing loss (HFHL) line, was selected for normal ABR thresholds to most stimuli (click, 8, and 16 kHz), but elevated thresholds for the 32-kHz stimulus.
After several generations (F4 for AFHL mice and F12 for HFHL mice) of selective breeding, males and females from each of the two selection lines were reciprocally mated with mice of the C3HeB/FeJ strain to produce (HFHL × C3HeB/FeJ) N1 and (AFHL × C3HeB/FeJ) N1 offspring. These N1 mice were then backcrossed to their respective NIH Swiss parental lines to produce [(AFHL × C3HeB/FeJ) × AFHL] N2 mice (AFHL-N2, n = 675) and [(HFHL × C3HeB/FeJ) × HFHL] N2 mice (HFHL-N2, n = 306). The two N2 populations were evaluated independently to identify quantitative trait loci (QTLs) responsible for their unique hearing phenotypes. The inbreeding of HFHL mice up to generation F12 ensured that residual genetic variation that could reduce the power of the study to detect any minor QTLs responsible for the less pronounced HFHL phenotype was eliminated.
Auditory brain stem response measurements
Hearing tests were performed as previously described (Noben-Trauth et al. 2010). Briefly, ABR measurements were made using a computer-aided evoked potential system (Intelligent Hearing System, IHS, Miami, FL). The Smart-EP, version 10, was modified for high-frequency capability and coupled to high-frequency transducers generating specific acoustic stimuli that enabled the system to measure and display the evoked brainstem responses of anesthetized mice. Subdermal needle electrodes were inserted at the vertex (active) and ventrolaterally to the right (reference) and the left (ground) ears. Stimuli were delivered to the right outer ear canal through a plastic tube channeled from the high-frequency transducer-. Mice were presented with click stimulus and with 8-, 16-, and 32-kHz tone pips from high to low intensity [100–10 dB sound pressure level (SPL) in 5-dB increments] at a rate of 19.1 times per second for a total of 350 sweeps per stimulus intensity. Sound pressure level thresholds were determined for each stimulus frequency by identifying the lowest intensity producing a recognizable ABR pattern on the computer screen. Peak amplitudes and latencies were obtained from plots of suprathreshold intensity for the different stimuli. Latencies were measured as the time lapse between the onset of stimulus and the apex of each wave. Amplitudes were calculated as the difference in signal intensity between the apex of a wave and the nadir of the next trough.
Distortion product otoacoustic emission measurements
Distortion product otoacoustic emissions (DPOAEs) were measured using a National Instruments (NI) LabView 8.6 software, operating a NI PCI-4461 Dynamic Signal Analyzer sound card installed in a PC. The system generated two pure tones, f1 and f2, at a fixed f2/f1 ratio of 1.25 and with presentation intensities of f2 = f1 − 10 dB SPL that were delivered separately by two Clarion SRU310H high frequency dome tweeters placed in the outer ear canal. These tones were varied to generate an f2 sweep from 5 to 55 kHz at intensities ranging from 15 to 85 dB SPL in 10-dB increments. The amplitude of the 2f1–f2 distortion product was plotted in decibel SPL against the f2 frequency that generated the distortion product.
Endocochlear potential measurements
Endocochlear potentials (EPs) were measured by inserting the tip of a small glass electrode through the round window and into the endolymph of the cochlea of an anesthetized mouse using a remote-controlled motorized micromanipulator. The voltage difference was amplified by a Warner dual-channel differential electrometer and routed to a PC-controlled data acquisition system (Axon Instruments) using AxoScope software, which displayed the measured output. EPs were recorded when the output indicated that a stable potential had been obtained.
Organ of Corti staining
To produce organ of Corti whole mounts, cochlear ducts were dissected from the surrounding bone in Leibovitz (Invitrogen) and fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 2 h at room temperature. The outer wall of the duct, Reissner’s membrane, and the tectorial membrane were removed and the tissue was permeabilized in 0.5% TritonX-100 for 30 min at room temperature. Each specimen was stained with rhodamine phalloidin (Invitrogen) diluted 1:100 in PBS for 30 min at room temperature. After washing in PBS, the organ of Corti was removed from the modiolos, mounted in ProLong® Gold antifade reagent (Invitrogen), and imaged using a Zeiss LSM confocal microscope. Image levels were adjusted using Adobe Photoshop.
Cochlear histology
In order to evaluate histology, ears were dissected from the temporal bone in PBS and perfused with 4% paraformaldehyde at room temperature for at least 12 h. The specimen was washed twice in PBS and decalcified for 3 weeks in 0.1 M EDTA, pH 8.0, in PBS. The inner ears were dehydrated with a graded series of ethanol, infiltrated with JB-4 polymer (Polysciences, Inc.), and embedded in the same polymer. Serial mid-modiolar sections were cut at 4-μm thickness using a tungsten carbide disposable blade on a RM2265 microtome (Leica). Sections were mounted on Superfrost Plus glass slides, stained with 0.1% Toluidine Blue O, and cleared in xylene. Slides were imaged on a DM5000B microscope (Leica) and photographed with a DFC500 digital camera (Leica). Image levels were adjusted with Adobe Photoshop software.
Genetic analysis
DNA was extracted from tail clips using the protocols and reagents from DNeasy® Blood and Tissue kits (Qiagen, Valencia, CA). Samples of DNA were adjusted to concentrations of approximately 100 ng/μl (40–130 ng/μl) and submitted for genotyping to The Partners Center for Personalized Genetic Medicine (Cambridge, MA). Three hundred AFHL N2 mice and several randomly selected P0 and F1 mice were genotyped at 662 single nucleotide polymorphisms (SNPs) spaced an average of 3.9 Mb apart along each of the 19 autosomes and the X chromosome. Of these 662 SNPs, 257 were polymorphic and informative in the AFHL N2 population. Similarly, 305 HFHL-N2 mice and randomly selected P0 and F1 mice were genotyped at 337 SNPs spaced an average of 6.5 Mbp apart. Only 145 of these SNPs were useful in the analysis. Furthermore, samples of DNA from an additional 355 AFHL N2 mice that had been phenotyped were diluted to approximately 10 ng/μl (1–20 ng/μl). These samples were genotyped using TaqMan® SNP Genotyping Assays with a StepOnePlus Real-Time PCR Thermocycler (Applied Biosystems) to confirm putative QTLs from the whole-genome scan of the AFHL-N2 cross. Finally, aliquots from all HFHL-N2 DNA samples were diluted to approximately 10 ng/μl and genotyped at additional loci in regions of particular interest using TaqMan® SNP Genotyping Assays.
For QTL intervals containing likely candidate genes with mutations known to produce a phenotype similar to what we observed, we sequenced the region containing the putative causal mutation in both NIH Swiss lines and in the C3HeB/FeJ strain. A sample of approximately 50–100 ng of genomic DNA from each strain/line was amplified via PCR with 50X Advantage cDNA Polymerase (Clontech Laboratories, Inc, Palo Alto, CA) mixed with standard concentrations of dNTP, PCR reaction buffer, and forward and reverse primers in molecular biology grade water. During PCR, samples were denatured (95°C for 1 min) and then subjected to 35 cycles consisting of denaturation (94°C for 30 s) alternating with extension (68°C for 2 min), followed by a final extension (68°C for 2 min). Reactions were inactivated by incubating the samples with shrimp alkaline phosphatase and exonuclease at 37°C for 60 min and 80°C for 5 min. The sequencing reactions were carried out using BigDye® Terminator v 3.1 using the manufacturer’s instructions (Applied Biosystems). Sequencing was performed on a 3730xl capillary sequencer (Applied Biosystems) and chromatograms analyzed using Sequencher v 4.5. Chromatograms from NIH Swiss AFHL and HFHL lines were compared with one another and with those of C3HeB/FeJ. Where appropriate, additional controls were evaluated as noted in “Results.”
QTL analysis
A whole-genome QTL analysis was performed using the ABR thresholds obtained from 300 AFHL N2 mice when they were 8 weeks old. Genotypes at 257 SNPs and the corresponding ABR thresholds for each of the four auditory stimuli (click, 8, 16, and 32 kHz) for each mouse were imported into Map Manager QTXb20. Map Manager QTXb20 was then used to perform least-squares linear regressions that generated likelihood ratio statistic (LRS) scores that were used to identify genomic regions associated with differences in ABR thresholds in the population (Manly et al. 2001). Significance thresholds for the LRS scores generated from the regressions were calculated by performing 1,000 permutations of the data (Churchill and Doerge 1994). LRS scores that exceeded the permutation values corresponding to the 37th, 95th, and 99.9th percentiles (equivalent to genome-wide p values of 0.63, 0.05, and 0.001) were considered suggestive, significant, or highly significant evidence of a QTL, respectively (Lander and Kruglyak 1995). For comparison with other studies, LRS scores were converted to LOD scores (LRS/2ln(10) = LOD) by dividing by 4.61. A similar analysis was also performed for 306 HFHL N2 mice, except that only 145 SNPs were used.
Once preliminary analyses were complete, interval mapping was performed for each chromosome for which a putative QTL was detected in the marker regression analysis. This allowed us to estimate the locations of the QTLs and determine support intervals for each (Manly and Olson 1999; Haley and Knott 1992). Once the initial QTLs were identified and mapped, we performed composite interval mapping to further clarify QTL positions and identify additional QTLs associated with each trait by entering the most significant QTL for each trait as background and rerunning the interval mapping procedure on chromosomes with markers that reached the suggestive level of significance or higher (Manly et al. 2001). We also tested for associations between each trait and each pair of marker loci to see if any gene interactions (i.e., epistatic effects) were present. Interaction tests were only considered significant if the total effect of the two loci were significant at the p = 10−5 level and the interaction was significant at the permutation-derived p = 0.01 level (Lander and Kruglyak 1995). If multiple interactions were detected that involved two sets of linked markers on the same two chromosomes, the combination of markers that gave the highest total LRS score was assumed to give the best estimate of the epistatic loci.
To confirm our results, an additional 355 AFHL-N2 mice that had been ABR tested and genotyped at loci in the region of each putative QTL were evaluated. These data were analyzed by least-squares linear regressions using Map Manager QTXb20. Permutation tests were used to determine significance levels.
Statistical analyses
Unless otherwise indicated, groups of data were compared by t tests if only two groups were present or by one-way ANOVA followed by Bonferroni-corrected posttests if more than two groups were present. Groups were evaluated to determine whether they had equal variances and Gaussian distributions. For comparisons between two groups with unequal variances, Welche’s correction was applied to the t test. For comparisons between groups with non-Gaussian distributions, we also performed non-parametric tests (Mann–Whitney for two groups, Kruskal–Wallis for more than two groups). However, the results of these non-parametric tests were not reported unless they differed qualitatively from the ANOVAs. GraphPad™ Prism 4.0b software was used to perform column statistic, plot the data, and compute p values for the comparisons.
Results
Auditory characteristics in NIH Swiss mice
To ascertain hearing function in the NIH Swiss population, we recorded ABRs of mice that were between 4 and 12 weeks of age. The mean thresholds for the click, 8-, and 16-kHz stimuli were between 17 and 22 dB SPL higher for NIH Swiss than for C3HeB/FeJ control mice (p < 0.01). The threshold shift was greatest at the 32 kHz stimulus, for which we detected a 35-dB SPL difference between NIH Swiss and C3HeB/FeJ mice (p < 0.001; Fig. 1A and Table 1). The most intriguing finding, however, was the wide threshold distributions at all four test stimuli that resulted in standard deviations much higher than those normally observed in inbred mice such as C3HeB/FeJ (p < 0.0001). The wide threshold distributions suggested that different ABR phenotypes were segregating in the parental population. To further investigate the variation in hearing phenotype, mice with the highest and lowest thresholds were selected and used to generate, via repeated brother × sister matings, two inbred lines that were designated AFHL and HFHL, respectively. After ten generations of inbreeding (F10), mice of the HFHL line exhibited ABR thresholds comparable to the control C3HeB/FeJ mice at the click, 8-, and 16-kHz stimuli (p > 0.05). In contrast, thresholds remained elevated by 24 dB SPL (p < 0.001) at the high-frequency (32 kHz) stimulus. Most importantly, smaller threshold variations were observed and standard deviations decreased to values similar to those of C3HeB/FeJ mice. Mice of the AFHL line also exhibited increasingly homogenous hearing thresholds during inbreeding until they consistently exhibited 40- to 50-dB SPL threshold shifts at all test frequencies in the F10 generation (Fig. 1A).

ABR and EP in AFHL and HFHL lines. A ABR thresholds of 8-week-old mice for click (c) and 8-, 16-, and 32-kHz stimuli. Graphs depict mean ABR thresholds (lines) and threshold distributions (circles) for NIH Swiss outbred mice (parental, n = 30), HFHL (HFHL F10, n = 20), and AFHL (AFHL F10, n = 14) mice after ten generations of inbreeding and C3HeB/FeJ control mice (n = 14). Thresholds of parental mice used to generate the AFHL (red symbol) and HFHL (green symbol) are shown. B ABR thresholds (mean±SD) for click (c) and 8-, 16-, and 32-kHz stimuli for HFHL mice at 4 (n = 10), 26 (n = 6), and 52 (n = 14) weeks of age. C ABR thresholds (mean±SD) for click (c) and 8-, 16-, and 32-kHz stimuli for AFHL mice at 4 (n = 9) and 26 (n = 5) weeks of age. D ABR wave I amplitudes (mean±SD) for click (n = 32), 8- (n = 25), 16- (n = 23), and 32-kHz (n = 5) stimuli for 4-week-old HFHL (green bar) and C3HeB/FeJ (n > 9, blue bar) mice. Amplitudes were calculated as the difference in signal intensity between each maxima and the next minima. ABR wave I, III, and V latencies (mean±SD) for the click (E), 16-kHz (F), and 32-kHz (G) stimulus for 4-week-old HFHL (n = 51, green triangle) and C3HeB/FeJ (n = 22, blue diamond) mice. Peak latencies were calculated as the time between the onset of the signal and the occurrence of each maximum. H Endocochlear potentials (mean±SD) for 8-week-old HFHL (n = 13) and AFHL (n = 11) mice. ns not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
To evaluate the progression of hearing loss in NIH Swiss mice, additional ABRs were measured at 26 and 52 weeks of age. At 26 weeks of age, mice of the HFHL line showed no significant increase in thresholds compared to 4-week-old mice (p > 0.05). However, at 52 weeks of age, a modest, albeit significant, increase in thresholds at all four stimuli was measured (p < 0.001, Fig. 1B). The greatest threshold shift (34 dB SPL) was observed at the 16-kHz stimulus, for which the threshold exceeded 60 dB SPL after 1 year. Notably, the standard deviations also increased over time. In mice of the AFHL line, thresholds increased at all tested frequencies to a profound hearing loss (thresholds >90 dB SPL) at 26 weeks of age (Fig. 1C and Table 1).
Additional information regarding the hearing phenotype of HFHL mice was obtained by measuring wave I amplitudes and peak latencies of the ABRs of 4-week-old HFHL and C3HeB/FeJ mice. HFHL mice had significantly reduced peak I amplitudes (p < 0.05) at suprathreshold intensities (60 dB SPL) for click, 8-, 16-, and 32-kHz (100 dB SPL) stimuli compared with the C3HeB/FeJ mice (Fig. 1D). Additionally, although peak latencies for waves I and III at the click and 16-kHz stimulus appeared normal for HFHL mice compared with the C3HeB/FeJ mice (Fig. 1E, F), significant delays in response to 100-dB SPL exposures were observed for peaks I, III, and V at the 32-kHz stimulus frequency (p < 0.001; Fig. 1G and Table 2). Wave V at the 8- and 16-kHz frequencies was also significantly delayed (p < 0.05; Table 2).
The presence of an adequate EP is critical for hair cell function. AFHL and HFHL mice exhibited EPs averaging at 91.9 ± 13.3 and 100.4 ± 11.7 mV, respectively (p > 0.05, t test; Fig. 1H). These values are within the range typically observed at the base of the cochlea in mice with normal hearing.
To evaluate outer hair cell function in the AFHL and HFHL lines, we measured DPOAEs. DPOAEs were essentially absent in AFHL mice by the time they reached 4 weeks of age (Fig. 2A, B). In contrast, at 4 and 26 weeks of age, HFHL mice had nearly normal DPOAE levels for all frequencies <30 kHz (Fig. 2C, D). However, for frequencies above 30 kHz, DPOAE levels were significantly reduced for HFHL mice compared with the C3HeB/FeJ controls. In fact, DPOAEs were basically absent at 32 kHz and above when input intensity dropped below 65 dB (Fig. 2E, F). HFHL mice retained relatively normal DPOAEs at frequencies below 20 kHz at 26 weeks of age, but showed some reduction in 2f1–f2 amplitudes at 52 weeks of age (Fig. 2D–F). These results suggest that HFHL mice have an outer hair cell defect that remains isolated to the basal portion of the cochlea.

DPOAEs in AFHL and HFHL lines. A Average DP gram of 4-week-old AFHL mice (n = 5) in response to 5- to 55-kHz f2 inputs of 75-dB SPL intensity. B I/O function for 4-week-old AFHL mice (n = 5) exposed to 16-kHz f2 inputs of varied intensities. C Average DP gram of 4-week-old HFHL (n = 31, green triangle) and C3HeB/FeJ mice (n = 25, blue diamond) in response to 5- to 55-kHz f2 inputs at 75-dB SPL intensity. D Average DP gram of 26-week-old HFHL (n = 18, green triangle) and C3HeB/FeJ mice (n = 6, blue diamond) in response to 5- to 55-kHz f2 inputs at 75-dB SPL intensity. E I/O function for 4- (n = 31), 26- (n = 18), and 52-week-old (n = 24) HFHL mice exposed to 16-kHz f2 inputs of varied intensities. F I/O function for 4- (n = 31), 26- (n = 18), and 52-week-old (n = 24) HFHL mice exposed to 32-kHz f2 inputs of varied intensities.
Cochlear morphology of AFHL and HFHL lines
To identify any morphological defects that might be responsible for the different auditory phenotypes, we examined the cochlear morphology of both HFHL and AFHL lines and C3H3B/FeJ control mice at 4 and 52 weeks of age (Fig. 3). Modiolar sections of the cochlea of HFHL and AFHL mice at 4 weeks of age showed a normal morphology of the organ of Corti and spiral ganglion (Fig. 3). At 52 weeks of age, we observed hair cell loss and degeneration of the spiral ganglion along the entire length of the cochlea in AFHL mice. In contrast, the organ of Corti of HFHL mice had a normal appearance, and degeneration of the spiral ganglion was observed only at the basal (high-frequency) portion of the cochlea. These changes in HFHL mice are most likely secondary effects since hearing loss was observed long before the onset of the histopathologies.

Inner ear histology in AFHL and HFHL lines. Toluidine blue-stained plastic sections through the mid-apical and basal portions of the cochlear ducts of 4- and 52-week-old HFHL and AFHL mice. The organ of Corti appears normal at 4 and 52 weeks in HFHL mice and at 4 weeks in AFHL mice. Hair cells are missing and the organ of Corti is highly degenerated in 52-week-old AFHL mice. In HFHL mice, spiral ganglia appear qualitatively normal in mid-apical and basal portions of the cochlea at 4 weeks and in the mid-apical portion at 52 weeks, but show some degradation in the basal portion of HFHL mice at 52 weeks. Also note the degeneration of nerve fibers in the habenula perforata at the base in 52-week-old HFHL mice. Spiral ganglia of AFHL mice are normal at 4 weeks, but are highly degraded throughout the cochlea at 52 weeks. Arrows indicate regions of degeneration. tm tectorial membrane, tC tunnel of Corti, sM scala media. Scale bar, 50 μm (upper row) and 100 μm (second to fourth rows).
Since hair bundle dysmorphology is commonly associated with hearing loss, we evaluated phalloidin-stained stereocilia of the inner (IHC) and outer hair cells (OHC) of AFHL and HFHL mice (Fig. 4). Examination of the surface of the organ of Corti at postnatal (P) day 5 and P7 revealed that the hair bundle morphology was normal in the HFHL line along the entire cochlea duct examined (Fig. 4A, B). In AFHL mice, however, OHC bundles have a narrow, compressed (U-shaped) appearance at P5 and P7 (Fig. 4A). Cumulatively, the audiology and morphology data suggest that the hearing defects in the AFHL and HFHL lines are of sensorineural origin.

Hair bundle morphology in HFHL and AFHL mice. A Phalloidin-stained P5 and P7 organs of Corti in HFHL, AFHL, and C3HeB/FeJ mice. Mid-apical hair bundles of HFHL mice appear qualitatively normal, but hair bundles of AFHL mice are more compressed and U-shaped than those of control C3HeB/FeJ mice. B Hair bundles of P6 HFHL mice appear normal at the base, the middle, and the apex of the cochlea. Scale bars, 5 μm.
(AFHL × HFHL) F1 complementation test
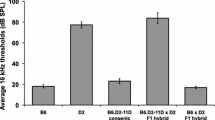
To test the proposition that alleles at different loci cause the HFHL and AFHL phenotypes, we determined the ABR thresholds of (AFHL × HFHL) F1 hybrids and [(AFHL × HFHL) × AFHL] N2 backcross mice at 8 weeks of age. Because (AFHL × HFHL) F1 mice are heterozygous at all loci for which the two lines segregate different alleles, this cross is expected to produce mice with a normal hearing phenotype if the AFHL and HFHL phenotypes are caused by recessive alleles at different loci. If, however, the two lines share the causative alleles at any loci responsible for either of the hearing phenotypes, the (AFHL × HFHL) F1 hybrids should express the respective phenotype. The (AFHL × HFHL) F1 mice exhibited an HFHL-like phenotype (Fig. 5A and Table 1). This indicates that the HFHL mice do not carry the AFHL alleles but that AFHL mice probably carry HFHL alleles at least at some loci. The presence of normal hair bundle morphology in the (AFHL × HFHL) F1 mice at P5 is consistent with the loss of AFHL alleles in the HFHL line (results not shown).

(AFHL × HFHL) F1 complementation test. A Click, 8-, 16-, and 32-kHz ABR thresholds of 8-week-old (HFHL × AFHL) F1 mice (n = 14). B Click, 8-, 16-, and 32-kHz ABR thresholds of 8-week-old (HFHL × AFHL) F1 × AFHL mice (n = 27). Each circle represents one measurement of one animal, and the line indicates the mean of this population.
In contrast, approximately 50% of the [(AFHL × HFHL) × AFHL] N2 mice are expected to express the AFHL phenotype if AFHL mice segregate alleles that are not present in the HFHL line. Mice of the [(AFHL × HFHL) × AFHL] N2 backcross displayed a bimodal distribution of ABR thresholds at the click, 8-, and 16-kHz stimulus and uniform distribution at the 32-kHz frequency. The AFHL phenotype was restored in ~50% of the [(AFHL × HFHL) × AFHL] N2 mice (Fig. 5B and Table 1), confirming that the alleles responsible for the AFHL phenotype were fixed in the AFHL line.
AFHL QTL analysis
N2 mice produced from each line were used to map the loci responsible for the different hearing phenotypes. We obtained ABR thresholds for 675 AFHL-N2 mice at 8 weeks of age. ABR thresholds of the AFHL-N2 population had complex but primarily bimodal frequency distributions (Fig. 6A–D), suggesting that a single major locus was responsible for much of the threshold variation in that population. The frequency distribution of click ABR thresholds is particularly interesting because this distribution appears to have three distinct maxima (Fig. 6A) at ~30, ~75, and ~100 dB SPL. The first and second maxima appear to lie at the means of separate Gaussian distributions, and the third peak occurs as a single spike at 100 dB SPL. Attempts to fit curves to this distribution were not satisfactory until the peak at 100 dB SPL was removed from the analysis. By doing so, we were able to fit the remainder of the histogram to the model for the sum of two Gaussian distributions (Fig. 6A; R 2 = 0.9272, n = 542) with means at 30.67 ± 7.45 and 81.19 ± 14.83 dB SPL. This distribution suggests that there may be two loci that have a large effect on click ABR thresholds in the AFHL population. Of these two loci, one major effect locus may be responsible for the primary loss of hearing (and the bimodal distribution seen on the histogram), whereas a second locus may cause the profound hearing loss (threshold ≥100 dB SPL) in mice homozygous for the defective allele at the first locus. This is consistent with the result that about half of the N2 population (324/675 = 48%) had normal hearing (<60 dB SPL) while almost one fourth (19.7%) was completely deaf.

QTL Analysis of AFHL-N2 cross. A ABR threshold distributions in 8-week-old AFHL-N2 mice (n = 675). Distributions are shown for the click and the 8-, 16-, and 32-kHz stimuli. Mice with thresholds of 100 dB SPL (n = 133) are not included in the histogram for the click stimulus to permit modeling of the frequency distribution. For all stimuli, histograms appear to exhibit bimodal distributions. Curves model the sum of two Gaussian distributions for the click (goodness-of fit R 2 = 0.9272, n = 542) and 32-kHz thresholds (goodness-of fit R 2 = 0.7488, n = 675). B–D Interval mapping plots for QTL affecting 32-kHz ABR thresholds on chromosome 7 (B), chromosome 8 (C), and chromosome 10 (D) in the AFHL-N2 population. Dashed lines indicate the permutation-derived genome-wide significance levels for suggestive, significant, and highly significant linkage. Bars indicate the 1.5 LOD support interval.
To identify the QTLs underlying hearing loss, we performed marker regression analyses on the hearing thresholds of 300 AFHL-N2 mice (Table 3). The preliminary genome-wide QTL scan for click stimulus thresholds identified two regions for which the LRS scores provided evidence of a QTL. One locus near rs13480652 on chromosome 10 was highly significant (LOD = 93.8) and explained approximately 77% of the threshold variation, while a second locus near rs3665356 on chromosome 14 was suggestive of a QTL (LOD = 1.6) and explained approximately 2% of the variation. Similarly, genome-wide scans of the 8- and 16-kHz thresholds detected a highly significant QTL near rs13480652 (LOD = 91.3 and 98.8, respectively) that explained the majority of the threshold variation (>75%) and a suggestive QTL near rs3665356 (LOD = 1.5 and 1.7, respectively) that explained only a small proportion (2–3%) of the variation.
Marker regression analysis of the 32-kHz thresholds identified a highly significant QTL on chromosome 10 at the rs6326263 locus (LOD = 40.0) with an effect size of 47%. In addition, a significant QTL was detected between rs31944466 and rs3700384 on chromosome 7 (LOD = 3.3), and a highly significant QTL was scored near rs13479916 on chromosome 8 (LOD = 4.6). The latter two QTLs accounted for 5% and 7% of the variation in 32-kHz thresholds, respectively. In addition, the analyses of the 32-kHz thresholds detected a suggestive QTL near rs3665356 (LOD = 1.8).
To more accurately identify the locations of each QTL in the AFHL line, we performed composite interval mapping analysis on each chromosome for which we identified a significant or highly significant QTL in the marker regression analysis (Fig. 6B–D). Because the effects of the chromosome 10 QTL were so strong, the rs6326263 locus was included as background in interval mapping of chromosomes 7 and 8. We also included the loci on chromosome 7 (rs31944466) and chromosome 8 (rs13479916) as background when performing interval mapping of chromosome 10 for the 32-kHz thresholds. The chromosome 7 and 8 background loci had negligible effects on the LOD score and effect size calculated when interval mapping chromosome 10. More importantly, the loci on chromosomes 7 and 8 maintained their significance (LOD = 3.9 and 4.6, respectively) even after correcting for background, indicating that these were probably not spurious QTLs. Based on composite interval mapping, the chromosome 10 QTL is located just proximal to rs6326263 (at 27 cM), with a 1.5 LOD support interval encompassing the region 22–33 cM from the centromere. The chromosome 7 QTL is located near rs31944466 (at 24 cM) and has a 1.5 LOD support interval that covers the proximal portion of the chromosome up to 37 cM. The chromosome 8 QTL is located close to rs13479916, with a 1.5 LOD support interval located 21–32 cM from the centromere. Composite interval mapping, with rs6326263 marker as background, was also performed on chromosome 14. The significance level of the LOD score did not improve, but the locus retained its status as a suggestive QTL. We did not detect any significant interactions among these QTLs affecting hearing thresholds.
To further ascertain the presence of the QTLs on chromosomes 7 and 8 that affected 32-kHz thresholds only, we genotyped an additional 355 AFHL-N2 mice at several SNPs near the putative locations of each QTL (rs8255275 and rs3680765 on chromosome 7; rs13479840, rs13479916, and rs13479929 on chromosome 8) and performed marker regression analysis on that population. We confirmed that the chromosome 7 QTL only affected the 32-kHz ABR thresholds (LOD = 3.9 at rs8255275), and this remained true even after correcting for the effects of the chromosome 10 locus (microsatellite D10MIt259 genotype was used to denote the chromosome 10 locus). The chromosome 8 QTL failed to reach the suggestive level of significance in the second sample of AFHL-N2 mice, but was still significant when evaluated using the entire population (LOD = 4.9 at rs13479916). We have designated the chromosome 7 and 8 QTLs Hfhl1 and Hfhl2 (high-frequency hearing loss 1 and 2), respectively.
HFHL QTL analysis
ABR thresholds obtained for 306 HFHL-N2 mice at 8 weeks of age had basically Gaussian distributions (Fig. 7A) at all four stimuli, suggesting that the differences were the result of many genes of small effect. However, the threshold distribution of the 32-kHz ABR thresholds is skewed, indicating that there may be at least one allele that has a larger effect. By marker regression analysis, we detected a highly significant QTL (LOD = 5.97) at marker rs6160140 on chromosome 7 that accounts for about 9% of the variation in 32-kHz ABR thresholds in the HFHL-N2 population. An additional locus on chromosome 9 (rs13480208) was suggestive of a QTL (LOD = 1.91) and accounts for about 3% of the variation in 32-kHz thresholds (Table 3).

QTL Analysis of HFHL-N2 cross. A ABR threshold distributions in 8-week-old HFHL-N2 mice. Distributions are shown for the click and the 8-, 16-, and 32-kHz stimuli. Histograms for the click, 8-, and 16-kHz stimuli were fitted with Gaussian distribution curves (goodness-of-fit R 2 = 0.9614, 0.9990, and 0.9716, respectively; n = 303–306). The histogram for the 32-kHz stimulus was fitted to the sum of two Gaussian distributions (R 2 = 0.9716, n = 306). B Interval mapping plot for QTL affecting 32-kHz ABR thresholds on chromosome 7 in the HFHL-N2 population (n = 305). Dashed lines indicate the permutation-derived genome-wide significance levels for suggestive, significant, and highly significant linkage. Bar indicates the 1.5 LOD support interval.
Closer inspection of the rs6160140 locus at chromosome 7 revealed that it did not exhibit the expected 1:1 ratio of heterozygotes to homozygotes (χ 2 = 6.27, p = 0.0123). Two other linked loci (rs13479325 and rs6279696) also did not have the expected genotypic ratio. However, an adjacent locus that also had a highly significant LOD score, rs6228386 (LOD = 5.2), did have a 1:1 genotypic ratio. Furthermore, two nearby loci that had been genotyped using TaqMan® SNP Genotyping Assays had the 1:1 genotypic ratio and also supported the presence of a highly significant QTL in the region (rs8255275 and rs3719301 with LOD = 5.53 and 5.37, respectively). Given the fact that rs6160140 lies between highly significant loci that did exhibit the theoretical genotypic ratio and that a few of the 145 loci tested are expected to deviate from the 1:1 genotypic ratio by chance alone, we chose not to eliminate rs6160140 and used it in all further analyses.
To more accurately identify the location of this highly significant QTL on chromosome 7, we performed composite interval mapping (Fig. 7B). The rs13480208 locus on chromosome 9 was used as background for the analysis of the chromosome 7 QTL, but this had only a negligible effect and confirmed the results from the interval mapping. Composite interval mapping indicated that the chromosome 7 QTL is located between rs6326263 and rs6160140 (at 27 cM). The 1.5 LOD support interval for the QTL encompasses the region 12–38 cM from the centromere of chromosome 7. The chromosome 7 QTL interval detected in the HFHL-N2 cross lies almost completely within the interval calculated for the chromosome 7 QTL mapped in the AFHL-N2 (Fig. 6A) cross, indicating that the QTL detected in these two independent crosses probably correspond to the same locus, Hfhl1. Composite interval mapping of the chromosome 9 QTL (with rs6160140 as background) did not affect the significance level of the QTL, but relocated it to the distal end of chromosome 9, near rs6152427. No significant interactions were detected.
Somewhat surprisingly given the HFHL phenotype, we also detected putative QTLs, with LOD scores at the suggestive level of significance that affected the click, 8-, and 16-kHz ABR thresholds. These putative QTLs were located at rs13476540 on chromosome 2 (LOD = 1.45), rs6354160 on chromosome 5 (LOD = 1.45), rs6268125 on chromosome 6 (LOD = 1.67), and rs6296891 on chromosome 8 (LOD = 1.73) and had minor effects, each accounting for only 1–3% of the variation (Table 3).
QTL effects
The effects of the three significant QTLs affecting 32-kHz ABR thresholds in the AFHL-N2 population are shown in Figure 8A. It is apparent that the chromosome 10 locus (rs6326263) alone is sufficient to cause profound deafness. The mean 32-kHz ABR thresholds for all groups homozygous for the AFHL allele at rs6326263 exceed 99 dB SPL, are not different (p > 0.05, ANOVA), and are significantly greater than the thresholds for all groups that are heterozygous for the AFHL allele at rs6326263 (p < 0.001). However, the Hfhl1 and Hfhl2 loci (represented by rs8255275 and rs13479916, respectively) appear to act additively when the wild-type allele is present at rs6326263. The mean 32-kHz ABR threshold for mice homozygous for AFHL alleles at both rs8255275 and rs13479916 (83 ± 17 dB SPL, n = 35) is significantly greater (p < 0.001) than the mean threshold for mice homozygous for AFHL alleles at either only rs8255275 (76 ± 19 dB SPL, n = 32) or only rs13479916 (73 ± 17 dB SPL, n = 38), both of which have significantly higher 32-kHz thresholds (p < 0.001) than mice that are heterozygous at all three loci (62 ± 14 dB SPL, n = 49).

QTL effects. A Shown are 32-kHz ABR thresholds (mean±SEM) by haplotype in the AFHL-N2 population (n = 588). Each column indicates the average ABR threshold for individuals possessing the haplotype shown on the X-axis. Haplotypes are composed of genotypes at loci representative of the QTL on chromosome 7 (C7), chromosome 8 (C8), and chromosome 10 (C10). rs8255275 genotypes represent the chromosome 7 QTL (Hfhl1), rs13479916 genotypes represent the chromosome 8 QTL (Hfhl2), and rs6326263 genotypes represent the chromosome 10 QTL. For each locus, a plus sign indicates heterozygosity and N indicates homozygosity for AFHL alleles. B Shown are 32-kHz ABR thresholds (mean±SEM) by genotype in the HFHL-N2 population (n = 288). rs8255275 genotypes were used to represent the chromosome 7 QTL. ns not significant. ***p < 0.001.
The effect of Hfhl1 on 32-kHz ABR thresholds in the HFHL-N2 population is also quite apparent (Fig. 8B). The mean 32-kHz ABR threshold in mice homozygous for the HFHL allele at the rs8255275 locus (51 ± 14 dB SPL, n = 131) is significantly higher (p < 0.001) than the 32-kHz ABR thresholds for mice heterozygous at rs8255275 (43 ± 10 dB SPL, n = 157).
Gene identification
Several hypomorphic variants that affect hearing and produce phenotypes similar to that of the AFHL line reside on chromosome 10 near the QTL detected here (Noben-Trauth and Johnson 2009). Two of these, ahl and ahl5, seemed to be particularly good candidates for contributing to the hearing loss in the AFHL line. The 753G→A transition of the Cdh23 gene (Cdh23 ahl) is common in inbred strains (Noben-Trauth et al. 2003), and the 343G→A transition of the Gipc3 gene (Gipc3 ahl5) was recently identified as the cause of progressive sensorineural hearing loss in Black Swiss mice (Charizopoulou et al. 2011). The Black Swiss heterogeneous strain was derived from a cross between the NIH Swiss stock and the C57BL/6 strain (Beck et al. 2000). To determine whether the Black Swiss and the AFHL line might contain the same variants, we performed an allelism test. AFHL mice × Black Swiss mice were crossed to produce (AFHL × BLSW) F1 offspring, and the ABR thresholds were measured. For all test stimuli, the (AFHL × BLSW) F1 mice exhibited impaired hearing. Average thresholds were 78.2, 86.6, 90.5, and 100 dB SPL for the click, 8-, 16-, and 32-kHz stimuli, respectively (Table 1). Since the Gipc3 ahl5 allele is recessive, our results suggested that the AFHL line did indeed harbor the Gipc3 ahl5 variant. DNA sequencing analyses confirmed that the AFHL line was homozygous for both the Cdh23 ahl and the Gipc3 ahl5 allele, whereas the HFHL line segregated the wild-type alleles at these loci.
Discussion
In this study, we identified a number of allelic loci that affect hearing in NIH Swiss outbred mice. The effects of these loci were highly variable, explaining from 1% to 78% of the observed variation in hearing thresholds of the N2 population. These results are consistent with previous studies in inbred and heterogeneous strains that detected QTLs that accounted for 8–82% of the variance (Johnson et al. 2000, 2008; Johnson and Zheng 2002; Noben-Trauth et al. 2010; Noguchi et al. 2006; Zheng et al. 2008). Interestingly, the QTLs identified here in the NIH Swiss population exert differential effects on the frequency range and on the onset and progression of the hearing deficit.
In the AFHL-N2 population, we identified a locus on chromosome 10 that had highly significant effects on all hearing thresholds. The AFHL population has both the Cdh23 753A mutation that is responsible for late-onset age-related hearing loss in a number of inbred strains (Noben-Trauth et al. 2003) and the more recently discovered mutation of the Gipc3 gene that leads to profound deafness in the Black Swiss stock (Charizopoulou et al. 2011). Mice with mutations in either of these genes have severe sensorineural hearing loss. They also develop irregular stereocilia bundles and, eventually, degeneration of the organ of Corti and spiral ganglia similar to the phenotype observed in the AFHL line (Di Palma et al. 2001; Charizopoulou et al. 2011). It is not surprising that the Black Swiss and NIH Swiss stocks share the Gipc3 ahl5 allele since Black Swiss mice were derived from the NIH Swiss stock (Beck et al. 2000). However, the NIH Swiss mice were previously reported to have a less severe phenotype than the Black Swiss mice (Drayton and Noben-Trauth 2006). This is surprising since the NIH Swiss mice have the Cdh23 ahl allele in addition to the Gipc3 ahl5 variant, and in other cases where the Cdh23 ahl allele was present in conjunction with a second locus polymorphism, the phenotype worsened (e.g., Johnson et al. 2001, 2008; Zheng et al. 2008). It is possible that two other loci, the ahl6 locus on chromosome 18 and a suggestive QTL on chromosome 13, that were also identified in the Black Swiss population have greater epistatic interactions with the Gipc3 ahl5 variant than with Cdh23 ahl, thus producing a more severe phenotype in Black Swiss mice (Drayton and Noben-Trauth 2006). It seems more likely, however, that the less severe phenotype observed in the NIH Swiss population was due to the influence of those NIH Swiss mice that only possessed a relatively mild HFHL phenotype.
The highly significant LOD scores along most of chromosome 10 in the AFHL population could indicate that loci on chromosome 10 in addition to Cdh23 ahl and Gipc3 ahl5 are contributing to the AFHL phenotype. However, we suggest that these LOD scores reflect the strength of the association of the phenotype with these two major effect QTLs and the relatively small amount of recombination that occurs with backcrossing.
We also detected two other loci in the AFHL population (Hfhl1 on chromosome 7 and Hfhl2 on chromosome 8) with significant effects. Both of these QTLs had substantially less impact on hearing thresholds than the chromosome 10 locus and, interestingly, only affected ABR thresholds at the 32-kHz stimulus. Other loci have been shown to affect primarily the basal (high-frequency) portion of the cochlea (Gao et al. 2007; Kermany et al. 2006; Noben-Trauth et al. 2003). However, those loci generally appear to be responsible for the degeneration of the organ of Corti concomitantly with or shortly after the loss of hearing (Gao et al. 2007; Kermany et al. 2006). Loci that appear to specifically affect high-frequency hearing are oftentimes responsible for progressive hearing loss, and the QTLs actually affect a greater range of hearing frequencies as degeneration of the organ of Corti progresses (Johnson et al. 2000; Zheng et al. 2008). Based on the phenotype of the HFHL population, Hfhl1 seems to be the first QTL whose specificity for affecting high-frequency hearing does not appear to be the result of the degeneration of the basal cochlea.
In contrast to the AFHL line, we did not detect a QTL on chromosome 10 that affected hearing in the HFHL line, although we did detect a QTL on chromosome 7. Sequencing indicated that the HFHL line does not carry the Gipc3 ahl5 or the Cdh23 ahl variants. The loss of the AFHL phenotype, together with the loss of these alleles, supports their role as the major cause of AFHL. Based on the complementation tests as well as the interval mapping data, the QTL on chromosome 7 in the HFHL line is probably the same locus that influences the 32-kHz ABR thresholds in the AFHL line (Hfhl1). Hfhl1 homozygotes have hearing thresholds ~10 dB SPL higher than heterozygotes, indicating that this locus has a relatively minor effect on hearing compared with other hearing loss QTLs (Johnson et al. 2000, 2008; Johnson and Zheng 2002; Noben-Trauth et al. 2010; Zheng et al. 2008). However, its effects are similar to those found in studies that detected multiple QTLs (Phl1 and Phl2) of moderate effect (Noguchi et al. 2006).
Despite its relatively mild phenotype and effect, the Hfhl1 locus is particularly interesting due to its frequency specificity. Mice of the HFHL line exhibit normal hearing at frequencies below ~30 kHz, but have poor hearing at higher frequencies. In addition, the phenotype is expressed at the onset of hearing, is completely penetrant, and is only mildly progressive. The frequency-specific nature of the hearing loss suggests that the defect may be the result of altered tonotopy or, at least, that there is a tonotopic component to the phenotype that restricts the defect to the basal region of the cochlea. Its unique phenotype makes the HFHL line an excellent model system for investigating frequency-specific hearing loss and tonotopy. Furthermore, the hearing phenotype of the HFHL line is created by a genetic complexity that may be representative of less severe and slowly progressing forms of human hearing loss.
It is interesting to note that Hfhl2 was not detected in the HFHL line despite the fact that we selected for precisely the phenotype that it would be expected to produce. The Hfhl2 locus may have been lost during inbreeding since Hfhl1 produces the same phenotype and both QTLs are only weakly cumulative. In support of this, the mean 32-kHz ABR thresholds of the HFHL line decreased approximately 8 dB SPL (n = 30, F9 generation; n = 20, F10 generation; t = 4.7, p < 0.0001) and the variance decreased by about 67% (F = 16.63, p < 0.0001) between generations F9 and F10. Furthermore, the AFHL and HFHL lines differ in the allele that they possess at one of the SNPs (rs13479929) located in the region of the QTL, indicating that genetic variation present in this region in the NIH Swiss population segregated between the two lines. It is also possible that the effect of Hfhl2 was too small to be detected in the HFHL population or that it was a false-positive QTL, especially since it was not significant when evaluated in the second sample of AFHL-N2 mice. However, gene interactions and genetic background have been shown to play a role in the expression of hearing phenotypes (Noben-Trauth et al. 1997; Noguchi et al. 2006). Hence, a more intriguing possibility is that the effects of Hfhl2 were not detected in the HFHL line because those effects are under epistatic control; that is, they are only observable in the presence of other allelic loci found in the AFHL population that are no longer present in the HFHL population.
Finally, our results are applicable to genetically diverse natural populations in which some individuals carry a single mutation that causes dramatic deficits in hearing, others possess intermediate deficits as a result of a number of loci of small effect or due to gene interactions, and still other individuals exhibit only marginally impaired hearing due to one or two alleles of small effect. The Hfhl2 QTL interval is syntenic with regions on human chromosomes 4 and 19, which harbor the deafness loci DFNB26 and DFNB65, respectively. Thus, our results also are relevant to human populations.
Abbreviations
- ABR:
-
Auditory-evoked brain stem response
- DPOAE:
-
Distortion product otoacoustic emission
- EP:
-
Endocochlear potential
- SNHL:
-
Sensorineural hearing loss
- QTL:
-
Quantitative trait locus
- HFHL:
-
High-frequency hearing loss
References
Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MFW, Fisher EMC (2000) Genealogies of mouse inbred strains. Nat Genet 24:23–25
Charizopoulou N, Lelli A, Schraders M, Ray K, Hildebrand MS, Ramesh A, Srisailapathy CRS, Oostrik J, Admiraal RJC, Neely HR, Latoche JR, Smith RJH, Northup JK, Kremer H, Holt JR, Noben-Trauth K (2011) Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat Comm 2:201
Chia R, Achilli F, Festing MFW, Fisher EMC (2005) The origins and uses of mouse outbred stocks. Nat Genet 37:1181–1186
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Dallos P (1996) Overview: cochlear neurobiology. In: Dallos P, Popper AN, Fay RF (eds) The cochlea. Springer, New York, pp 1–43
Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K (2001) Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet 27:103–107
Drayton M, Noben-Trauth K (2006) Mapping quantitative trait loci for hearing loss in Black Swiss mice. Hear Res 212:128–139
Dror AA, Avraham KB (2009) Hearing loss: mechanisms revealed by genetics and cell biology. Annu Rev Genet 43:411–437
Gao J, Maison SF, Wu X, Hirose K, Jones SM, Bayazitov I, Tian Y, Mittleman G, Matthews DB, Zakharenko SS, Liberman MC, Zuo J (2007) Orphan glutamate receptor d1 subunit required for high frequency hearing. Mol Cell Biol 27:4500–4512
Haley CS, Knott SA (1992) A simple regression technique for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69:315–324
Hudspeth AJ (1982) Extracellular current flow and the site of transduction by vertebrate hair cells. J Neurosci 2:1–10
Johnson KR, Zheng QY (2002) Ahl2, a second locus affecting age-related hearing loss in mice. Genomics 80:461–464
Johnson KR, Zheng QY, Erway LC (2000) A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics 70:171–180
Johnson KR, Zheng QY, Bykhovskaya Y, Spirina O, Fichel-Ghodsian N (2001) A nuclear–mitochondrial DNA interaction affecting hearing impairment in mice. Nat Genet 27:191–194
Johnson KR, Longo-Guess C, Gagnon LH, Yu H, Zheng QY (2008) A locus on distal chromosome 11 (ahl8) and its interactions with Cdh23 ahl underlie the early onset, age-related hearing loss of DBA/2J mice. Genomics 92:219–225
Kermany MH, Parker LL, Guo Y-K, Miller D, Swanson DJ, Yoo T-J, Goldowitz D, Zuo J (2006) Identification of 17 hearing impaired mouse strains in TMGC ENU-mutagenesis screen. Hear Res 220:76–86
Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247
Manly KF, Olson JM (1999) Overview of QTL mapping software and introduction to Map Manager QT. Mamm Genome 10:327–334
Manly KF, Cudmore RH Jr, Meer JM (2001) Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome 12:930–932
Mann ZF, Kelley MW (2011) Development of tonotopy in the auditory periphery. Hear Res. doi:10.106/j.heares.2011.01.011
Noben-Trauth K, Zheng QY, Johnson KR, Nishina PM (1997) Deafness susceptibility locus that interacts with deaf waddler (dfw). Genomics 44:266–272
Noben-Trauth K, Zheng QY, Johnson KR (2003) Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet 35:21–23
Noben-Trauth K, Johnson KR (2009) Inheritance patterns of progressive hearing loss in laboratory strains of mice Brain Res. 1277:42–51
Noben-Trauth K, Latoche JR, Neely HR, Bennett B (2010) Phenotype and genetics of progressive sensorineural hearing loss (Snhl1) in the LXS set of recombinant inbred strains of mice. PLoS ONE 5:1–8
Noguchi Y, Kurima K, Makishima T, Hrabé de Angelis M, Fuchs H, Frolenkov G, Kitamura K, Griffith AJ (2006) Multiple quantitative trait loci modify cochlear hair cell degeneration in the Beethoven (Tmc1 Bth) mouse model of progressive hearing loss DFNA36. Genetics 173:2111–2119
Rice MC, O’Brien SJ (1980) Genetic variance of laboratory outbred Swiss mice. Nature 283:157–161
Zheng QY, Ding D, Yu H, Salvi RJ, Johnson KR (2008) A locus on distal chromosome 10 (ahl4) affecting age-related hearing loss in A/J mice. Neurobiol Aging 30:1693–1705
Acknowledgments
We thank M’Hamed Grati and Dennis Drayna for comments on the manuscript. The Division of Intramural Research at NIDCD supported the study.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Keller, J.M., Neely, H.R., Latoche, J.R. et al. High-Frequency Sensorineural Hearing Loss and Its Underlying Genetics (Hfhl1 and Hfhl2) in NIH Swiss Mice. JARO 12, 617–631 (2011). https://doi.org/10.1007/s10162-011-0270-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-011-0270-7