
Abstract
Efferent inhibition of cochlear hair cells is mediated by α9α10 nicotinic cholinergic receptors (nAChRs) functionally coupled to calcium-activated, small conductance (SK2) potassium channels. Before the onset of hearing, efferent fibers transiently make functional cholinergic synapses with inner hair cells (IHCs). The retraction of these fibers after the onset of hearing correlates with the cessation of transcription of the Chrna10 (but not the Chrna9) gene in IHCs. To further analyze this developmental change, we generated a transgenic mice whose IHCs constitutively express α10 into adulthood by expressing the α10 cDNA under the control of the Pou4f3 gene promoter. In situ hybridization showed that the α10 mRNA is expressed in IHCs of 8-week-old transgenic mice, but not in wild-type mice. Moreover, this mRNA is translated into a functional protein, since IHCs from P8-P10 α10 transgenic mice backcrossed to a Chrna10 −/− background (whose IHCs have no cholinergic function) displayed normal synaptic and acetylcholine (ACh)-evoked currents in patch-clamp recordings. Thus, the α10 transgene restored nAChR function. However, in the α10 transgenic mice, no synaptic or ACh-evoked currents were observed in P16-18 IHCs, indicating developmental down-regulation of functional nAChRs after the onset of hearing, as normally observed in wild-type mice. The lack of functional ACh currents correlated with the lack of SK2 currents. These results indicate that multiple features of the efferent postsynaptic complex to IHCs, in addition to the nAChR subunits, are down-regulated in synchrony after the onset of hearing, leading to lack of responses to ACh.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Efferent inhibition of cochlear hair cells is mediated by the release of acetylcholine (ACh) from neurons originating in the superior olivary complex of the brainstem. In the mature cochlea, the medial olivocochlear (OC) efferent pathway projects to outer hair cells (OHCs) where large synaptic contacts are formed (Guinan 1996). Activation of this pathway reduces cochlear sensitivity through the action of ACh on nicotinic receptors (nAChRs) at the base of OHCs. Significant progress has been made in defining the cellular mechanisms of hair cell inhibition: α9 and α10 nAChR subunits arrange into a pentameric assembly with a likely (α9)2(α10)3 stoichiometry (Elgoyhen et al. 1994, 2001; Lustig et al. 2001; Plazas et al. 2005; Sgard et al. 2002) and activation of the α9α10 nAChR leads to an increase in intracellular Ca2+ and the subsequent opening of small conductance Ca2+-activated K+ (SK2) channels, thus leading to hyperpolarization of hair cells (Dulon et al. 1998; Fuchs and Murrow 1992; Housley and Ashmore 1991; Oliver et al. 2000).
Although adult inner hair cells (IHCs) receive very few (if any) direct axosomatic contacts from efferent fibers, before the onset of hearing [until about postnatal (P) day 12 in rats and mice], a transient efferent innervation is found on IHCs (Simmons 2002; Katz et al. 2004). These transient efferent axosomatic synapses with IHCs most likely play a role in the modulation of the Ca2+ spiking activity, a characteristic of immature IHCs, which may drive rhythmic or bursting activity of neurons at higher levels of the auditory pathway (Glowatzki and Fuchs 2000). Previous studies have suggested that this transient efferent innervation may play a role in the ultimate functional maturation of cochlear hair cells (Simmons 2002). Most impressively, surgical lesion of the efferent nerve supply causes kittens to fail to develop normal hearing (Walsh et al. 1998).
With maturation of the cochlea, a number of changes in the expression of voltage-gated channels tend to reduce IHC spiking (Kros et al. 1998; Marcotti et al. 2003a, b). These changes signal the transformation from a developing epithelium with active formation of synaptic contacts to a sensing epithelium where receptor potentials represent the mechanical input in a graded fashion. These changes are accompanied by the loss of direct efferent innervation to IHCs, and this is directly correlated to the cessation of transcription in IHCs of the gene coding for the α10 (Chrna10) but not the α9 (Chrna9) nAChR subunit (Elgoyhen et al. 1994, 2001; Morley and Simmons 2002; Simmons 2002). In fact, Chrna9 continues to be transcribed into adult stages (Elgoyhen et al. 1994). To further analyze this critical developmental change near the onset of hearing, we generated a transgenic mice whose IHCs constitutively express α10 into adulthood by expressing the α10 cDNA under the control of the mouse Pou4f3 gene promoter, a hair cell transcription factor (Erkman et al. 1996). We reasoned that if the lack of responses to ACh after the onset of hearing was due to the cessation in transcription of Chrna10, constitutive expression of the α10 subunit would result in functional receptors. However, ACh sensitivity was lost on schedule, as in wild-type animals. Thus, we found that expression of the α10 nAChR subunit into adult ages is not sufficient to sustain cholinergic function, and efferent innervation, of IHCs after the onset of hearing.
Materials and methods
Generation of Pou4f3-α10 transgenic mice
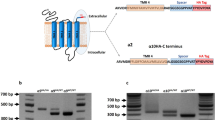
The Pou4f3-α10 transgenic mice were generated using a construct like that previously engineered to drive hair cell expression of Cre recombinase (Sage et al. 2006). As shown in Figure 1A, a 1.5-kb fragment containing the cDNA encoding the α10 nAChR (Elgoyhen et al. 2001) was subcloned into pSP73 (Promega Corp, Madison, WI, USA). A 9-kb portion of the 5′ upstream region of the mouse Pou4f3 gene (L. Erkman and G. Rosenfeld, University of California at San Diego, La Jolla) was introduced upstream of the α10 cDNA and used to drive α10 nAChR subunit expression. A β-globin intronic sequence of 600 bp was blunt-ligated to the 5′ end of the α10 insert and a Flag tag (encoding the sequence DYKDDDDK) was inserted before the stop codon of the α10 sequence. A human growth hormone polyA initiation sequence of 580 was ligated to the 3′ end of the α10 insert. The construct was verified by sequencing. The Pou4f3-α10 transgene was released by ClaI digestion and used to generate the Pou4f3-α10 transgenic mouse line (with a B6CB5 background, a hybrid between C57BL/6J and CBA/J strains) by using standard pronuclear injection techniques (Young et al. 1998). All animal work was conducted using procedures reviewed and approved by the Institutional Animal Care and Use Committees of INGEBI and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Research Animals.

Genetic engineering, genotyping, and RNA expression of the Pou4f3-α10 transgene. A Map of the transgene construct. Pou4f3, 9-kb portion of the 5′ upstream promoter region of the Pou4f3 gene; intron, 600-bp β-globin intronic sequence; α10 cDNA, entire coding region plus 5′ and 3′ untranslated regions of the α10 nAChR subunit with a FLAG tag before the stop codon; polyA, 580-bp human growth hormone initiation sequence. B Routine genotyping of Pou4f3-α10 transgenic mice performed by PCR from isolated tail biopsy tissue genomic DNA samples and amplimers that anneal at the Pou4f3 and the intronic sequences (black arrows in A). A representative gel indicating no band in a wild-type mice and a positive band in a Pou4f3-α10 transgenic. C RT-PCR from transgenic cochleae indicating a positive band of 446 bp in transgenic mice using amplimers that anneal at the α10 cDNA and the FLAG sequence (gray arrows in A). Lane a positive; lane b control minus reverse transcriptase; lane c control minus oligo dT; lane d control minus RNA.
Genotyping
For transgenic genotyping, primers tgα10 β-globin (5′-CATGAGGGTCCATGGTGATAC-3′) and tgα10 Pou4f3 (5′-GCATCAGGCTCTCAGATGGCG-3′) were used, producing a 494-bp fragment (Fig. 1B). Polymerase chain reaction (PCR) was performed using 94°C for 2 min followed by 94°C for 30 s, 58°C for 1 min, and 72°C for 1.5 min for 30 cycles with 20 ng of genomic DNA obtained from tail biopsies, buffer Mix D (Epicentre, Madison, WI, USA), and Qiagen Taq polymerase (Qiagen, Valencia, CA, USA). The genotyping of the Chrna10 −/− mice was performed as previously described (Vetter et al. 2007).
Reverse transcriptase PCR
Eight-week-old mice were killed, the cochleae removed, and immediately frozen in liquid nitrogen. For each genotype, total RNA was extracted using Trizol reagent (Invitrogen, Buenos Aires, Argentina) following the manufacturer’s instructions after grinding the tissue in a TH-1 homogenizer (OMNI, Marietta, Gainsville) and centrifuging at 12,000×g at 4°C to remove bone fragments. A total of 1 μg of RNA was used to reverse transcribe using Superscript II reverse transcriptase (Invitrogen) and oligo dT (Invitrogen). One microliter of this reaction was used to amplify the transgenic α10 cDNA using one amplimer that anneals to the FLAG sequence (5′-CCATGGTCACATTCTCCACA-3′) and another to the α10 cDNA sequence (5′-CTTGTCATCGTCGTCCTTGTAGTC-3′). In transgenic mice, a 446-bp fragment was obtained.
In situ hybridization
In situ hybridization experiments used previously published protocols (Hiel et al. 1996). Briefly, the temporal bones of 8-week-old mice were fixed, decalcified, and embedded for cryosectioning. Postfixed, acetylated, and dehydrated 14-μm cryosections were hybridized with 35S-labeled riboprobe (989 bp; 1.2 × 106 cpm/slide) for 16 h at 56°C. After high-stringency washes and dehydration, tissue sections were coated with photographic emulsion NBT-2 (Eastman Kodak, Rochester, NY, USA) and developed for 2–5 weeks at 4°C. Relative expression levels for α10 mRNA in OHCs and IHCs were compared by counting grains in a box placed over selected regions of cochlear cross-sections. Background counts were repeatedly collected, averaged, and subtracted for each cross-section. Labeled sections from base, middle, and apical cochlear turns were obtained from one wild-type (five sections) and two α10 Pou4f3-α10 transgenic mice (six sections).
Electrophysiological recordings from cochlear hair cells
Mice were killed by decapitation. All experimental protocols were carried out in accordance with the AVMA Guidelines on Euthanasia (June 2007). Apical turns of the organ of Corti were excised from mice and used within 3 h. Day of birth was considered postnatal day 0, P0. Cochlear preparations were mounted under a Leica DMLFS microscope (Leica Microsystems, Wetzlar, Germany) and viewed with differential interference contrast using a 40× water immersion objective and a Hamamatsu C7500-50 camera with contrast enhancement (Hamamatsu, Hamamatsu City, Japan). Methods to record from IHCs were essentially as described previously (Glowatzki and Fuchs 2000; Oliver et al. 2000).
Briefly, IHCs were identified visually with the 40× objective and during recordings by the size of their capacitance (7 to 12 pF) and their characteristic voltage-dependent Na+ and K+ currents (Kros et al. 1998). Some cells were removed to access IHCs, but mostly, the pipette moved through the tissue under positive pressure. The extracellular solution was as follows (in mM): 155 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 5.6 d-glucose, and 10 HEPES buffer, pH 7.4. The pipette solution contained (in mM): 150 KCl, 3.5 MgCl2, 0.1 CaCl2, 5 ethyleneglycol-bis(β-aminoethyl ether)-N,N,N′,N′-teraacetic acid (EGTA), 5 HEPES buffer, 2.5 Na2ATP, pH 7.2 (KCl-EGTA saline). Solutions containing ACh or high K+ were applied by a gravity-fed multichannel glass pipette (∼150-μM tip diameter) positioned about 300 μM from the recorded cell. All working solutions containing either ACh or elevated K+ or both were made up in a saline containing low Ca2+ (0.5 mM) and no Mg2+ so as to optimize the experimental conditions for measuring currents flowing through the α9α10 receptors (Weisstaub et al. 2002).
Recording pipettes, 1.2-mm I.D, had resistances of 5–8 MΩ. Currents in IHCs were recorded in the whole-cell patch-clamp mode with an Axopatch 200B amplifier, low-pass-filtered at 2–10 kHz, and digitized at 5–20 kHz with a Digidata 1322A board (Molecular Devices, California, USA). Recordings were made at room temperature (22–25°C). Holding potentials were not corrected for liquid junction potentials or for the voltage drop across the uncompensated series resistance.
Results
IHCs of Pou4f3-α10 mice express α10 mRNA after the onset of hearing
It has been reported that the Pou4f3 transcription factor is expressed in hair cells from early embryonic until adult stages (Erkman et al. 1996) and that Cre expression driven by the Pou4f3 promoter starts as early as embryonic day 13.5 in a transgenic mouse (Sage et al. 2006). Thus, the use of the Pou4f3 promoter has become a useful tool for transgenic expression of genes in cochlear hair cells. The generation of a transgenic mouse in which the expression of the α10 cDNA is driven by Pou4f3 is explained in “Materials and methods”, the construct shown in Figure 1A and genotyping in Figure 1B. In order to assess Pou4f3-α10-driven expression of α10 RNA, reverse transcriptase PCR (RT-PCR) was performed from total RNA extracted from 8-week-old mice. As shown in Figure 1C, a 446-bp fragment was obtained in Pou4f3-α10 transgenic mice. Since one of the amplimers anneals to the FLAG sequence, this fragment can only derive from the RNA that has been correctly transcribed from the α10 transgenic cDNA. Amplification from genomic DNA is precluded since no band was observed in the control reaction without reverse transcriptase (lane b). Lanes c and d are control reactions, minus oligo dT and minus RNA, respectively.
To establish a hair cell localization of the transgenic α10 RNA, in situ hybridization was performed using a α10 riboprobe. As previously reported (Elgoyhen et al. 2001; Morley and Simmons 2002) and as shown in Figure 2A, wild-type mice express α10 in the OHC but not in the IHC region after the onset of hearing. However, in Pou4f3-α10 transgenic mice, silver grains were observed both in OHCs and IHCs of 8-week-old mice (Fig. 2B), indicating that the signal observed by RT-PCR most likely arises from the expression of the transgenic α10 RNA in the hair cells. Although in OHCs one cannot distinguish the transgenic versus the endogenous expression, in the IHCs, the signal can only arise from α10 RNA derived from the successful expression of the Pou4f3-α10 transgene, since wild-type IHC do not express α10 at this adult developmental stage (Fig. 2A; Elgoyhen et al. 2001; Morley and Simmons 2002).

Transgenic α10 RNA is found in the IHC region after the onset of hearing. A In situ hybridization showing that silver grains are only observed in the OHC region of 8-week-old wild-type mice. B In Pou4f3-α10 transgenic cochleae of 8-week-old mice, staining is observed both in the OHC and IHC region. Grain counts were collected from five wild-type and six Pou4f3-α10 transgenic cochlear sections. Wild-type IHC grain counts were not different from background (∼50 grains), while there were 212 ± 14 grains over wild-type OHCs after subtracting background. In the Pou4f3-α10 transgenic cochlear sections grain counts over IHCs were 308 ± 60, not significantly different (p = 0.59, unpaired two-tailed t test) from those over OHCs, 259 ± 55 (n = 6) after subtracting background (∼50). Thus, there was consistently and significantly enhanced expression of α10 mRNA in adult Pou4f3-α10 transgenic IHCs. TM tectorial membrane, DCs Deiter’s cells. A representative in situ from the basal turn of the cochlea is shown.
Backcross of the Pou4f3-α10 to a α10 nAChR knockout background restores ACh responses and synaptic currents before the onset of hearing
In order to learn whether the transgenic α10 RNA is effectively translated into a functional protein, Pou4f3-α10 transgenic mice were backcrossed into a Chrna10 null background. It has been reported that Chrna10 −/− mice lack responses to ACh and the synaptic cholinergic currents observed in normal neonatal mice before the onset of hearing (Vetter et al. 2007). Therefore, we reasoned that responses to ACh in Pou4f3-α10 × Chrna10 −/− would only be observed if the α10 transgene was translated into a α10 protein subunit that restored normal function when co-assembled with the endogenous α9 subunit.
As previously reported (Vetter et al. 2007) and a representative trace shown in Figure 3A, IHCs isolated from Chrna10 +/+ mice at P8-9, a developmental stage when IHCs are transiently innervated by OC terminals (Glowatzki and Fuchs 2000; Katz et al. 2004; Simmons 2002), robustly respond to ACh, exhibiting inward currents at −90 mV (250 ± 57 pA, n = 3). In contrast, no response to 1 mM ACh (n = 0/7 cells tested) is detected in IHCs from Chrna10 −/− mice (Fig. 3A, middle panel). As shown in Figure 3A (right panel), the backcrossing of the Pou4f3-α10 transgenic mice into the α10 null background restored the normal inward responses to 1 mM ACh (V hold −90 mV) at P8-9 in all IHCs tested. The shape and magnitude of the response observed in Pou4f3-α10 × Chrna10 −/− (298 ± 24 pA, n = 3 cells, 3 mice) did not differ from that observed in wild-type mice.

Functional responses in a Pou4f3-α10 × α10 nAChR knockout backcross. A Representative responses to 1 mM ACh of IHCs from P8-9 mice, in Chrna10 +/+ (n = 3 cells, 3 mice), Chrna10 −/− (n = 7 cells, 3 mice), and Chrna10 −/− × Pou4f3-α10 mice (n = 3 cells, 3 mice), V hold = −90 mV. Note the lack of responses in α10 knockout mice and restoration of responses when Chrna10 −/− are crossed with the Pou4f3-α10 transgenic mice. B Same as in A, but at V hold = −40 mV. C Superfusion of the cells with high potassium (40 mM at V hold of −90 mV) causes a change in the holding current due to the change in the K+ equilibrium potential in the three genotypes. In the case of Chrna10 +/+ (n = 3 cells, 3 mice), synaptic currents appear on top of the holding current because of the release of ACh from depolarized efferent terminals. Moreover, 1 mM ACh produces an inward current on top of the change in the holding current. No synaptic activity and responses to ACh can be observed in IHCs from Chrna10 −/− (n = 8 cells, 3 mice), and this is restored in the Chrna10 −/− × Pou4f3-α10 background (n = 3 cells, 3 mice). D Synaptic currents enclosed by the boxes shown in C, plotted at an extended timescale.
IHCs from Chrna10 +/+ mice also exhibit outward currents at −40 mV (281 ± 47 pA, n = 3; Fig. 3B), indicating functional coupling to SK2 channels (Katz et al. 2004; Vetter et al. 2007), whereas no response is found at −40 mV in IHCs from Chrna10 −/− mice (n = 0/8 cells tested; Fig. 3B, middle panel). As shown in Figure 3B (right panel), the backcrossing of the Pou4f3-α10 transgenic mice into the α10 null background restored the normal outward responses to 1 mM ACh (325 ± 48 pA, n = 3 cells, 3 mice) at P8-9 in all IHCs tested.
Finally, when the preparation is superfused with a buffer containing 40 mM K+ to depolarize the efferent terminals, thus increasing the frequency of ACh release (Glowatzki and Fuchs 2000; Katz et al. 2004), synaptic currents are observed in IHCs of Chrna10 +/+ mice (n = 3/3 cells tested; Fig. 3C, left panel), but not in IHCs from Chrna10 −/− mice (n = 0/8 cells tested; Fig. 3C, middle panel; Vetter et al. 2007). Even when adding 1 mM ACh in the presence of 40 mM K+, a procedure that enhances small responses to ACh (due to the change in the K+ equilibrium potential and the concomitant increase in the driving force for K+ ions at the holding voltage of −90 mV; Katz et al. 2004), no responsive IHCs are observed in Chrna10 −/− mice (Fig. 3C, middle panel). When the preparation was superfused with a buffer containing 40 mM K+ to depolarize the efferent terminals, synaptic currents were observed in all Pou4f3-α10 × Chrna10 −/− IHCs (Fig. 3C, right panel), and the addition of 1 mM ACh in the presence of 40 mM K+ produced an inward current similar to that observed in IHCs of Chrna10 +/+ mice. Figure 3D shows boxes in C at an expanded timescale.
Thus, the experiments described so far demonstrate that the Pou4f3-α10 transgene is indeed transcribed into RNA and then translated into a functional α10 protein that can assemble with the endogenous α9 subunit, leading to normal ACh responses and synaptic currents.
IHCs of Pou4f3-α10 mice fail to respond to ACh and to elicit synaptic currents after the onset of hearing
Since IHCs continue to express the α9 mRNA after the onset of hearing (Elgoyhen et al. 1994) and the Pou4f3-α10 transgenic mice shown here constitutively express α10 mRNA even after the onset of hearing, one would expect to find functional α9α10 receptors after this critical developmental period if the lack of responses of IHCs to ACh is solely due to the cessation in the expression of Chrna10.
As shown in Figure 4A, B (upper panels), P9-10 IHCs of Pou4f3-α10 transgenic mice responded to 100 μM ACh with outward currents at −40 mV and inward currents at −90 mV, with overall shape and magnitude (411 ± 67 pA, n = 9 cells, 5 mice and 367 ± 43 pA, n = 9 cells, 5 mice, respectively) which did not significantly differ (p > 0.05) to that observed for wild-type mice (366 ± 63 and 272 ± 43 for −40 and −90 mV, respectively, n = 10 cells, 6 mice, not shown). Moreover, IHCs of P9-10 Pou4f3-α10 transgenic mice exhibited synaptic currents when exposed to 40 mM K+ saline to depolarize the efferent terminals (Fig. 4C, D, upper panel), and the addition of 1 mM ACh in the presence of 40 mM K+ produced an inward current at −90 mV (Fig. 4C, upper panel), as has been previously described for wild-type mice (Vetter et al. 2007).

Lack of ACh responses in IHCs of Pou4f3-α10 transgenic mice after the onset of hearing. A, B Responses to ACh of IHCs from Pou4f3-α10 transgenic mice at a V hold of −40 and −90 mV, respectively. Note normal responses before the onset of hearing (upper panels, n = 9 cells, 5 mice) and lack of responses after the onset of hearing (lower panels, n = 9 cells, 4 mice). C Superfusion of the cells with high potassium (40 mM at V hold of −90 mV) causes a change in the holding current due to the change in the K+ equilibrium potential. Before the onset of hearing (upper panels, n = 9 cells, 5 mice), synaptic currents appear on top of the holding current because of the release of ACh from depolarized efferent terminals, and 1 mM ACh produces an inward current on top of the shift in the holding current. No synaptic activity and responses to 1 mM ACh can be observed in IHCs of Pou4f3-α10 transgenic mice after the onset of hearing (lower panels, n = 9 cells, 4 mice). D Synaptic currents enclosed by the boxes shown in C, plotted at an expanded timescale.
However, as observed in Figure 4 (lower panels), the constitutive expression of the α10 subunit was not sufficient to maintain functional α9α10 receptors after the onset of hearing. Thus, at P17-20, IHCs from Pou4f3-α10 mice failed to respond to 1 mM ACh both at −40 and −90 mV (Fig. 4A, B, n = 9 cells, 4 mice), failed to exhibit synaptic currents at 40 mM K+ (Fig. 4C, D), and failed to respond to 1 mM ACh under conditions in which the driving force for K+ was increased (Fig. 4C).
IHCs of Pou4f3-α10 mice lack functional SK currents after the onset of hearing
The inhibitory nature of the OC synapse in immature IHCs is due to the activation of an SK2 channel after Ca2+ influx through the α9α10-containing nAChRs (Glowatzki and Fuchs 2000; Gomez-Casati et al. 2005; Katz et al. 2004). After the onset of hearing, both Chrna10 and SK2 gene expression is down-regulated, and this is correlated with the disappearance of ACh-evoked responses (Katz et al. 2004). Thus, it appears that the regulation of the transcription of both the Chrna10 and SK2 genes is tightly orchestrated. We therefore evaluated the presence of functional SK2 channels in Pou4f3-α10 transgenic mice by promoting Ca2+ influx through voltage-dependent Ca2+ channels. When immature P9 IHCs from wild-type mice were depolarized for 4 s from a holding potential of −84 mV (Fig. 5A) using 1 mM EGTA as the intracellular Ca2+ buffer and 1.3 mM extracellular Ca2+, a slowly activating outward current was evident (Fig. 5B, left panel, black traces). Extracellular application of 300 nM apamin, a selective SK2 channel blocker (Kohler et al. 1996), abolished this slowly activating outward K+ current (gray traces in Fig. 5B and traces shown in Fig. 5C, left panels: here, records in the presence of apamin were subtracted from control records, leaving only the apamin sensitive current; n = 9 cells, 9 animals), suggesting that this current was due to the activation of SK2 channels as a result of Ca2+ influx through voltage-gated calcium channels. After the onset of hearing, this current is no longer evident (Fig. 5B, C, middle panels, n = 5 cells, 5 animals). As observed in wild-type mice, IHCs of Pou4f3-α10 transgenic mice also did not exhibit Ca2+-activated SK2 currents after the onset of hearing (n = 4 cells, 4 mice; Fig. 5B, C, right panels). Panels D and E show the magnitude of the apamin-sensitive currents +16 mV and the amplitude of the apamin-sensitive tail currents measured at 2 s during the depolarizing step (a, in Fig. 5B) and 0.5 s after it (b, in Fig. 5B), respectively. Whereas at P9 the magnitude of the SK component was significantly different from zero (312 ± 72 and 72 ± 21 at a and b, respectively, n = 9 cells, 9 animals, p ≤ 0.01, Student’s t test), at P18, those values did not differ from zero (p ≥ 0.3) both for wild-type (n = 5 cells, 5 animals) and Pou4f3-α10 mice (n = 4 cells, 4 mice).

Lack of functional SK currents in Pou4f3-α10 transgenic mice after the onset of hearing. A Protocol used to measure the slowly activating outward currents in IHCs. Currents were elicited by 4-s depolarizing voltage steps in 20-mV increments from −84 to 16 mV starting from a holding potential of −84 mV. B Representative outward currents recorded in IHCs in the absence (black traces) or presence (gray traces) of 300 nM apamin in the extracellular solution. Left panel, wild-type responses at P9 (n = 9 cells, 9 mice); middle panel, wild-type responses at P18 (n = 5 cells, 5 animals); right panel, responses of Pou4f3-α10 transgenic mice at P18 (n = 4 cells, 4 animals). C Subtraction of the currents recorded in the presence of apamin from the total currents reveals the presence of SK2 currents in wild-type mice at P9 and the lack of SK currents at P18 both in wild-type and Pou4f3-α10 transgenic mice. D, E Magnitude of the apamin-sensitive currents at +16 mV and the amplitude of the tail currents measured at 2 s during the depolarizing step (a in B) and 0.5 s after it (b in B), respectively.
Discussion
Responses of IHCs to ACh prior to the onset of hearing is strictly dependent upon the expression of both the α9 and the α10 nAChR subunits, as demonstrated by the generation of subunit specific null mutant mice (Vetter et al. 2007). Since the expression of the α10, but not that of the α9 subunit, is developmentally regulated (Elgoyhen et al. 2001; Katz et al. 2004), the present work tested the hypothesis that the lack of cholinergic currents in IHCs after the onset of hearing might result from the cessation of the transcription of the Chrna10 gene. To that end, we generated the Pou4f3-α10 line of transgenic mice, which drove expression of the α10 nAChR subunit after the onset of hearing. Nevertheless, this was not sufficient for the formation of functional α9α10 channels, leading to either ACh responses or efferent synaptic currents after P12.
The absence of functional responses after the onset of hearing in the transgenic mice could have alternative explanations. Lack of transcription of the transgene or misexpression of the transgenic RNA can result from the generation of transgenic mice (Haruyama et al. 2009; Su et al. 2004). This is precluded in the case of the Pou4f3-α10 line, since transgenic RNA was present in the cochlea as assessed by RT-PCR and it was localized to the IHC region as demonstrated by in situ hybridization. Alternatively, the α10 cDNA plasmid used to engineer the transgene construct could have led to a non-functional α10 protein subunit, particularly since a FLAG tag was added before the stop codon of the cDNA. However, this was not the case since the Pou4f3-α10 transgene rescued the α10 null phenotype, demonstrated by the presence of normal responses to ACh and synaptic currents in the Pou4f3-α10 × Chrma10−/− backcross, thus indicating that in vivo, the transgenic α10 subunit efficiently assembles with the endogenous α9 to form functional channels. Importantly, taken together, these results indicate that lack of cholinergic responses of IHCs after the onset of hearing goes beyond the transcription of the Chrna10 gene.
The possibility exists that after the onset of hearing, genes other than Chrna10 also cease transcription and/or translation and that these genes lead to proteins which form part of a macromolecular synaptic complex that includes, but extends beyond the nAChR and which is necessary for assembly, trafficking and/or anchorage of the nAChR to the plasma membrane at the base of the IHC. For example, RIC-3, a transmembrane protein which acts as a molecular chaperone, is required for efficient receptor folding, assembly, and functional expression of the α7 nAChR (Millar 2008). Similar chaperon proteins have not been described in the case of α9α10 receptors. However, it is known that activation of the α9α10 nAChR leads to an increase in intracellular Ca2+ and the subsequent opening of small conductance Ca2+-activated K+ SK channels, thus leading to hyperpolarization of hair cells (Dulon et al. 1998; Fuchs and Murrow 1992; Housley and Ashmore 1991; Oliver et al. 2000). Moreover, SK channels and α9α10 are known to co-localize in the same functional microdomain, and through such close coupling, the gating kinetics of the SK channels determine the time course of synaptic action, outlasting the driving calcium signals (Oliver et al. 2000). In addition, through the generation of a KCNN2 (gene coding for the SK2 protein) knockout mice, it has been demonstrated recently that the KCNN2 gene is solely responsible for encoding this class of small conductance, calcium-activated potassium channel in cochlear hair cells (Johnson et al. 2007; Kong et al. 2008) and that it cannot be replaced by the later developmental arrival of rapidly activating, iberiotoxin-sensitive “BK”-type potassium channels shown in mammals (Hafidi et al. 2005; Kros et al. 1998; Langer et al. 2003) and birds (Fuchs and Sokolowski 1990). The present results demonstrate that as observed in wild-type mice, the Pou4f3-α10 transgenic also lack functional SK2 currents after the onset of hearing. This observation, together with the fact that KCNN2 knockout mice totally lack ACh responses in hair cells (Johnson et al. 2007; Kong et al. 2008), might indicate the SK2 protein as fundamentally required for the assembly, trafficking, and/or anchorage of the nAChR macromolecular synaptic complex. Alternatively, proteins known to form a macromolecular complex with SK2 channels, such as calmodulin, protein kinase CK2, and protein phosphatase 2A (Bildl et al. 2004), might also be developmentally regulated and be the linking molecules of the SK2 channel with the nAChR macromolecular complex.
Although the lack of ACh responses in the KCNN2 knockout mice points towards the SK2 protein as a key player, the fact that in these mice a concomitant OC fiber degeneration is also observed (Kong et al. 2008; Murthy et al. 2009) does not allow an unequivocal conclusion. Thus, lack of pre- and postsynaptic cross talk in the absence of innervation, rather than lack of the SK2 protein as a stabilizing component of the macromolecular synaptic complex, could also lead to the same result. The need of presynaptic neuronal input for the correct assembly of the postsynaptic apparatus has been described at the neuromuscular junction where motor nerve terminals seem to organize postsynaptic differentiation by releasing a proteoglycan called agrin. Agrin activates a receptor tyrosine kinase called muscle-specific kinase on the myotube surface, which leads to clustering of nAChRs and other postsynaptic components through association with the cytoplasmic linker protein rapsyn (Sanes and Lichtman 2001).
Finally, we cannot preclude the possibility of a developmental regulation of the translation of the α10 mRNA, which might prevent α10 protein synthesis after the onset of hearing. Emerging studies show that translational control in eukaryotic cells is critical for gene regulation during nutrient deprivation and stress, development and differentiation, nervous system function, aging, and disease (Sonenberg and Hinnebusch 2009). For example, microRNAs are major regulators of gene expression and function at the posttranscriptional level (Carthew and Sontheimer 2009). Further work is required in order to dissect the alternative possibilities proposed as the underlying mechanisms for the developmental regulation of the cholinergic responses of IHCs.
References
Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B. Protein kinase CK2 is coassembled with small conductance Ca(2+)-activated K+ channels and regulates channel gating. Neuron 43:847–858, 2004.
Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655, 2009.
Dulon D, Luo L, Zhang C, Ryan AF. Expression of small-conductance calcium-activated potassium channels (SK) in outer hair cells of the rat cochlea. Eur. J. Neurosci 10:907–915, 1998.
Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. α9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79:705–715, 1994.
Elgoyhen AB, Vetter D, Katz E, Rothlin C, Heinemann S, Boulter J. Alpha 10: A determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc. Natl. Acad. Sci. U. S. A 98:3501–3506, 2001.
Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O’Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature 381:603–606, 1996.
Fuchs PA, Sokolowski BH. The acquisition during development of Ca-activated potassium currents by cochlear hair cells of the chick. Proc. Biol. Sci 241:122–126, 1990.
Fuchs PA, Murrow BW. Cholinergic inhibition of short (outer) hair cells of the chick’s cochlea. J. Neurosci 12:800–809, 1992.
Glowatzki E, Fuchs P. Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science 288:2366–2368, 2000.
Gomez-Casati ME, Fuchs PA, Elgoyhen AB, Katz E. Biophysical and pharmacological characterization of nicotinic cholinergic receptors in cochlear inner hair cells. J. Physiol 566:103–118, 2005.
Guinan JJ. Physiology of olivocochlear efferents. In: Dallos P, Popper A, Fay R (eds) The Cochlea. New York, Springer, pp. 435–502, 1996.
Hafidi A, Beurg M, Dulon D. Localization and developmental expression of BK channels in mammalian cochlear hair cells. Neuroscience 130:475–484, 2005.
Haruyama N, Cho A, Kulkarni AB. Overview: Engineering transgenic constructs and mice. Current Protocols in Cell Biology, Chapter 19, Unit 19.10, 2009.
Hiel H, Elgoyhen A, Drescher D, Morley B. Expression of nicotinic acetylcholine receptor mRNA in the adult rat peripheral vestibular system. Brain. Res 738:347–352, 1996.
Housley GD, Ashmore JF. Direct measurement of the action of acetylcholine on isolated outer hair cells of the guinea pig cochlea. Proc. R. Soc. Lond. B 244:161–167, 1991.
Johnson SL, Adelman JP, Marcotti W. Genetic deletion of SK2 channels in mouse inner hair cells prevents the developmental linearization in the Ca2+ dependence of exocytosis. J. Physiol 583:631–646, 2007.
Katz E, Elgoyhen AB, Gomez-Casati ME, Knipper M, Vetter DE, Fuchs PA, Glowatzki E. Developmental regulation of nicotinic synapses on cochlear inner hair cells. J. Neurosci 24:7814–7820, 2004.
Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science 273:1709–1714, 1996.
Kong JH, Adelman JP, Fuchs PA. Expression of the SK2 calcium-activated potassium channel is required for cholinergic function in mouse cochlear hair cells. J. Physiol 586:5471–5485, 2008.
Kros CJ, Ruppersberg JP, Rusch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature 394:281–284, 1998.
Langer P, Grunder S, Rusch A. Expression of Ca2+-activated BK channel mRNA and its splice variants in the rat cochlea. J. Comp. Neurol 455:198–209, 2003.
Lustig LR, Peng H, Hiel H, Yamamoto T, Fuchs P. Molecular cloning and mapping of the human nicotinic acetylcholine receptor α10 (CHRNA10). Genomics 73:272–283, 2001.
Marcotti W, Johnson SL, Holley MC, Kros CJ. Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J. Physiol 548:383–400, 2003a.
Marcotti W, Johnson SL, Rusch A, Kros CJ. Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J. Physiol 552:743–761, 2003b.
Millar NS. RIC-3: A nicotinic acetylcholine receptor chaperone. Br. J. Pharmacol 153(Suppl 1):S177–S183, 2008.
Morley BJ, Simmons DD. Developmental mRNA expression of the alpha10 nicotinic acetylcholine receptor subunit in the rat cochlea. Brain Res. Dev. Brain Res 139:87–96, 2002.
Murthy V, Maison SF, Taranda J, Haque N, Bond CT, Elgoyhen AB, Adelman JP, Liberman MC, Vetter DE. SK2 channels are required for function and long-term survival of efferent synapses on mammalian outer hair cells. Mol. Cell. Neurosci 40:39–49, 2009.
Oliver D, Klocker N, Schuck J, Baukrowitz T, Ruppersberg JP, Fakler B. Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron 26:595–601, 2000.
Plazas PV, Katz E, Gomez-Casati ME, Bouzat C, Elgoyhen AB. Stoichiometry of the α9α10 nicotinic cholinergic receptor. J Neurosci 25:10905–10912, 2005.
Sage C, Huang M, Vollrath MA, Brown MC, Hinds PW, Corey DP, Vetter DE, Chen ZY. Essential role of retinoblastoma protein in mammalian hair cell development and hearing. Proc. Natl. Acad. Sci. U. S. A 103:7345–7350, 2006.
Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci 2:791–805, 2001.
Sgard F, Charpentier E, Bertrand S, Walker N, Caput D, Graham D, Bertrand D, Besnard F. A novel human nicotinic receptor subunit, α10, that confers functionality to the α9-subunit. Mol. Pharmacol 61:150–159, 2002.
Simmons DD. Development of the inner ear efferent system across vertebrate species. J. Neurobiol 53:228–250, 2002.
Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 136:731–745, 2009.
Su M, Hu H, Lee Y, d’Azzo A, Messing A, Brenner M. Expression specificity of GFAP transgenes. Neurochem. Res 29:2075–2093, 2004.
Vetter DE, Katz E, Maison SF, Taranda J, Turcan S, Ballestero J, Liberman MC, Elgoyhen AB, Boulter J. The alpha10 nicotinic acetylcholine receptor subunit is required for normal synaptic function and integrity of the olivocochlear system. Proc. Natl. Acad. Sci. U. S. A 104:20594–20599, 2007.
Walsh E, McGee J, McFadden S, Liberman M. Long-term effects of sectioning the olivocochlear bundle in neonatal cats. J. Neurosci 18:3859–3869, 1998.
Weisstaub N, Vetter D, Elgoyhen A, Katz E. The alpha9/alpha10 nicotinic acetylcholine receptor is permeable to and is modulated by divalent cations. Hearing. Res 167:122–135, 2002.
Young JI, Otero V, Cerdan MG, Falzone TL, Chan EC, Low MJ, Rubinstein M. Authentic cell-specific and developmentally regulated expression of pro-opiomelanocortin genomic fragments in hypothalamic and hindbrain neurons of transgenic mice. J. Neurosci 18:6631–6640, 1998.
Acknowledgments
The authors want to thank the laboratory of Marcelo Rubinstein at INGEBI for generating the transgenic mouse line. This work was supported by the National Institutes of Deafness and other Communication Disorders (NIDCD) grant R01DC001508 to P.A.F. and A.B.E, an International Research Scholar Grant from the Howard Hughes Medical Institute, The National Organization for Hearing Research, a Research Grant from ANPCyT (Argentina), and a Grant from the University of Buenos Aires (Argentina) to A.B.E., NIDCD R01DC006258 to D.E.V, and a CONICET grant to EK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Julián Taranda and Jimena A. Ballestero equally contributed.
Rights and permissions
About this article
Cite this article
Taranda, J., Ballestero, J.A., Hiel, H. et al. Constitutive Expression of the α10 Nicotinic Acetylcholine Receptor Subunit Fails to Maintain Cholinergic Responses in Inner Hair Cells After the Onset of Hearing. JARO 10, 397–406 (2009). https://doi.org/10.1007/s10162-009-0173-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-009-0173-z