
Abstract
Sepsis is a major cause of acute kidney injury (AKI) among patients in the intensive care unit. However, the numbers of basic science papers for septic AKI account for only 1% of all publications on AKI. This may be partially attributable to the specific pathophysiology of septic AKI as compared to that of the other types of AKI because it shows only modest histological changes despite functional decline and often requires real-time functional analysis. To increase the scope of research in this field, this article reviews the basic research information that has been reported thus far on the subject of septic AKI, mainly from the viewpoint of functional dysregulation, including some knowledge acquired with multiphoton intravital imaging. Moreover, the efficacy and limitation of the potential novel therapies are discussed. Finally, the author proposes several points that should be considered when designing the study, such as monitoring the long-term effects of the intervention and reflecting the clinical settings for identifying the molecular mechanisms and for challenging the intervention effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute kidney injury (AKI) is an important challenge that negatively impacts patient survival in an intensive care unit (ICU). Although there was a 9% decline in the in-hospital mortality rate among patients treated with renal replacement therapy in the ICU from 2007 (44.9%) to 2016 (36.1%) in Japan, the mortality rate of sepsis patients remains high at > 50% [1]. Moreover, translational research for AKI has not yet been successful, and no specific treatment has been established in the clinical setting. The search result for the keywords “acute, kidney, injury, and rats” in PubMed showed over 6000 scientific articles, and “mice” instead of “rats” provides an additional above 3800 papers (June 2020). Sepsis is a major cause of AKI; however, when the term “sepsis” was added to either search, it decreased the results to < 270 (< 5%) and < 400 (< 11%), respectively. These numbers (rats + mice) account for about only 1% of all AKI publications. Septic AKI can lead to mortality in patients in the intensive care; therefore, this topic is relevant and should focus on more basic research. Septic AKI involves an infection that critically influences the development of AKI as compared to the other types of AKI. Thus, this review aimed to distinguish septic AKI from other types of AKI, discuss the basic scientific outcomes based on functional changes (Fig. 1), and introduce potential novel treatments.

Causes of septic oliguria/anuria. Fluid resuscitation could resolve dehydration and glomerular filtration rate (GFR) reduction, but not tubular leakage (depicted by small yellow circles). Rather, continuous fluids under tubular leakage accelerate a positive balance. GFR reduction and tubular occlusion could deteriorate each other
Reduction in the glomerular filtration rate (GFR)
Glomerular hemodynamics
Changes in the GFR, from the viewpoint of hemodynamic change, are currently investigated on relatively big animals, such as dogs and sheep, because it is easier to create ICU-like experimental settings for these animals as compared to that for smaller animals, such as rodents, and to perform continuous inulin/inulin-like compound-used clearance analyses. The former may be essential for the replication of a hyperdynamic state-like human septic AKI [2]. Changes in the cardiac output were accompanied by changes in the renal blood flow in septic animals [3]. Thus, animals in the hyperdynamic states typically show increased renal blood flow despite reduced renal function in septic AKI. One hypothesis for decline in GFR under conditions of increased renal blood flow is the gain in the ratio of the efferent-to-afferent arteriole diameter under efferent vasodilation that increases plasma flow to the peritubular capillaries. The vasoconstrictors that showed higher affinity for the efferent arterioles, such as angiotensin II, might prevent AKI in the animals; thus, this is an area of focus in human AKI research [4, 5]. It is noteworthy that angiotensin II infusions initiated at 2 h after Escherichia coli infusions ameliorated both oliguria and serum creatinine levels, despite reduced renal blood flow in sheep [6]. Studies on sheep or pigs also reported that angiotensin II did not worsen the ATP level [7], medullary hypoxia [8], and mitochondrial respiration [9] during sepsis, suggesting that the vasoconstrictive effects of angiotensin II on the efferent arteriole might not induce reductions in blood supply to the peritubular capillaries. In a rodent sepsis model, lipopolysaccharides (LPS), an endotoxin, introduced at a dosage designed to induce AKI, constricted the afferent arteriole and decreased the ratio of the efferent-to-afferent arteriole diameter [10], thereby reducing the GFR.
Changes in glomerular cells
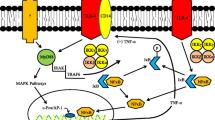
In addition to the afferent/efferent arteriole-dependent intraglomerular pressure changes, Xu et al. [11] reported that endotoxemia caused ultrastructural alterations in the glomerular endothelium. The endothelial surface layer (ESL), also called the glycocalyx, is a barrier composed of negatively charged proteoglycans and glycoproteins. The ESL covers the endothelial fenestrae and limits the proteins from permeating the fenestrae; however, breakdown of this layer promotes permeability and allows access to even relatively large proteins, such as albumin [12]. Xu et al. [11] reported that endotoxemia produced TNF-α that degraded the ESL in the glomerular endothelium that was consistent with the observations reported in the vasculature of other organs [13]. Furthermore, LPS/TNF-α induced glomerular endothelial swelling and fenestrae density reduction at 24 h. Although an increase in albumin filtration increases colloidal osmotic pressure elevation in the Bowman’s capsule, the “benefit” of the GFR might be imperceptible owing to the fenestrae alteration that occurs during endotoxemia. The physiological turnover rate of the ESL components was approximately 5 days [14]; thus, the alterations in the glomerular endothelium were expected to be reversible after the end of the cytokine storm.
Prerenal cause
The prerenal causes of GFR reduction were often resolved by fluid and vasopressor treatment although this might not be the case if sepsis increased the systemic vascular permeability. The optimization of fluid resuscitation for sepsis or septic AKI in clinical studies has been debated in multiple review articles [15,16,17,18,19]. However, few studies have compared the efficacy of fluid use in septic AKI in basic studies [20,21,22,23,24]. Most interventional studies against septic AKI, especially those performed on rodents did not reflect a clinical setting because a small animal ICU setting is difficult to create/maintain.
Reduction of tubular flow rate
AKI could occur with oliguria and without a rise in the serum creatinine level. The unresponsiveness of serum creatinine could result from the dilution of creatinine by fluids [25, 26] or via reduced production of creatinine during sepsis [27]. However, the underlying mechanism of development of oliguria in spite of the maintenance of renal circulation with fluid resuscitation and additional vasoactive agents was unknown until recently. We addressed this question using intravital imaging combined with multiphoton microscopy to find that that the filtrate flow rate had reduced in the proximal tubules, while the GFR was maintained at a normal level (Figs. 2 and 3) because of the paracellular leakage of the tubular fluid into the interstitium in rats and mice in the LPS model. The LPS bound to TLR4 in the proximal tubules and subsequent signaling disrupted the tight junctions among the proximal tubular cells, resulting in paracellular leakage [28, 29]. The following experiments revealed that leakage at the proximal tubules affected recovery from oliguria when renal circulation was maintained with fluid resuscitation [29]. The GFR was restored through fluid resuscitation; however, tubular flow did not reach the bladder because of leakage at the proximal tubules (Fig. 1). Mechanistically, LPS/TLR4 stimulated p38 MAP kinase (transiently; < 6 h) and NF-κB (relatively continuously; until 24 h), disrupting the localization of ZO-1 and claudin 2 and reducing occludin expression [29]. Intravenously injected LPS conjugated with fluorophore first (− 20 min) was bound to S1 segments of the proximal tubules [30, 31]; thereafter, it was distributed to both the S1 and S2 segments overtime (1 h) [31]. In a later phase (6, 24, or 48 h), the distribution of the LPS showed heterogeneity; some proximal tubules accumulated LPS, and the others exhibited mild accumulation. However, the cause of heterogeneity remains unclear. These tubules with considerable accumulation of LPS might be downstream S2 segment tubules; however, until now, there was no definitive examination for this speculation. Some tubules highly accumulated LPS, while others showed a reduction in the tubular flow rate or cell swelling [31], resulting in tubular occlusion and halted tubular flow. Continuous intravenous fluid infusion occasionally reopened the occluded tubular lumen that was accompanied by the delivery of tubular fluid to the lumen of the distal nephron, indicating recovery of the tubular flow [31]. Hato et al. [32] reported with elegant fluorescence lifetime microscopy that LPS changes the nucleotide metabolism in the S2 segments of the proximal tubules with increased reactive oxygen species production that was possibly induced by LPS binding to the S1 segment. It is noteworthy that fluorescence lifetime image analysis showed that LPS decreased the metabolic heterogeneity between S1 and S2, albeit with heterogeneity in the tubular flow rate.

Time-lapse imaging after an intravenous bolus injection of Lucifer yellow, a fluorophore that is freely filtered from the glomeruli. Lucifer yellow was injected into a mouse that had received saline or LPS at either 6 or 24 h earlier. Green fluorescence at 0 s in the image is autofluorescence derived from proximal tubules. Distal nephron (indicated by yellow arrows) does not produce detectable green autofluorescence. a Several proximal tubules showed Lucifer yellow in the lumen within 20 s after injection. Lucifer yellow flowed into some distal nephrons at 30 s. At 180 s, Lucifer yellow flowed out from some distal nephrons. b Atto 565-conjugated LPS (red) was injected in the mouse used in these images. Atto565-LPS accumulated in a part of the proximal tubules, presumably at the S2 segment. Several proximal tubules showed Lucifer yellow in the lumen within 20 s after injection in the mice 6 h after LPS injection. The inflow rates of Lucifer yellow into these proximal tubules were similar to that in healthy control mice, indicating preserved GFR in these nephrons. At 50 s after injection, Lucifer yellow-derived fluorescence was detected in the proximal tubules that took up LPS (red), but not in distal nephron in the imaging window. Lucifer yellow was detected in the distal tubular lumen at 180 s after the injection. c Atto 565-conjugated albumin (red) was injected in the mouse used in these images. The Atto 565-albumin was detected in the peritubular capillaries and glomerulus (white asterisk). At 33 s after following the Lucifer yellow injection 24 h after LPS injection, Lucifer yellow was detected in the Bowman’s space of glomerulus, some proximal tubular lumens, and peritubular capillaries. The slower appearance of Lucifer yellow and more flow into the peritubular capillary, compared to higher images, suggest poor hemodynamics, including GFR reduction. Male C57/BL6J mice were purchased from CLEA (Tokyo, Japan) and housed in our colony until they were of proper age (> 55 week-old) for the induction of AKI consistently. The detailed intravital imaging setting, including microscopy setting, has been described previously [29, 31, 107]. PT proximal tubules, DCT/CD distal convoluted tubules, and cortical collecting duct

Representative multiphoton unlabeled images of the murine kidney (720-nm excitation laser wavelength) in each stage of septic AKI in the mice aged > 55 week. The left image is from a healthy control mouse. The lower images were enlarged images of the ensquared part in each upper image. The tubular lumen is visible as a dark lumen in each “blue” tubule. The blue color was derived from the autofluorescence of the tubules. There are some bright spots containing strong green fluorescence in the image in the middle; these could be derived from the non-degradable metabolites in the aged tubules (the mouse was 57-week-old). The middle is from a mouse that received LPS (5 mg/kg) 6 h before image acquisition. The tubular lumen is visible, while the tubular flow rate is slowed (see images in Fig. 2b). The single nephron GFR in the mouse was at similar levels as those in LPS-untreated normal mice that were evaluated based on how quickly the Lucifer yellow dye flowed into the first segments of the proximal tubules. The right image is from a mouse that received LPS (5 mg/kg) 24 h before image acquisition. The mouse showed reduced GFR, and the tubular lumen was almost occluded in the image. Male C57/BL6J mice were purchased from CLEA (Tokyo, Japan) and housed in our colony until they were of proper age (> 55 week-old). The detailed intravital imaging setting is described in our previous publication [29, 31, 107]
Microcirculation failure
The renal capillary network was often destroyed during non-septic AKI [33], and angiogenesis was indispensable for efficient tubular recovery [34, 35]. In septic AKI, microcirculation failure, rather than histological capillary destruction, caused changes in the oxygen supply to the tubules. Rodent septic AKI models elicited heterogeneity of the red blood cells (RBCs) in each capillary, irrespective of the total renal perfusion or GFR [22, 31, 36]. The microcirculation failure in each capillary could be intermittent or could last for > 10 s [36]. There appeared to be several causes for RBC flow cessation, such as clotting, leukocyte attachment [31], and neutrophil extracellular entrapment (Fig. 4). The capillary occlusion or cessation of the RBC flow-induced reactive nitrogen species production and mitochondrial membrane potential reduction in the adjacent tubules of the endotoxemic rodents [31, 36]. Intravital imaging of the multiphoton laser microscopy carefully analyzed the co-localization between the microcirculation failure of the peritubular capillaries and LPS-accumulated “leaking” in the proximal tubules; however, the occurrence in the capillary network was random [31].

A 3D image of the neutrophil extracellular entrapped in the live kidney of a mouse that received LPS 10 min before capturing. Green represents released/dead cell DNA (Sytox green), and magenta represents the autofluorescence of the tubules. Saturated Sytox green fluorescence changes to yellow in the nuclei. Male C57/BL6J mice were purchased from CLEA (Tokyo, Japan) and housed in our colony until they were of proper age (12 week of age). The image was acquired as previously described [114, 115]
Histopathological changes
As noted above, AKI is currently diagnosed via the GFR or urine output, and it is important to ensure patient survival during the acute phase. The pathological features of AKI differed, depending on the AKI cause. Renal ischemia/reperfusion injury that is widely employed as an AKI model induces dramatic changes in the renal histology. Conditions related to renal ischemia/reperfusion injury include acute tubular necrosis, cell sloughing, cast formation, perivascular inflammation [37], capillary rarefaction [35], tertiary lymphoid tissue formation [38], tubular ferroptosis [39], and intrarenal denervation [40]. However, these pathological features were not always observed in septic AKI, wherein only modest histological changes in the kidneys were observed in human sepsis and animal models of sepsis [41, 42].
Therapeutic candidates against septic AKI
Standard supportive treatments, such as fluids and vasoconstrictors, are used to restore the GFR; however, they do not address the changes in the tubules in the septic AKI cases. In addition, there are concerns surrounding fluid resuscitation [43,44,45,46,47]. Therapies that specifically target the reduction in the GFR/tubular flow rate and tubular injury in AKI do not currently exist in clinical settings, although several basic research papers have reported on their efficacy. The problems regarding this failed translation include physical barriers in the laboratory that are not discussed in this review [48]. Instead, this review evaluates several candidate treatments that target relatively multiple factors.
Ischemic preconditioning
Ischemic preconditioning is a possible strategy against perioperative end-organ damage, including AKI. Repeated short-time hypoxic/ischemic stress to the kidney has caused a tolerance to AKI in multiple experimental models, including sepsis [49,50,51], ischemia/reperfusion [52,53,54], and cisplatin models [55] in rodents. This method did not always work for large animals, such as dogs [56] and pigs [57,58,59]. Remote ischemic preconditioning involves repeated occlusion of the limbs and might be applicable to both, basic and clinical studies. However, the effect was inconsistent; for example, it showed beneficial effects on sepsis in mice [60] and sheep [61], but not on ischemia/reperfusion in mice [62] and pigs [63]. Moreover, the causes of AKI did not appear to affect the results. Remote ischemic preconditioning before LPS injection did not ameliorate decreases in blood pressure, systemic cytokine storms, or the urinary concentration of the tissue inhibitor of metalloproteinases-2*insulin-like growth factor-binding protein-7 in a single-center RCT [64]. Conversely, remote ischemic preconditioning before the induction of anesthesia safely improved the long-term kidney function after living-donor renal transplantation in a double-blind RCT [65].
Endotoxin preconditioning
Endotoxin preconditioning involves the injection of a single low-dose endotoxin, including LPS, to protect the organs from subsequent events. Experiments on pigs have reported that endotoxin preconditioning suppressed the systemic oxygen demand despite unchanged global hemodynamics in response to the Salmonella abortus equi endotoxin [66]. Although the precise mechanism is yet to be clarified, studies on mice have demonstrated that the macrophages were the key cell type required to induce the anti-inflammatory phenotype [67]. Moreover, the protection of the kidneys involved inducing macrophage clustering around the Bowman’s capsule and the S1 segment of the proximal tubules following LPS administration in mice [62]. The beneficial effects of the endotoxin preconditioning largely relied on changes in the macrophage primarily focused on the anti-inflammatory phenotype; therefore, it has been reported that endotoxin preconditioning prevents organ damage associated with inflammation in several organs, such as ischemia-induced neuronal damage [68, 69] and pancreatic damage [70]. However, owing to the modification effects of immune cell function, there are reports of deteriorating disease conditions during sepsis [71] and lupus nephritis [72]. Chen et al. [71] demonstrated that super-low-dose LPS (ng order per kg body weight) exacerbated bacteremia and mortality and reduced neutrophil extracellular trap formation after cecum ligation and puncture in mice. Moreover, low-dose LPS (μg order per kg body weight) improved bacteremia and mortality rate and induced the neutrophil extracellular trap. Currently, no human studies have examined the effects of endotoxin preconditioning on sepsis/AKI because there are multiple concerns and steps that need to be overcome in basic research before clinical application. For example, the LPS dosage used for the human studies was 1–2 ng/kg that was considered a “super-low” priming dosage. The fact that a rodents’ response to LPS was much weaker than that of a human further complicates this issue. Another problem was the influence of risk factors on the protective effects, given that most studies have used young animals, and an older immune response could be different [73]. For instance, a comparison between a 2-month-old and a 12-month-old ICR mice showed that the beneficial effects of endotoxin preconditioning were weaker in the older mice [74].
Cholinergic anti-inflammatory pathway stimulation
Metz et al. developed a concept that has been termed the cholinergic anti-inflammatory pathway [75], mediated in a β2 adrenergic/α7 nicotinic acetylcholine receptor-dependent manner in the spleen [76, 77]. Several strategies have been used to stimulate the cholinergic anti-inflammatory pathway [78,79,80,81,82,83]. Although the precise neuronal network responsible for each stimulation remains debated, there are multiple neuronal networks capable of inducing the cholinergic anti-inflammatory pathway, such as the central C1 neurons [82], the peripheral vagus [81, 83, 84], the sympathetic [85], and the sciatic nerves [86]. Nicotine was administered for reno-protection; it stimulated this pathway and protected the kidneys from ischemia/reperfusion injury in mice [87]; however, this treatment could worsen the survival rate during sepsis owing to the suppression of the host defenses [84, 88,89,90]. Nicotine administration was considered too risky for humans. Several methods were alternatively proposed to stimulate the cholinergic anti-inflammatory pathway. First, electrical vagus nerve stimulation could be performed via a portable device that has already been developed and is awaiting clinical application. The efficacy of vagus nerve stimulation on experimental AKI has been confirmed using the ischemia/reperfusion [81] and cisplatin [91] models. Either afferent or efferent nervous stimulation attenuated AKI via which both nerves were involved in the splenic α7 nicotinic acetylcholine receptor-dependent pathway (although afferent seemed to induce protection partially through an unknown pathway). Second, noninvasive ultrasound exposure reportedly stimulates the peripheral neurons and induces the cholinergic anti-inflammatory pathway. The features of the ultrasonography examination could be used for application to the targeted organ at a variable strength [92]. Splenic ultrasonography modulation suppressed LPS-induced reduction of noradrenaline and acetylcholine and cytokine production expression in the spleen [92] and attenuated AKI in the cecum ligation and puncture [79] and ischemia/reperfusion [83] models. Third, electroacupuncture could be another tool to stimulate this pathway. Electroacupuncture at ST36 acupoint, known as Zusanli, induced anti-inflammatory responses [86] and protected the kidneys in septic models [93, 94]. This protection was attenuated by treatment with reserpine [86, 93], D1 receptor antagonist [86], α7nAChR antagonists [95,96,97,98], vagotomy [95, 99], and adrenalectomy [93], but not splenectomy [86]. Therefore, electroacupuncture appeared to induce this protection via the cholinergic and adrenergic pathways.
Atrial natriuretic peptide
Atrial natriuretic peptide (ANP) is secreted from the atrium following reduced cardiac function. Endogenous ANP secretion is increased during human septic shock [100] and in the ovine hyperdynamic endotoxin model [101]. Its recombinant peptide at a low dosage was used against AKI, based on empirical observations. However, this effect has not been confirmed with strong evidence from multicenter, high-quality, large-sample RCTs [102]. In rodents, ANP suppressed renal ischemia-/reperfusion-induced [103,104,105,106] and LPS model injuries [107]. We employed a suppressor dosage of ANP combined with fluid resuscitation in the LPS model of rats and demonstrated that a 2-h treatment with ANP during the early phase (2–4 h after LPS) improved urine flow, GFR, tubular flow leakage, and survival rate [107]; however, the efficacy considerably lowered with treatment in a relatively later phase (18–20 h after LPS). This study also revealed that, in fluid-administered mice, endogenously secreted ANP stimulated its receptor GCA in the endothelial cells and prevented vascular permeability gain and GFR reduction after LPS; further, exogenously administered ANP stimulated GCA in the proximal tubular cells and prevented LPS-induced tubular leakage. The limitations in basic research involved the use of young rodents and the sole purpose of the LPS model. ANP (the human recombinant type used in LPS study) is already approved and has been used for acute heart failure treatment in Japan for the previous 25 years.
Meditation, exposure to cold, and breathing techniques
Pickkers et al. performed several studies using the endotoxin challenge in humans [108,109,110,111] and demonstrated that a training program, composed of meditation, cold exposure, and special breathing, increased adrenaline/noradrenaline in the plasma and created a tolerance against experimental endotoxemia [110]. These data reported the sympathetic nervous system (plasma adrenaline and noradrenaline) stimulation, increased leukocytes in the plasma, acute respiratory alkalosis (normalized immediately after cessation of the special breathing), relatively high lactate, and decreases in oxygen saturation in the trained individuals following LPS administration. The limitation of this method is that the training protocol is too difficult for subjects with AKI risk factors. From the article [110], the following are the examples of this difficulty: “standing in the snow barefoot for up to 30 min and lying bare-chested in the snow for 20 min; daily dipping/swimming in ice-cold water (0–1 °C) for several minutes (including complete submersions); and hiking up a snowy mountain (elevation: 1590 m) bare-chested, wearing nothing but shorts and shoes at temperatures ranging from − 5 to − 12 °C (wind chill: − 12 to − 27 °C)” [110].
The current limitations of these therapeutic candidates are summarized in Table 1.
Conclusion and message
Septic AKI is associated with considerable hospital-based mortality; once a novel treatment method has been developed, it could save thousands of lives. Recent developments using artificial intelligence have enabled the prediction of “new” antibiotics [112]. However, this might not be the case for AKI because there is lack of knowledge for the formation of a specific drug/chemical structure to train artificial intelligence, necessitating further basic research. For future basic studies to fight against septic AKI, the following three points should be considered in the study design.
-
As much as possible, studies to find the pathogenesis/molecular mechanism should additionally examine the effect of factors existing in the clinical setting. Young rodent tests might show different results than older mice. Use of rodents might benefit the modification of genes (and are less expensive); however, the hemodynamic pattern does not reflect the clinical contribution of the target molecule that might be less or more when in actual clinical use. Septic patients are usually under antibiotic therapy and fluid administration [113], and this might mask the pathogenesis/molecular targets.
-
Intervention studies must confirm either mortality rate or CKD development because these are the objectives of the therapy. Attenuating the injury only at one time point does not guarantee that the treatment could prevent patient mortality.
-
As far as possible, intervention studies should reflect the clinical setting. Anti-immune therapy might suppress host defenses. Thus, even if it works against sterile inflammation, it might worsen bacteremia and increase mortality.
References
Miyamoto Y, Iwagami M, Aso S, Yasunaga H, Matsui H, Fushimi K, et al. Temporal change in characteristics and outcomes of acute kidney injury on renal replacement therapy in intensive care units: analysis of a nationwide administrative database in Japan, 2007–2016. Crit Care. 2019;23(1):172. https://doi.org/10.1186/s13054-019-2468-8.
Hollenberg SM, Dumasius A, Easington C, Colilla SA, Neumann A, Parrillo JE. Characterization of a hyperdynamic murine model of resuscitated sepsis using echocardiography. Am J Respir Crit Care Med. 2001;164(5):891–5. https://doi.org/10.1164/ajrccm.164.5.2010073.
Langenberg C, Bellomo R, May C, Wan L, Egi M, Morgera S. Renal blood flow in sepsis. Crit Care. 2005;9(4):R363–R374374. https://doi.org/10.1186/cc3540.
Khanna A, Ostermann M, Bellomo R. Angiotensin II for the treatment of vasodilatory shock. N Engl J Med. 2017;377(26):2604. https://doi.org/10.1056/NEJMc1714511.
Sacha GL, Lam SW, Bauer SR. Did the beneficial renal outcomes with vasopressin VANISH? Ann Transl Med. 2016;4(Suppl 1):S67. https://doi.org/10.21037/atm.2016.10.59.
Wan L, Langenberg C, Bellomo R, May CN. Angiotensin II in experimental hyperdynamic sepsis. Crit Care. 2009;13(6):R190. https://doi.org/10.1186/cc8185.
May CN, Ishikawa K, Wan L, Williams J, Wellard RM, Pell GS, et al. Renal bioenergetics during early gram-negative mammalian sepsis and angiotensin II infusion. Intensive Care Med. 2012;38(5):886–93. https://doi.org/10.1007/s00134-012-2487-2.
Lankadeva YR, Kosaka J, Evans RG, Bellomo R, May CN. Urinary oxygenation as a surrogate measure of medullary oxygenation during angiotensin II therapy in septic acute kidney injury. Crit Care Med. 2018;46(1):e41–e4848. https://doi.org/10.1097/CCM.0000000000002797.
Correa TD, Jeger V, Pereira AJ, Takala J, Djafarzadeh S, Jakob SM. Angiotensin II in septic shock: effects on tissue perfusion, organ function, and mitochondrial respiration in a porcine model of fecal peritonitis. Crit Care Med. 2014;42(8):e550–e55959. https://doi.org/10.1097/CCM.0000000000000397.
Lugon JR, Boim MA, Ramos OL, Ajzen H, Schor N. Renal function and glomerular hemodynamics in male endotoxemic rats. Kidney Int. 1989;36(4):570–5. https://doi.org/10.1038/ki.1989.232.
Xu C, Chang A, Hack BK, Eadon MT, Alper SL, Cunningham PN. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014;85(1):72–81. https://doi.org/10.1038/ki.2013.286.
Salmon AH, Ferguson JK, Burford JL, Gevorgyan H, Nakano D, Harper SJ, et al. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol. 2012;23(8):1339–500. https://doi.org/10.1681/ASN.2012010017.
Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217–23. https://doi.org/10.1038/nm.2843.
Potter DR, Jiang J, Damiano ER. The recovery time course of the endothelial cell glycocalyx in vivo and its implications in vitro. Circ Res. 2009;104(11):1318–25. https://doi.org/10.1161/CIRCRESAHA.108.191585.
Marik P, Bellomo R. A rational approach to fluid therapy in sepsis. Br J Anaesth. 2016;116(3):339–49. https://doi.org/10.1093/bja/aev349.
Mohsenin V. Practical approach to detection and management of acute kidney injury in critically ill patient. J Intensive Care. 2017;5:57. https://doi.org/10.1186/s40560-017-0251-y.
Perner A, Prowle J, Joannidis M, Young P, Hjortrup PB, Pettila V. Fluid management in acute kidney injury. Intensive Care Med. 2017;43(6):807–15. https://doi.org/10.1007/s00134-017-4817-x.
Finfer S, Myburgh J, Bellomo R. Intravenous fluid therapy in critically ill adults. Nat Rev Nephrol. 2018;14(9):541–57. https://doi.org/10.1038/s41581-018-0044-0.
Marik PE, Byrne L, van Haren F. Fluid resuscitation in sepsis: the great 30 mL/kg hoax. J Thorac Dis. 2020;12(Suppl 1):S37–S47. https://doi.org/10.21037/jtd.2019.12.84.
Johannes T, Mik EG, Nohe B, Raat NJ, Unertl KE, Ince C. Influence of fluid resuscitation on renal microvascular PO2 in a normotensive rat model of endotoxemia. Crit Care. 2006;10(3):R88. https://doi.org/10.1186/cc4948.
Johannes T, Mik EG, Ince C. Nonresuscitated endotoxemia induces microcirculatory hypoxic areas in the renal cortex in the rat. Shock. 2009;31(1):97–103. https://doi.org/10.1097/SHK.0b013e31817c02a5.
Legrand M, Bezemer R, Kandil A, Demirci C, Payen D, Ince C. The role of renal hypoperfusion in development of renal microcirculatory dysfunction in endotoxemic rats. Intensive Care Med. 2011;37(9):1534–42. https://doi.org/10.1007/s00134-011-2267-4.
Aksu U, Bezemer R, Demirci C, Ince C. Acute effects of balanced versus unbalanced colloid resuscitation on renal macrocirculatory and microcirculatory perfusion during endotoxemic shock. Shock. 2012;37(2):205–9. https://doi.org/10.1097/SHK.0b013e31823ca89c.
Ergin B, Zafrani L, Kandil A, Baasner S, Lupp C, Demirci C, et al. Fully balanced fluids do not improve microvascular oxygenation, acidosis and renal function in a rat model of endotoxemia. Shock. 2016;46(1):83–91. https://doi.org/10.1097/SHK.0000000000000573.
Pickering JW, Ralib AM, Endre ZH. Combining creatinine and volume kinetics identifies missed cases of acute kidney injury following cardiac arrest. Crit Care. 2013;17(1):R7. https://doi.org/10.1186/cc11931.
Endre ZH, Pickering JW. Biomarkers and creatinine in AKI: the trough of disillusionment or the slope of enlightenment? Kidney Int. 2013;84(4):644–7. https://doi.org/10.1038/ki.2013.168.
Doi K, Yuen PS, Eisner C, Hu X, Leelahavanichkul A, Schnermann J, et al. Reduced production of creatinine limits its use as marker of kidney injury in sepsis. J Am Soc Nephrol. 2009;20(6):1217–21. https://doi.org/10.1681/ASN.2008060617.
Eadon MT, Hack BK, Xu C, Ko B, Toback FG, Cunningham PN. Endotoxemia alters tight junction gene and protein expression in the kidney. Am J Physiol Renal Physiol. 2012;303(6):F821–F830830. https://doi.org/10.1152/ajprenal.00023.2012.
Nakano D, Kitada K, Wan N, Zhang Y, Wiig H, Wararat K, et al. Lipopolysaccharide induces filtrate leakage from renal tubular lumina into the interstitial space via a proximal tubular Toll-like receptor 4-dependent pathway and limits sensitivity to fluid therapy in mice. Kidney Int. 2020. https://doi.org/10.1016/j.kint.2019.11.024.
Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, et al. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol. 2011;22(8):1505–16. https://doi.org/10.1681/ASN.2011020203.
Nakano D, Doi K, Kitamura H, Kuwabara T, Mori K, Mukoyama M, et al. Reduction of tubular flow rate as a mechanism of oliguria in the early phase of endotoxemia revealed by intravital imaging. J Am Soc Nephrol. 2015;26(12):3035–44. https://doi.org/10.1681/ASN.2014060577.
Hato T, Winfree S, Day R, Sandoval RM, Molitoris BA, Yoder MC, et al. Two-photon intravital fluorescence lifetime imaging of the kidney reveals cell-type specific metabolic signatures. J Am Soc Nephrol. 2017;28(8):2420–30. https://doi.org/10.1681/ASN.2016101153.
Menshikh A, Scarfe L, Delgado R, Finney C, Zhu Y, Yang H, et al. Capillary rarefaction is more closely associated with CKD progression after cisplatin, rhabdomyolysis, and ischemia-reperfusion-induced AKI than renal fibrosis. Am J Physiol Renal Physiol. 2019;317(5):F1383–F13971397. https://doi.org/10.1152/ajprenal.00366.2019.
Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens. 2004;13(1):1–7. https://doi.org/10.1097/00041552-200401000-00001.
Zhang Y, Nakano D, Guan Y, Hitomi H, Uemura A, Masaki T, et al. A sodium-glucose cotransporter 2 inhibitor attenuates renal capillary injury and fibrosis by a vascular endothelial growth factor-dependent pathway after renal injury in mice. Kidney Int. 2018;94(3):524–35. https://doi.org/10.1016/j.kint.2018.05.002.
Wu L, Gokden N, Mayeux PR. Evidence for the role of reactive nitrogen species in polymicrobial sepsis-induced renal peritubular capillary dysfunction and tubular injury. J Am Soc Nephrol. 2007;18(6):1807–15. https://doi.org/10.1681/ASN.2006121402.
Nakano D, Nishiyama A. Multiphoton imaging of kidney pathophysiology. J Pharmacol Sci. 2016;132(1):1–5. https://doi.org/10.1016/j.jphs.2016.08.001.
Sato Y, Mii A, Hamazaki Y, Fujita H, Nakata H, Masuda K, et al. Heterogeneous fibroblasts underlie age-dependent tertiary lymphoid tissues in the kidney. JCI Insight. 2016;1(11):e87680. https://doi.org/10.1172/jci.insight.87680.
Mishima E, Sato E, Ito J, Yamada KI, Suzuki C, Oikawa Y, et al. Drugs repurposed as antiferroptosis agents suppress organ damage, including AKI, by functioning as lipid peroxyl radical scavengers. J Am Soc Nephrol. 2020;31(2):280–96. https://doi.org/10.1681/ASN.2019060570.
Hasegawa S, Susaki EA, Tanaka T, Komaba H, Wada T, Fukagawa M, et al. Comprehensive three-dimensional analysis (CUBIC-kidney) visualizes abnormal renal sympathetic nerves after ischemia/reperfusion injury. Kidney Int. 2019;96(1):129–38. https://doi.org/10.1016/j.kint.2019.02.011.
Langenberg C, Bagshaw SM, May CN, Bellomo R. The histopathology of septic acute kidney injury: a systematic review. Crit Care. 2008;12(2):R38. https://doi.org/10.1186/cc6823.
Langenberg C, Gobe G, Hood S, May CN, Bellomo R. Renal histopathology during experimental septic acute kidney injury and recovery. Crit Care Med. 2014;42(1):e58–67. https://doi.org/10.1097/CCM.0b013e3182a639da.
Byrne L, Obonyo NG, Diab SD, Dunster KR, Passmore MR, Boon AC, et al. Unintended consequences: fluid resuscitation worsens shock in an ovine model of endotoxemia. Am J Respir Crit Care Med. 2018;198(8):1043–54. https://doi.org/10.1164/rccm.201801-0064OC.
Jacob M, Saller T, Chappell D, Rehm M, Welsch U, Becker BF. Physiological levels of A-, B- and C-type natriuretic peptide shed the endothelial glycocalyx and enhance vascular permeability. Basic Res Cardiol. 2013;108(3):347. https://doi.org/10.1007/s00395-013-0347-z.
Payen D, de Pont AC, Sakr Y, Spies C, Reinhart K, Vincent JL, et al. A positive fluid balance is associated with a worse outcome in patients with acute renal failure. Crit Care. 2008;12(3):R74. https://doi.org/10.1186/cc6916.
Montomoli J, Donati A, Ince C. Acute kidney injury and fluid resuscitation in septic patients: are we protecting the kidney? Nephron. 2019;143(3):170–3. https://doi.org/10.1159/000501748.
Ostermann M, Liu K, Kashani K. Fluid management in acute kidney injury. Chest. 2019;156(3):594–603. https://doi.org/10.1016/j.chest.2019.04.004.
Liu KD, Humphreys BD, Endre ZH. The ten barriers for translation of animal data on AKI to the clinical setting. Intensive Care Med. 2017;43(6):898–900. https://doi.org/10.1007/s00134-017-4810-4.
Yang CC, Ma MC, Chien CT, Wu MS, Sun WK, Chen CF. Hypoxic preconditioning attenuates lipopolysaccharide-induced oxidative stress in rat kidneys. J Physiol. 2007;582(Pt 1):407–19. https://doi.org/10.1113/jphysiol.2006.122747.
Olguner CG, Koca U, Altekin E, Ergur BU, Duru S, Girgin P, et al. Ischemic preconditioning attenuates lipid peroxidation and apoptosis in the cecal ligation and puncture model of sepsis. Exp Ther Med. 2013;5(6):1581–8. https://doi.org/10.3892/etm.2013.1034.
Jia P, Wu X, Dai Y, Teng J, Fang Y, Hu J, et al. MicroRNA-21 is required for local and remote ischemic preconditioning in multiple organ protection against sepsis. Crit Care Med. 2017;45(7):e703–e710710. https://doi.org/10.1097/CCM.0000000000002363.
Nishioka S, Nakano D, Kitada K, Sofue T, Ohsaki H, Moriwaki K, et al. The cyclin-dependent kinase inhibitor p21 is essential for the beneficial effects of renal ischemic preconditioning on renal ischemia/reperfusion injury in mice. Kidney Int. 2014;85(4):871–9. https://doi.org/10.1038/ki.2013.496.
Jankauskas SS, Pevzner IB, Andrianova NV, Zorova LD, Popkov VA, Silachev DN, et al. The age-associated loss of ischemic preconditioning in the kidney is accompanied by mitochondrial dysfunction, increased protein acetylation and decreased autophagy. Sci Rep. 2017;7:44430. https://doi.org/10.1038/srep44430.
Livingston MJ, Wang J, Zhou J, Wu G, Ganley IG, Hill JA, et al. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy. 2019;15(12):2142–62. https://doi.org/10.1080/15548627.2019.1615822.
Spath MR, Bartram MP, Palacio-Escat N, Hoyer KJR, Debes C, Demir F, et al. The proteome microenvironment determines the protective effect of preconditioning in cisplatin-induced acute kidney injury. Kidney Int. 2019;95(2):333–49. https://doi.org/10.1016/j.kint.2018.08.037.
Kosieradzki M, Ametani M, Southard JH, Mangino MJ. Is ischemic preconditioning of the kidney clinically relevant? Surgery. 2003;133(1):81–90. https://doi.org/10.1067/msy.2003.93.
Behrends M, Walz MK, Kribben A, Neumann T, Helmchen U, Philipp T, et al. No protection of the porcine kidney by ischaemic preconditioning. Exp Physiol. 2000;85(6):819–27.
Orvieto MA, Zorn KC, Mendiola FP, Gong EM, Lucioni A, Mikhail AA, et al. Ischemia preconditioning does not confer resilience to warm ischemia in a solitary porcine kidney model. Urology. 2007;69(5):984–7. https://doi.org/10.1016/j.urology.2007.01.100.
Hernandez DJ, Roberts WB, Miles-Thomas J, Magheli A, Saha S, Schaeffer EM, et al. Can ischemic preconditioning ameliorate renal ischemia-reperfusion injury in a single-kidney porcine model? J Endourol. 2008;22(11):2531–6. https://doi.org/10.1089/end.2008.0145.
Pan T, Jia P, Chen N, Fang Y, Liang Y, Guo M, et al. Delayed remote ischemic preconditioning ConfersRenoprotection against septic acute kidney injury via exosomal miR-21. Theranostics. 2019;9(2):405–23. https://doi.org/10.7150/thno.29832.
Orbegozo Cortes D, Su F, Santacruz C, Hosokawa K, Donadello K, Creteur J, et al. Ischemic conditioning protects the microcirculation, preserves organ function, and prolongs survival in sepsis. Shock. 2016;45(4):419–27. https://doi.org/10.1097/SHK.0000000000000526.
Zhang Y, Nakano D, Nishiyama A. Remote ischemic preconditioning of the femoral artery and vein does not protect against renal ischemia/reperfusion-induced injury in anesthetized mice. J Urol Nephrol Open Access. 2016;2(2):1–5. https://doi.org/10.15226/2473-6430/2/2/00110.
Bedir S, Ma Y, Antonelli J, Cadeddu JA, Gahan JC. Ineffectiveness of remote ischemic renal preconditioning in a porcine solitary-kidney model. J Endourol. 2015;29(5):590–4. https://doi.org/10.1089/end.2014.0563.
Zwaag J, Beunders R, Warle MC, Kellum JA, Riksen NP, Pickkers P, et al. Remote ischaemic preconditioning does not modulate the systemic inflammatory response or renal tubular stress biomarkers after endotoxaemia in healthy human volunteers: a single-centre, mechanistic, randomised controlled trial. Br J Anaesth. 2019;123(2):177–85. https://doi.org/10.1016/j.bja.2019.03.037.
Veighey KV, Nicholas JM, Clayton T, Knight R, Robertson S, Dalton N, et al. Early remote ischaemic preconditioning leads to sustained improvement in allograft function after live donor kidney transplantation: long-term outcomes in the REnal Protection Against Ischaemia-Reperfusion in transplantation (REPAIR) randomised trial. Br J Anaesth. 2019;123(5):584–91. https://doi.org/10.1016/j.bja.2019.07.019.
Klaus S, Heringlake M, Block K, Nolde J, Staubach K, Bahlmann L. Metabolic changes detected by microdialysis during endotoxin shock and after endotoxin preconditioning. Intensive Care Med. 2003;29(4):634–41. https://doi.org/10.1007/s00134-002-1602-1.
Hato T, Winfree S, Kalakeche R, Dube S, Kumar R, Yoshimoto M, et al. The macrophage mediates the renoprotective effects of endotoxin preconditioning. J Am Soc Nephrol. 2015;26(6):1347–62. https://doi.org/10.1681/ASN.2014060561.
Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35(11):2576–81. https://doi.org/10.1161/01.STR.0000143450.04438.ae.
Rosenzweig HL, Minami M, Lessov NS, Coste SC, Stevens SL, Henshall DC, et al. Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J Cereb Blood Flow Metab. 2007;27(10):1663–744. https://doi.org/10.1038/sj.jcbfm.9600464.
Obermaier R, Drognitz O, Grub A, von Dobschuetz E, Schareck W, Hopt UT, et al. Endotoxin preconditioning in pancreatic ischemia/reperfusion injury. Pancreas. 2003;27(3):e51–e5656. https://doi.org/10.1097/00006676-200310000-00020.
Chen K, Geng S, Yuan R, Diao N, Upchurch Z, Li L. Super-low dose endotoxin pre-conditioning exacerbates sepsis mortality. EBioMedicine. 2015;2(4):324–33. https://doi.org/10.1016/j.ebiom.2015.03.001.
Ondee T, Surawut S, Taratummarat S, Hirankarn N, Palaga T, Pisitkun P, et al. Fc gamma receptor IIB deficient mice: a lupus model with increased endotoxin tolerance-related sepsis susceptibility. Shock. 2017;47(6):743–52. https://doi.org/10.1097/SHK.0000000000000796.
Nikolich-Zugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018;19(1):10–9. https://doi.org/10.1038/s41590-017-0006-x.
Sun Y, Li H, Yang MF, Shu W, Sun MJ, Xu Y. Effects of aging on endotoxin tolerance induced by lipopolysaccharides derived from Porphyromonas gingivalis and Escherichia coli. PLoS ONE. 2012;7(6):e39224. https://doi.org/10.1371/journal.pone.0039224.
Metz CN, Tracey KJ. It takes nerve to dampen inflammation. Nat Immunol. 2005;6(8):756–7. https://doi.org/10.1038/ni0805-756.
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–8. https://doi.org/10.1038/nature01339.
Vida G, Pena G, Kanashiro A, Thompson-Bonilla Mdel R, Palange D, Deitch EA, et al. beta2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. FASEB J. 2011;25(12):4476–85. https://doi.org/10.1096/fj.11-191007.
Kox M, van Eijk LT, Verhaak T, Frenzel T, Kiers HD, Gerretsen J, et al. Transvenous vagus nerve stimulation does not modulate the innate immune response during experimental human endotoxemia: a randomized controlled study. Arthritis Res Ther. 2015;17:150. https://doi.org/10.1186/s13075-015-0667-5.
Gigliotti JC, Huang L, Bajwa A, Ye H, Mace EH, Hossack JA, et al. ultrasound modulates the splenic neuroimmune axis in attenuating AKI. J Am Soc Nephrol. 2015;26(10):2470–81. https://doi.org/10.1681/ASN.2014080769.
Bonaz B, Sinniger V, Hoffmann D, Clarencon D, Mathieu N, Dantzer C, et al. Chronic vagus nerve stimulation in Crohn's disease: a 6-month follow-up pilot study. Neurogastroenterol Motil. 2016;28(6):948–53. https://doi.org/10.1111/nmo.12792.
Inoue T, Abe C, Sung SS, Moscalu S, Jankowski J, Huang L, et al. Vagus nerve stimulation mediates protection from kidney ischemia-reperfusion injury through alpha7nAChR+ splenocytes. J Clin Invest. 2016;126(5):1939–52. https://doi.org/10.1172/JCI83658.
Abe C, Inoue T, Inglis MA, Viar KE, Huang L, Ye H, et al. C1 neurons mediate a stress-induced anti-inflammatory reflex in mice. Nat Neurosci. 2017;20(5):700–7. https://doi.org/10.1038/nn.4526.
Inoue T, Abe C, Kohro T, Tanaka S, Huang L, Yao J, et al. Non-canonical cholinergic anti-inflammatory pathway-mediated activation of peritoneal macrophages induces Hes1 and blocks ischemia/reperfusion injury in the kidney. Kidney Int. 2019;95(3):563–76. https://doi.org/10.1016/j.kint.2018.09.020.
van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, et al. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis. 2005;191(12):2138–48. https://doi.org/10.1086/430323.
Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334(6052):98–101. https://doi.org/10.1126/science.1209985.
Torres-Rosas R, Yehia G, Pena G, Mishra P, del Rocio T-B, Moreno-Eutimio MA, et al. Dopamine mediates vagal modulation of the immune system by electroacupuncture. Nat Med. 2014;20(3):291–5. https://doi.org/10.1038/nm.3479.
Sadis C, Teske G, Stokman G, Kubjak C, Claessen N, Moore F, et al. Nicotine protects kidney from renal ischemia/reperfusion injury through the cholinergic anti-inflammatory pathway. PLoS ONE. 2007;2(5):e469. https://doi.org/10.1371/journal.pone.0000469.
Pabst MJ, Pabst KM, Collier JA, Coleman TC, Lemons-Prince ML, Godat MS, et al. Inhibition of neutrophil and monocyte defensive functions by nicotine. J Periodontol. 1995;66(12):1047–55. https://doi.org/10.1902/jop.1995.66.12.1047.
Matsunaga K, Klein TW, Friedman H, Yamamoto Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J Immunol. 2001;167(11):6518–24. https://doi.org/10.4049/jimmunol.167.11.6518.
Steiner AA, Oliveira DL, Roberts JL, Petersen SR, Romanovsky AA. Nicotine administration and withdrawal affect survival in systemic inflammation models. J Appl Physiol. (1985). 2008;105(4):1028–34. https://doi.org/10.1152/japplphysiol.90619.2008.
Uni R, Inoue T, Nakamura Y, Fukaya D, Hasegawa S, Wu CH, et al. Vagus nerve stimulation even after injury ameliorates cisplatin-induced nephropathy via reducing macrophage infiltration. Sci Rep. 2020;10(1):9472. https://doi.org/10.1038/s41598-020-66295-0.
Cotero V, Fan Y, Tsaava T, Kressel AM, Hancu I, Fitzgerald P, et al. Noninvasive sub-organ ultrasound stimulation for targeted neuromodulation. Nat Commun. 2019;10(1):952. https://doi.org/10.1038/s41467-019-08750-9.
Villegas-Bastida A, Torres-Rosas R, Arriaga-Pizano LA, Flores-Estrada J, Gustavo-Acosta A, Moreno-Eutimio MA. Electrical stimulation at the ST36 acupoint protects against sepsis lethality and reduces serum TNF levels through vagus nerve- and catecholamine-dependent mechanisms. Evid Based Complement Alternat Med. 2014;2014:451674. https://doi.org/10.1155/2014/451674.
Harpin D, Simadibrata CL, Mihardja H, Barasila AC. Effect of electroacupuncture on urea and creatinine levels in the wistar sepsis model. Med Acupunct. 2020;32(1):29–37. https://doi.org/10.1089/acu.2019.1369.
Song Q, Hu S, Wang H, Lv Y, Shi X, Sheng Z, et al. Electroacupuncturing at Zusanli point (ST36) attenuates pro-inflammatory cytokine release and organ dysfunction by activating cholinergic anti-inflammatory pathway in rat with endotoxin challenge. Afr J Tradit Complement Altern Med. 2014;11(2):469–74. https://doi.org/10.4314/ajtcam.v11i2.35.
Geng Y, Chen D, Zhou J, Jiang H, Zhang H. Role of Cholinergic anti-inflammatory pathway in treatment of intestinal ischemia-reperfusion injury by electroacupuncture at Zusanli. Evid Based Complement Alternat Med. 2017;2017:6471984. https://doi.org/10.1155/2017/6471984.
Liu J, Li C, Peng H, Yu K, Tao J, Lin R, et al. Electroacupuncture attenuates learning and memory impairment via activation of alpha7nAChR-mediated anti-inflammatory activity in focal cerebral ischemia/reperfusion injured rats. Exp Ther Med. 2017;14(2):939–46. https://doi.org/10.3892/etm.2017.4622.
Jiang T, Wu M, Zhang Z, Yan C, Ma Z, He S, et al. Electroacupuncture attenuated cerebral ischemic injury and neuroinflammation through alpha7nAChR-mediated inhibition of NLRP3 inflammasome in stroke rats. Mol Med. 2019;25(1):22. https://doi.org/10.1186/s10020-019-0091-4.
Zhang L, Huang Z, Shi X, Hu S, Litscher D, Wang L, et al. Protective effect of electroacupuncture at zusanli on myocardial injury in septic rats. Evid Based Complement Alternat Med. 2018;2018:6509650. https://doi.org/10.1155/2018/6509650.
Schneider F, Lutun P, Couchot A, Bilbault P, Tempe JD. Plasma cyclic guanosine 3′–5′ monophosphate concentrations and low vascular resistance in human septic shock. Intensive Care Med. 1993;19(2):99–104. https://doi.org/10.1007/bf01708370.
Hinder F, Booke M, Traber LD, Matsumoto N, Nishida K, Rogers S, et al. Nitric oxide synthase inhibition during experimental sepsis improves renal excretory function in the presence of chronically increased atrial natriuretic peptide. Crit Care Med. 1996;24(1):131–6. https://doi.org/10.1097/00003246-199601000-00022.
Yamada H, Doi K, Tsukamoto T, Kiyomoto H, Yamashita K, Yanagita M, et al. Low-dose atrial natriuretic peptide for prevention or treatment of acute kidney injury: a systematic review and meta-analysis. Crit Care. 2019;23(1):41. https://doi.org/10.1186/s13054-019-2330-z.
Shaw SG, Weidmann P, Hodler J, Zimmermann A, Paternostro A. Atrial natriuretic peptide protects against acute ischemic renal failure in the rat. J Clin Invest. 1987;80(5):1232–7. https://doi.org/10.1172/JCI113197.
Chujo K, Ueno M, Asaga T, Sakamoto H, Shirakami G, Ueki M. Atrial natriuretic peptide enhances recovery from ischemia/reperfusion-induced renal injury in rats. J Biosci Bioeng. 2010;109(6):526–30. https://doi.org/10.1016/j.jbiosc.2009.11.021.
Koga H, Hagiwara S, Kusaka J, Matsumoto S, Nishida T, Yokoi I, et al. Human atrial natriuretic peptide attenuates renal ischemia-reperfusion injury. J Surg Res. 2012;173(2):348–53. https://doi.org/10.1016/j.jss.2010.10.011.
Mitaka C, Si MK, Tulafu M, Yu Q, Uchida T, Abe S, et al. Effects of atrial natriuretic peptide on inter-organ crosstalk among the kidney, lung, and heart in a rat model of renal ischemia-reperfusion injury. Intensive Care Med Exp. 2014;2(1):28. https://doi.org/10.1186/s40635-014-0028-8.
Kitamura H, Nakano D, Sawanobori Y, Asaga T, Yokoi H, Yanagita M, et al. Guanylyl cyclase A in both renal proximal tubular and vascular endothelial cells protects the kidney against acute injury in rodent experimental endotoxemia models. Anesthesiology. 2018;129(2):296–310. https://doi.org/10.1097/ALN.0000000000002214.
Leijte GP, Kiers D, van der Heijden W, Jansen A, Gerretsen J, Boerrigter V, et al. Treatment with acetylsalicylic acid reverses endotoxin tolerance in humans in vivo: a randomized placebo-controlled study. Crit Care Med. 2019;47(4):508–16. https://doi.org/10.1097/CCM.0000000000003630.
Kiers D, Leijte GP, Gerretsen J, Zwaag J, Kox M, Pickkers P. Comparison of different lots of endotoxin and evaluation of in vivo potency over time in the experimental human endotoxemia model. Innate Immun. 2019;25(1):34–45. https://doi.org/10.1177/1753425918819754.
Kox M, van Eijk LT, Zwaag J, van den Wildenberg J, Sweep FC, van der Hoeven JG, et al. Voluntary activation of the sympathetic nervous system and attenuation of the innate immune response in humans. Proc Natl Acad Sci USA. 2014;111(20):7379–84. https://doi.org/10.1073/pnas.1322174111.
Koch RM, Kox M, Thijs EJM, Rahamat-Langendoen JC, van de Veerdonk FL, Gerretsen J, et al. Development of endotoxin tolerance does not influence the response to a challenge with the mucosal live-attenuated influenza vaccine in humans in vivo. Front Immunol. 2017;8:1600. https://doi.org/10.3389/fimmu.2017.01600.
Stokes JM, Yang K, Swanson K, Jin W, Cubillos-Ruiz A, Donghia NM, et al. A deep learning approach to antibiotic discovery. Cell. 2020;180(4):688–702.e13. https://doi.org/10.1016/j.cell.2020.01.021.
Miyaji T, Hu X, Yuen PS, Muramatsu Y, Iyer S, Hewitt SM, et al. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int. 2003;64(5):1620–31. https://doi.org/10.1046/j.1523-1755.2003.00268.x.
Nakano D, Kobori H, Burford JL, Gevorgyan H, Seidel S, Hitomi H, et al. Multiphoton imaging of the glomerular permeability of angiotensinogen. J Am Soc Nephrol. 2012;23(11):1847–56. https://doi.org/10.1681/ASN.2012010078.
Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. https://doi.org/10.1038/ncomms7673.
Acknowledgements
We thank Enago Group for editing drafts of this manuscript for English language.
Funding
This work was supported by Organization Collaboration Research Promotion Fund of Kagawa University Research Promotion Program 2019 (KURPP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Ethical standards
All experiments were approved by the Institutional Animal Care and Use Committee of Kagawa University and followed standard guidelines for the humane care and use of animals in scientific research.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Nakano, D. Septic acute kidney injury: a review of basic research. Clin Exp Nephrol 24, 1091–1102 (2020). https://doi.org/10.1007/s10157-020-01951-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-020-01951-3