
Abstract
Background
The clinical significance of miR-17 in patients with acute myeloid leukemia (AML) remains unknown.
Methods
Real-time quantitative reverse transcription-polymerase chain reaction (qPCR) was performed to detect the miR-17 expression in 115 de novo AML patients, 31 patients at complete remission (CR) time, 8 patients at relapse time and 30 normal controls.
Results
MiR-17 was upregulated in de novo AML compared with normal controls. Patients with high expression of miR-17 had less CEBPA double mutation, less favorable ELN-risk and lower CR rate. The level of miR-17 was significantly decreased at CR phase and was returned to primary level even higher when in relapse phase. In addition, Cox regression analysis revealed that miR-17 expression retained independent prognostic significance for overall survival (OS). Moreover, the gene-expression profile analysis of miR-17 in AML obtained from TCGA database was involved in multiple biological functions and signal pathways. Among the differential expressed genes (DEGs), we identified FGL2, PLAUR, SLC2A3, GPR65, CTSS, TLR7, S1PR3, OGFRL1, LILRB1, IL17RA, SIGLEC10, SLAMF7, PLXDC2, HPSE, TCF7 and MYCL as potential direct targets of miR-17 according to in silico analysis.
Conclusions
High expression of miR-17 in de novo AML patients pointed to dismal clinical outcome and disease recurrence, which could serve as novel prognostic biomarker for AML patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the most common type of leukemia in adults, acute myeloid leukemia (AML) is a malignant clone disease of the hematopoietic system. Although there have been great advances in the treatment of AML, patients have highly heterogenous clinical course and their long-term prognosis is still dismal [1]. Therefore, identifying novel prognostic markers to improve the existing molecular-based stratification and risk-adapted therapy for AML patients is urgently required.
It is well known that microRNAs (miRNAs) have been implicated in both biological and pathological processes such as cell growth, apoptosis, migration and invasion. There is an increasing body of evidence unraveling the molecular mechanism of miRNAs as either oncogenes or tumor suppressors in human cancers, which is mostly depending on the function of their different target genes and specific tumor microenvironment [2]. Aberrantly expressed miRNA patterns with clinical prognostic significance have been reported in AML patients [3]. For instance, in a cohort of 176 AML patients from TCGA database, the data of miRNAs sequencing and gene microarray were analyzed to identify risk miRNAs with prognostic value. Among the 705 miRNAs that were studied, upregulated miR-520a, 599, 606, 137 and 362 predicted unfavorable outcomes of AML patients [4]. In another study of 187 cytogenetically normal AML (CN-AML) patients, miR-181a overexpression was strongly correlated with better survival [5].
The miR-17-92 cluster, known as OncomiR-1, is a highly conserved polycistronic transcript among vertebrates, which is located on the open reading frame 25 of chromosome 13 (C13orf25) and yields six mature miRNAs (miR-17, 18a, 19a, 20a, 19b, and 92a) [6]. Accumulating evidence have revealed that the members of miR-17-92 family are very often dysregulated and play critical roles in a wide range of cancer types including osteosarcoma, hepatocellular carcinoma, renal cancer, ovarian cancer, lymphoma, retinoblastoma and so on [7]. Whereas, the expression and prognostic significance of miR-17 in AML has not been fully investigated. In current study, we aimed to evaluate the expression and clinical significance of miR-17 in de novo AML patients.
Materials and methods
Patients and samples
In accordance with the Declaration of Helsinki Principles, written informed consents were obtained from all patients. A total of 115 newly diagnosed AML patients and 30 healthy controls were enrolled in this study. Bone marrow (BM) samples were collected from the 30 healthy controls, 115 AML patients at primary diagnosis, 31 patients at complete remission (CR), and 8 patients at relapse.
RNA extraction, reverse transcription and real-time quantitative PCR
Briefly, RNA was extracted from Ficoll-separated BM mononuclear cells with Trizol (Thermo Fisher Scientific, USA) following the protocol of the manufacture. MiR-17 expression was determined using the TaqMan MicroRNA Assay (Thermo Fisher Scientific, USA). Reverse transcription was performed to synthesize cDNA using the TaqMan MicroRNA Reverse Transcription kit (Thermo Fisher Scientific, USA). The quantification of miR-17 expression was conducted using the TaqMan PCR Master Mix (Assay ID: 000,393, Thermo Fisher Scientific, USA). RNU6B were selected as endogenous controls. Relative miR-17 expression level was calculated by 2−△△CT method. All experiments were run in triplicate.
Statistical analysis
Pearson Chi-square analysis and Fisher’s exact test were carried to compared the differences of categorical variables between two groups, while Mann–Whitney U test and T test were carried to compared the differences of continuous variables between two groups. Wilcoxon singed rank test were conducted to compared the differences of matched patients. The effect of miR-17 expression on clinical outcome were evaluated by Kaplan–Meier and Cox regression model. P < 0.05 was considered as statistically significant. SPSS 25.0 software was conducted for statistical analyzes.
Results
The upregulated miR‐17 expression in BM of AML patients
We detected the expression of miR-17 in BM from 30 healthy donors and 115 AML patient (including 57 CN-AML patients). As shown in Fig. 1, the relative level of miR-17 in AML patients ranged from 3.49 to 59.62 (median 8.34), which was significantly higher than that in normal controls (median 3.62, range 0.42–14.01, P < 0.0001). In addition, significant overexpression of BM miR-17 expression was also presented in CN-AML patients.

BM miR-17 expression in controls and AML patients. The distributions of miR‐17 expression in controls, whole-cohort AML and CN‐AML were presented with scatter plots. The median level of miR‐17 expression in each group was shown with horizontal line. AML acute myeloid leukemia; CN‐AML cytogenetically normal AML. **P < 0.01; ***P < 0.001; NSP > 0.05. Data are shown as mean from three independent experiments
Correlation of BM miR-17 level with clinical features in AML
To analyze the clinical relevance of miR-17 expression with AML, the whole-cohort patients were divided into high and low miR-17 expression groups based on the its median level. The association of the clinical and genetic characteristics with miR-17 expression was shown in Table 1. The low miR-17 expression group had more patients with CEBPA double mutation (P = 0.029) and favorable ELN-risk (P = 0.047) than the high miR-17 group. There were no strikingly differences in sex, age, white blood cells, hemoglobin, platelets, BM blasts, FAB subtypes and common fusion genes between the high and low miR-17 expression groups.
Low expression of miR-17 is predictive for favorable survival
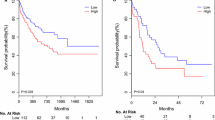
The AML patients with low miR-17 expression had higher chance for remission (P = 0.002; Table 1). 61.97% of patients with CR after induction therapy had lower miR-17 expression, and 68.18% of patients who did not achieve CR had higher miR-17 expression (Fig. 2). We further analyzed the prognostic value of miR-17 expression in AML patients. Kaplan–Meier analysis showed that the median OS time was shorter in the whole-cohort AML patients with high miR-17 expression than those with low miR-17 expression (P < 0.001; Fig. 3a). Similar result was obtained in CN-AML patients (P = 0.035; Fig. 3b). To further validate the adverse prognostic significance of miR-17 expression in AML, data from TCGA database was set as a validation cohort. A total of 188 AML patients were classified into two groups according to median level of miR-17 expression. Consistent with our results, the low expression of miR-17 was significantly correlated with longer OS (P = 0.032, Fig. 3c). Moreover, in the univariate and multivariate Cox analysis, miR-17 expression retained independent prognostic significance for OS even in the presence of other covariates, such as age and IDH1 mutation (Table 2).

Correlation between miR-17 expression and CR state. The distributions of AML patients according to the miR-17 expression and CR state. CR complete remission

The impact of BM miR-17 expression on survival in AML patients. a, b Kaplan–Meier survival analysis based on high or low level of miR-17 expression in whole-cohort AML and CN-AML patients. c Kaplan–Meier survival analysis of the AML patients from TCGA dataset as a validation cohort
miR-17 expression in the surveillance of AML
To monitor the dynamic alteration of miR-17 expression in the follow-up of AML patients, qPCR was conducted to assess miR-17 expression in 31 patients out of the 71 patients achieved CR. The expression of miR-17 significantly decreased after successful induction chemotherapy (P < 0.001, Fig. 4a). Moreover, the dynamic change of miR-17 expression was also detected in eight relapsed patients. The results indicated that miR-17 expression level was returned to primary level even higher when in relapse phase, which might predict disease recurrence in AML patients (Fig. 4b).

miR-17 expression in the follow-up of AML patients in different stages. a miR-17 expression levels in 31 AML patients achieving a CR before and after treatment. b Dynamic alteration of miR-17 expression in the 8 paired AML patients with different clinical stages. Data are shown as mean ± SD from three independent experiments
Biological insights in AML of miR-17 profiles
To further unveil the biological function of miR-17 in AML patients, we analyzed differential gene expression based on miR-17 level using the high throughput sequencing data of the AML cohort from TCGA database [8]. We found that 429 differentially expressed genes (DEGs) associated with miR-17 expression, among which 310 and 119 genes were negatively and positively correlated with miR-17 expression, respectively (Fig. 5). To screen for the target genes of miR-17, the online applications Targetscan, miRWalk and miRanda website tools were employed in silico analysis. Among the predicted target genes, miR-17 expression was inversely correlated with the expression of FGL2, PLAUR, SLC2A3, GPR65, CTSS, TLR7, S1PR3, OGFRL1, LILRB1, IL17RA, SIGLEC10, SLAMF7, PLXDC2, HPSE, TCF7 and MYCL. Gene Ontology (GO) analysis revealed that the DEGs associated with miR-17 expression were involved in immune response, complement activation, leukocyte migration, receptor-mediated endocytosis, phagocytosis, engulfment, positive regulation of B cell activation, immunoglobulin production and so on (Fig. 6a). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that the enriched pathways of the DEGs were involved in osteoclast differentiation, hematopoietic cell lineage, acute myeloid leukemia, transcriptional mis-regulation in cancer, cytokine-cytokine receptor interaction, pathways in cancer and so on (Fig. 6b). To further identify more proteins which belong to same pathway or have similar function, the 429 DEGs were clustered into 20 groups on the basis of their enrichment score and then rendered as a network plot (Fig. 6c). Furthermore, a protein–protein interaction (PPI) network of the DEGs was constructed and identified fourteen molecular complex detection (MCODE) modules (Fig. 7). Interestingly, seven targets of (PTAFR, TLR7, S1PR3, GPR65, CTSS, PLAUR and SLC2A3) were identified as hub genes of the MCODE modules. Therefore, the gene-expression profiling signature suggests that miR-17 plays important roles in regulating biological functions and signal pathways, which further confirms its clinical significance in AML patients.

DEGs between AML patients with high and low miR-17 expression. The rows indicate the genes and the columns represent the patients. The patients were sorted in order from left to right by increasing levels of miR-17. Pink and blue represent expression levels above and below median gene expression (white), respectively. DEGs: differentially expressed genes

Functional enrichment analysis of DEGs. a GO and b KEGG analysis for hub genes by DAVID. c Enriched terms were colored by cluster ID, connected by edges and rendered as a network plot using the Cytoscape tool. GO Gene Ontology, KEGG Kyoto Encyclopedia of Genes and Genomes

PPI network and MCODE components identified to be associated with the analysis of DEGs. a Overall PPI network of the DEGs. b Individual modules selected from the PPI network using the MCODE method. Each color represents each MCODE network. PPI protein–protein interaction; MCODE molecular complex detection
Discussion
In current study, we explored the prognostic value of miR-17 in de novo AML for the first time. The results showed that miR-17 frequently overexpressed in AML patients, which was significantly related to poor CR rate and shorter OS. Cases with high miR-17 levels had a lower frequency of CEBPA double mutation and less favorable risk according to ELN risk stratification. The follow-up of 31 patients achieved CR and 8 relapse patients revealed that miR-17 expression significantly decreased after successful induction chemotherapy and returned to primary level even higher when in relapse phase. Moreover, the gene expression profile of miR-17 involved in multiple biological functions and signal pathways. By bioinformatics analysis, we found FGL2, PLAUR, SLC2A3, GPR65, CTSS, TLR7, S1PR3, OGFRL1, LILRB1, IL17RA, SIGLEC10, SLAMF7, PLXDC2, HPSE, TCF7 and MYCL as direct targets of miR-17 among the DEGs. These results suggested that miR-17 might function as independent prognostic biomarker and predict disease recurrence for AML patients.
The miR-17∼92 cluster has been reported to be crucial for vertebrate development such as lymphocyte maturation [9], skeletal development [10], epithelial proliferation and branching [11]. Targeted deletion of miR-17∼92 in mice leads to early neonatal lethality with ventricular septal defects, lung hypoplasia, and B lymphopoiesis inhibition [12]. Accordingly, the miR-17∼92 cluster is involved in cardiovascular, neurodegenerative and immune diseases [13,14,15]. Other than the involvement in normal development described above, the majority of the previous studies have demonstrated the potential oncogenic role of miR-17∼92 cluster in various cancers. For instance, in retinoblastoma, miR-17∼92 acted as an RB-collaborating gene to promote retinoblastoma, in part by regulating p21Cip1 and p57Kip [16]. In colon cancer, miR-17 induced epithelial-mesenchymal transition and the formation of a stem cell-like population through the modulation of CYP7B1 expression [17]. In lung cancer, miR-17-5p was overexpressed and correlated with poor survival of patients [18]. In B-cell chronic lymphocytic leukemia (CLL), the miR-17∼92 cluster members were highly amplified, among which four (miR-17, miR-20a, miR-18a and miR-19b) and five (miR-17, miR-20a, miR-18a, miR-19a and miR-19b) members were significantly induced by CD154 and stromal cell culture, respectively [19]. Moussay et al. proved circulating miR-20a was a reliable classifier to distinguish CLL patients from healthy controls (AUC = 0.920) and associated well with the disease severity (P = 0.0242) [19]. The miR-17/92 cluster was also found overexpressed in B-cell Lymphomas, and the individual members miR-19a/19b were required and sufficient for the B-cell lymphoma tumorigenesis [20, 21].The study performed by Meenhuis A found miR-17/20 in combination with two other miRNAs promoted expansion and replating capacity of myeloid progenitors by targeting sequestosome 1–regulated pathways [22]. In another study, Li et al. showed miR-17/20 was particularly amplified in MLL-rearrangement AML patients [23]. However, there is some evidence that miR-17 also possesses antitumor properties. Aberrant low expression of miR-17, consistent with the high frequency loss of heterozygosity and deletions at 13q31.3, has been reported in several types of cancers [24, 25]. For example, in prostate cancer, miR-17 has been demonstrated to attenuate androgen receptor signaling and cell growth by targeting proto-oncogenic transcriptional activator PCAF [26]. In oral squamous cell carcinoma, miR-17 functioned as a tumor suppressor via regulating KPNA2/PI3K/AKT axis [27].
Here, we reported that miR-17 predicted poor prognosis and added to the prognostic value of various previously identified molecular indicators in AML, such as TP53, CEBPA, NPM1, FLT3-ITD and so on. In our study, APL patients were not enrolled, considering that it has shifted to a highly curable AML subtype with its own typically different genetic characteristics, risk stratification and target-therapy. The role of miR-17 as prognostic factor was not restricted to CN-AML but proved in a highly mixed population of AML including patients with old age and cytogenetic abnormalities. There was significant difference only in CEBPA double mutation between the high and low miR-17 groups. Though the regulation of CEBPA expression by miR-17 had been reported in several studies [28, 29], how miR-17 influences CEBPA double mutation is not fully illuminated. In line with our results, previous study found no significant association between miR-17 and NPM1 or FLT3 mutation status in AML patients [30]. In addition, there was evidence showing that miR-17 ~ 92 cluster contributed to the MEIS1/HOXA9-mediated transformation of MLL leukemia [31]. MLL is involved in over 100 different recurrent rearrangements, of which greater than 70 translocation partner genes (TPGs) have now been identified [32, 33]. Despite the vast number of partner genes, only nine TPGs (AF4, AF9, ENL, AF10, AF6, ELL, AF1P, AF17 and SEPT6) seem to be predominantly recombined to MLL. In our study, we detected MLL-partial tandem duplications (MLL-PTD) and eleven MLL-related fusion (MLL-AF9, MLL-AF4, MLL-ENL, MLL-AF10, MLL-SEPT6, MLL-ELL, MLL-AF17, MLL-AF1q, MLL-AF1p, MLL-AF6, MLL-AFX). In accordance with previous reports [34, 35], two of the four (50%) cases with MLL-related abnormalities were classified as M5 category indicating the commonly association with monocytic leukemias. The four MLL-related aberrations (including two MLL-PTD mutations and two MLL-related fusions) were exclusively found in high miR-17 group but unfortunately outside the significance level. The presence of distinct MLL lesions in AML is widely considered as an independent dismal prognostic factor despite improved regimen options like allogeneic hematopoietic stem cell transplantation appealing for novel effective regimens [36, 37]. Interestingly, the colony forming ability of MLL-fusion containing cells could be dramatically abolished in response to treatment with antagomir-17 [31]. Whether, therapy targeting miR-17 can benefit AML patients especially with the MLL-related aberrations or M5 subtype needs further study.
To derive biological insights into the oncogenic mechanism of miR-17 in AML, gene-expression profiling signature characteristic of miR-17 was analyzed. Bioinformatics data showed that those miR-17-associated genes were involved in hematopoietic cell lineage, transcriptional mis-regulation and pathways in AML. We found a negative correlation between miR-17 expression with sixteen miR-17-targeted genes among the DEGs. Notably, there were seven targets of miR-17 (PTAFR, TLR7, S1PR3, GPR65, CTSS, PLAUR and SLC2A3) identified as hub genes in the MCODE modules. Among them, PTAFR is a platelet-activating factor and involved in a wide range of human functions. The membrane expression of PTAFR, resulted from cell maturation and differentiation, was a marker of mature cells and rarely observed in AML blasts [38]. Fiedler ERC et al. have proved that PAFR could sensitize CML cells to the dasatinib treatment after binging with PAF [39]. Another previous study revealed PTAFR as a promising target for tumor repopulation induced by radiotherapy in solid tumor [40]. TLR7, namely Toll‐like receptor 7, was enriched into Toll-like receptor signaling pathway, inflammatory response and innate immune response. Ge F et al. have demonstrated TLR7 as a novel DNA methylation prognostic signature for AML patients who might benefit from TLR7‐based immunosuppressive therapy [41]. S1PR3, as an inflammation-activated S1P receptor, not only governed the myeloid fate in normal hematopoiesis via the TNFα–NF-κB axis, but also predicted prognosis in human AML [42]. GPR65, a member of purine receptor family, has been reported to suppress hematopoiesis by establishing repressive chromatin, repressing Gata2 transcription and thus inhibiting GATA-2-GPCR circuit [43]. CTSS, as lysosomal globular proteases, participated in various biological processes including immune response, neutrophil degranulation, proteolysis and so on. As summarized in a systematic review, the prognostic value of CTSS remains controversial in leukemia [44]. PLAUR, also known as CD87, has shown poor survival benefits in leukemias, with the potential to be used as a target for fusion protein therapy of PLAUR-expressing AML [45, 46]. SLC2A3, a regulator of early embryonic development, contributes to glucose transport and participates in multiple pathways [47]. Available evidence proved that low expression of SLC2A3 predicted poor response of demethylation and vitamin C, which had a worse effect on OS in AML [48]. Taken together, the gene-expression profiling associated with miR-17 might provide mechanistic insights into the clinical prognostic significance of miR-17 in AML.
In conclusion, our study revealed that miR-17 was upregulated in de novo AML patients, and its high expression pointed to dismal clinical outcome and disease recurrence. MiR-17 expression might serve as a novel independent prognostic biomarker for AML patients. Further studies are required to explore the biological roles of miR-17 and its target genes in AML.
References
Marcucci G, Haferlach T, Dohner H (2011) Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol 29:475–486
Jorge AL, Pereira ER, Oliveira CS et al (2021) MicroRNAs: understanding their role in gene expression and cancer. Einstein 19:5996
Wallace JA, O’Connell RM (2017) MicroRNAs and acute myeloid leukemia: therapeutic implications and emerging concepts. Blood 130:1290–1301
Gao HY, Wang W, Luo XG et al (2018) Screening of prognostic risk microRNAs for acute myeloid leukemia. Hematology 23:747–755
Schwind S, Maharry K, Radmacher MD et al (2010) Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28:5257–5264
Mendell JT (2008) miRiad roles for the miR-17-92 cluster in development and disease. Cell 133:217–222
Mogilyansky E, Rigoutsos I (2013) The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ 20:1603–1614
Ley TJ, Miller C, Ding L et al (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368:2059–2074
Lai M, Gonzalez-Martin A, Cooper AB et al (2016) Regulation of B-cell development and tolerance by different members of the miR-17∼92 family microRNAs. Nat Commun 7:12207
Marcelis CL, Hol FA, Graham GE et al (2008) Genotype-phenotype correlations in MYCN-related Feingold syndrome. Hum Mutat 29:1125–1132
Carraro G, El-Hashash A, Guidolin D et al (2009) miR-17 family of microRNAs controls FGF10-mediated embryonic lung epithelial branching morphogenesis through MAPK14 and STAT3 regulation of E-Cadherin distribution. Dev Biol 333:238–250
Ventura A, Young AG, Winslow MM et al (2008) Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132:875–886
Small EM, Frost RJ, Olson EN (2010) MicroRNAs add a new dimension to cardiovascular disease. Circulation 121:1022–1032
Tsitsiou E, Lindsay MA (2009) microRNAs and the immune response. Curr Opin Pharmacol 9:514–520
Hébert SS, De Strooper B (2009) Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci 32:199–206
Conkrite K, Sundby M, Mukai S et al (2011) miR-17~92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev 25:1734–1745
Xi XP, Zhuang J, Teng MJ et al (2016) MicroRNA-17 induces epithelial-mesenchymal transition consistent with the cancer stem cell phenotype by regulating CYP7B1 expression in colon cancer. Int J Mol Med 38:499–506
Chen Q, Si Q, Xiao S et al (2013) Prognostic significance of serum miR-17-5p in lung cancer. Med Oncol 30:353
Willimott S, Wagner SD (2012) Stromal cells and CD40 ligand (CD154) alter the miRNome and induce miRNA clusters including, miR-125b/miR-99a/let-7c and miR-17-92 in chronic lymphocytic leukaemia. Leukemia 26:1113–1116
Ota A, Tagawa H, Karnan S et al (2004) Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Can Res 64:3087–3095
Mu P, Han YC, Betel D et al (2009) Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 23:2806–2811
Meenhuis A, van Veelen PA, de Looper H et al (2011) MiR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood 118:916–925
Li Z, Lu J, Sun M et al (2008) Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA 105:15535–15540
Shao J, Li Y, Wu Q et al (2002) High frequency loss of heterozygosity on the long arms of chromosomes 13 and 14 in nasopharyngeal carcinoma in Southern China. Chin Med J 115:571–575
Zhang X, Ladd A, Dragoescu E et al (2009) MicroRNA-17-3p is a prostate tumor suppressor in vitro and in vivo, and is decreased in high grade prostate tumors analyzed by laser capture microdissection. Clin Exp Metas 26:965–979
Gong AY, Eischeid AN, Xiao J et al (2012) miR-17-5p targets the p300/CBP-associated factor and modulates androgen receptor transcriptional activity in cultured prostate cancer cells. BMC Cancer 12:492
Wangzhou K, Fu W, Li M et al (2021) microRNA-17 is a tumor suppressor in oral squamous cell carcinoma and is repressed by LSD1. Oral diseases
An X, Ma K, Zhang Z et al (2016) miR-17, miR-21, and miR-143 enhance adipogenic differentiation from porcine bone marrow-derived mesenchymal stem cells. DNA Cell Biol 35:410–416
Calura E, Pizzini S, Bisognin A et al (2016) A data-driven network model of primary myelofibrosis: transcriptional and post-transcriptional alterations in CD34+ cells. Blood Cancer J 6:439
Faraoni I, Laterza S, Ardiri D et al (2012) MiR-424 and miR-155 deregulated expression in cytogenetically normal acute myeloid leukaemia: correlation with NPM1 and FLT3 mutation status. J Hematol Oncol 5:26
Mian YA, Zeleznik-Le NJ (2016) The miR-17∼92 cluster contributes to MLL leukemia through the repression of MEIS1 competitor PKNOX1. Leuk Res 46:51–60
Meyer C, Kowarz E, Hofmann J et al (2009) New insights to the MLL recombinome of acute leukemias. Leukemia 23:1490–1499
Meyer C, Hofmann J, Burmeister T et al (2013) The MLL recombinome of acute leukemias in 2013. Leukemia 27:2165–2176
Bernard OA, Berger R (1995) Molecular basis of 11q23 rearrangements in hematopoietic malignant proliferations. Genes Chromosomes Cancer 13:75–85
Bower M, Parry P, Carter M et al (1994) Prevalence and clinical correlations of MLL gene rearrangements in AML-M4/5. Blood 84:3776–3780
Pigneux A, Labopin M, Maertens J et al (2015) Outcome of allogeneic hematopoietic stem-cell transplantation for adult patients with AML and 11q23/MLL rearrangement (MLL-r AML). Leukemia 29:2375–2381
Zotova OV, Lukianova AS, Valchuk MO et al (2021) 11q23/MLL rearrangements in adult acute leukemia. Exp Oncol 43:229–233
Donnard M, Guglielmi L, Turlure P et al (2002) Membrane and intracellular platelet-activating factor receptor expression in leukemic blasts of patients with acute myeloid and lymphoid leukemia. Stem cells (Dayton, Ohio) 20:394–401
Fiedler ERC, Bhutkar A, Lawler E et al (2018) In vivo RNAi screening identifies Pafah1b3 as a target for combination therapy with TKIs in BCR-ABL1(+) BCP-ALL. Blood Adv 2:1229–1242
da Silva IA, Chammas R, Lepique AP et al (2017) Platelet-activating factor (PAF) receptor as a promising target for cancer cell repopulation after radiotherapy. Oncogenesis 6:296
Ge F, Zhang P, Niu J et al (2020) NDRG2 and TLR7 as novel DNA methylation prognostic signatures for acute myelocytic leukemia. J Cell Physiol 235:3790–3797
Xie SZ, Kaufmann KB, Wang W et al (2021) Sphingosine-1-phosphate receptor 3 potentiates inflammatory programs in normal and leukemia stem cells to promote differentiation. Blood cancer discovery 2:32–53
Gao X, Wu T, Johnson KD et al (2016) GATA Factor-G-protein-coupled receptor circuit suppresses hematopoiesis. Stem Cell Rep 6:368–382
Hadad EH, Ahmadzadeh A, Abooali A et al (2020) Prognostic role and therapeutic susceptibility of cathepsin in various types of solid tumor and leukemia: a systematic review. J Cell Physiol 235:7709–7730
Ramage JG, Vallera DA, Black JH et al (2003) The diphtheria toxin/urokinase fusion protein (DTAT) is selectively toxic to CD87 expressing leukemic cells. Leuk Res 27:79–84
Graf M, Reif S, Hecht K et al (2005) High expression of urokinase plasminogen activator receptor (UPA-R) in acute myeloid leukemia (AML) is associated with worse prognosis. Am J Hematol 79:26–35
Pliszka M, Szablewski L (2021) Glucose transporters as a target for anticancer therapy. Cancers (Basel) 13
Liu J, Hong J, Han H et al (2020) Decreased vitamin C uptake mediated by SLC2A3 promotes leukaemia progression and impedes TET2 restoration. Br J Cancer 122:1445–1452
Funding
This study has been supported by Changzhou Sci&Tech Program (Grant No. CJ20200118, CJ20210075), Young Talent Development Plan of Changzhou Health Commission (CZQM2020023), Key Project of Medical Research of Jiangsu Provincial Health Commission (ZD2021043) and the Foundation of 333 Project of Jiangsu Province (BRA2018014).
Author information
Authors and Affiliations
Contributions
YC, YL and LS analyzed the data and drafted the manuscript. XY, HC, HY, YY, WD and YG contributed to statistical analysis. YL, WG and XZ participated in study design and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Cao, Y., Liu, Y., Shang, L. et al. Overexpression of miR-17 predicts adverse prognosis and disease recurrence for acute myeloid leukemia. Int J Clin Oncol 27, 1222–1232 (2022). https://doi.org/10.1007/s10147-022-02161-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10147-022-02161-5