
Abstract
Aneurysmal subarachnoid hemorrhage (SAH) is associated with a high mortality rate and may leave surviving patients severely disabled. After the initial hemorrhage, clinical outcome is further compromised by the occurrence of delayed cerebral ischemia (DCI). Overweight and obesity have previously been associated with protective effects in the post-bleeding phase. The aim of this study was to assess the effects of a patient’s body mass index (BMI) and leptin levels on the occurrence of DCI, DCI-related cerebral infarction, and clinical outcome. In total, 263 SAH patients were included of which leptin levels were assessed in 24 cases. BMI was recorded along disease severity documented by the Hunt and Hess and modified Fisher scales. The occurrence of clinical or functional DCI (neuromonitoring, CT Perfusion) was assessed. Long-term clinical outcome was documented after 12 months (extended Glasgow outcome scale). A total of 136 (51.7%) patients developed DCI of which 72 (27.4%) developed DCI-related cerebral infarctions. No association between BMI and DCI occurrence (P = .410) or better clinical outcome (P = .643) was identified. Early leptin concentration in serum (P = .258) and CSF (P = .159) showed no predictive value in identifying patients at risk of unfavorable outcomes. However, a significant increase of leptin levels in CSF occurred from 326.0 pg/ml IQR 171.9 prior to DCI development to 579.2 pg/ml IQR 211.9 during ongoing DCI (P = .049). In our data, no association between obesity and clinical outcome was detected. After DCI development, leptin levels in CSF increased either by an upsurge of active transport or disruption of the blood-CSF barrier. This trial has been registered at ClinicalTrials.gov (NCT02142166) as part of a larger-scale prospective data collection. BioSAB: https://clinicaltrials.gov/ct2/show/NCT02142166
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) remains a devastating disease affecting around 9/100,000 people each year [8]. After SAH, the main predictors of outcome are the initial clinical grades either defined by the Hunt and Hess (H&H) or WFNS scale and the amount of subarachnoid blood load stratified according to the modified Fisher scale (mFisher) [7, 37]. Despite general advancements in critical care management for SAH patients, outcome in those with higher clinical grade or blood load remains poor [9, 29]. The initial aneurysm rupture results in a steep increase of intracranial pressure and a sudden drop in cerebral perfusion pressure [30]. It is currently assumed that this initial increase in intracranial pressure and consecutive drop in cerebral perfusion pressure has irreversibly initiated a deleterious cascade coined with the umbrella term early brain injury.
During the first 2 weeks post-hemorrhage, patients remain susceptible to ischemic strokes in which cerebral vasospasm plays an undisputed role alongside many other contributing factors [38]. This delayed cerebral ischemia (DCI) can eventually result in cerebral infarctions, further compromising long-term clinical outcome [39].
Obesity is an established risk factor for cardio- and cerebrovascular disease, surgical complications, and nosocomial infections [2]. Contrary to this association, there is an “obesity paradox” where an increased body mass index (BMI) was associated with an overall lower mortality and complication rate, after ischemic stroke [22, 34] and intracerebral hemorrhage [5, 21]. SAH patients suffering from obesity do not fare worse or may even profit from obesity by unknown mechanisms in regard to DCI development, clinical outcome, and overall rate of complications [6, 28, 36]. A single trial in SAH patients described a lower risk of DCI and DCI-related infarction associated with elevated BMI [33]. Nonetheless, the results of a systematic review addressing the obesity paradox in SAH remained inconclusive as most trials suffered restrictions in design, resulting in limited external validity [31]. In addition, previous studies have exclusively relied on BMI as an obesity variable, even though its sensitivity for high body fat mass has been shown to be only 50% [26].
Leptin, initially discovered as a regulator of food intake and energy expenditure, is emerging as a pleiotropic molecule involved in various physiological and pathological conditions [13, 15]. Under normal physiological circumstances, this peptide has an inhibitory effect on appetite via its modulation of the hypothalamic satiety center. Leptin is, however, also part of a broader neuronal circuit regulating weight and governing energy homeostasis. Crossing the blood–brain barrier, leptin acts on receptors within the central nervous system and exerts an anti-apoptotic effect, increases neuronal survival, and can induce neurogenesis as well as angiogenesis [24]. As part of the cytokine superfamily, leptin has structural and functional similarities with pro-inflammatory cytokines such as interleukin-1, -6, and -12, hence the name adipokine. Furthermore, the leptin receptor (OB-R) is related to class I cytokine receptors, including a common signal-transducing component from the IL-6-related family of cytokines [3]. In obesity, leptin resistance develops, leading to an inability to detect satiety despite sufficient available energy stores. Serum leptin concentrations correlate positively with the percentage of body fat, illustrating the insensitivity of most people suffering from obesity, to endogenous leptin production [4]. This makes leptin an interesting potential obesity variable for assessing the obesity paradox in SAH patients. In the present study, we therefore set out to investigate the role of body fat content for DCI occurrence and long-term outcome after SAH in a prospective manner and based not only on BMI but also on the levels of leptin in cerebrospinal fluid (CSF) and serum. The hypothesis under which this study was designed was that if the obesity paradox holds true, and leptin is an easily available marker for total body fat mass, its concentrations should be inversely related to the risk of DCI and unfavorable outcomes.
Methods
Patient population and study design
Patients with aneurysmal subarachnoid hemorrhage presenting at our institution between 2010 and 2018 were screened for eligibility. This observational data collection was approved by the local ethics committee (EK 062/14) and the trial was retrospectively registered (NCT02142166). This manuscript is composed and written according to the STROBE statement for reporting of observational studies. Patients were included in case of confirmed aneurysm rupture on CT or conventional angiography, aged ≥ 18 years. Patients were excluded in case of foreseeable early mortality due to direct brain stem injury (on imaging or uni-/bilateral fixed pupils). Basic demographic data alongside BMI were recorded for each patient on admission based on weight (kilograms) and height (meters). BMI was categorized according the WHO criteria [42] with the addition of an obesity subdivision resulting in five categories: underweight (< 18.5 kg/m2), normal weight (≥ 18.5 to < 25 kg/m2), overweight (≥ 25 to < 30 kg/m2), obesity grade 1 (≥ 30 to < 35 kg/m2), and obesity grade 2 (≥ 35 kg/m2). This subdivision was added to allow a more precise risk stratification. Initial clinical grade based on the Hunt and Hess grading scale and the amount of subarachnoid blood on CT scanning according to the modified Fisher scale were noted. Additionally, the length of ICU stay and of ventilation was documented for each patient.
Cerebral infarctions diagnosed during DCI (as defined below) or as the first sign of ongoing DCI were registered as DCI-related infarction. Embolic stroke due to halted anticoagulation in patients with cardiac arrhythmias or as a result of surgical or endovascular aneurysm treatment or endovascular DCI rescue treatment was identified based on history and presentation on imaging and excluded. Cases in which multiple DCI-related infarctions triggered withdrawal of technical life-support were labeled as DCI-related mortality. Clinical outcome was assessed by the extended Glasgow Outcome Scale (GOSE) after 12 months by an independent assessor via a structured telephone interview with the patient, his or her next-of-kin or caretaker. Patients were excluded in case of missing long-term outcome data.
Patient management
All patients were treated according to a standardized treatment protocol consistent over the inclusion period. [1, 43] Aneurysms were secured via endovascular coiling or surgical clipping within 48 h after admission, after which patients were monitored in a neurosurgical intensive care unit. All patients were routinely treated with oral nimodipine in a 6 × 60 mg/day dose. DCI occurrence was either determined by clinical deterioration not attributable to other causes (i.e., hydrocephalus, electrolyte imbalance, seizure, and infection) [40], or functional deterioration defined by a new CT perfusion deficit. Oxygenation (ptiO2 < 10 mmHg or ≥ 10 mmHg with continuous decrease) or metabolic crisis (lactate/pyruvate ratio ≥ 40 or < 40 with continuous increase) as measured by invasive neuromonitoring probes (Neurovent PTO®, Raumedic®, Helmbrechts, Germany, and 71 High Cut-Off Brain Microdialysis Catheter, μdialysis®, Stockholm, Sweden) were indicators to perform CT perfusion imaging. First-tier treatment was initiated after diagnosis of DCI and consisted of induced euvolemic arterial hypertension (≥ 180 mmHg) by means of intravenous noradrenaline infusion. In refractory cases, second-tier endovascular rescue treatment was considered by either transluminal balloon-angioplasty or continuous intra-arterial spasmolysis [43]. Nutrition was administered according to the guidelines by the European Society for Clinical Nutrition and Metabolism (ESPEN) for ICU patients [35].
Data collection and subgroup analyses
From patients treated between 2014 and 2016, a sample subgroup of 24 patients was selected and included to be representative for the entire cohort based on age, gender, H&H, and mFisher, with the added prerequisite of having an external ventricular drainage (EVD) in place for cerebrospinal fluid (CSF) collection. Leptin levels were determined in serum and CSF samples during three a priori defined sampling periods: early (d0-3), pre-DCI (measurement 2–4 days prior to DCI diagnosis), and during ongoing DCI. Systemic (ng/ml) and CSF leptin (pg/ml) levels were collected once in every epoch, for each individual patient, resulting in three sample pairs (serum and CSF) per patient. Sampling was done in the afternoon (between 12:00 and 17:00) together with routine blood drawing and leptin concentrations were measured in CSF and plasma by means of a sandwich enzyme-linked immune-assay (Human Leptin Kit, Meso Scale Discovery®, Rockville, USA).
Pairs of patient subgroups were defined based on dichotomization of the initial Hunt and Hess clinical grading (good grade H&H1-2 vs. poor grade H&H3-5), modified Fisher scale (mFisher1-2 vs. mFisher3-4), and clinical outcome (unfavorable: GOSE1-4 vs. favorable: GOSE5-8). Additional subgroups were created based on the occurrence of DCI (noDCI vs. DCI) and DCI-related cerebral infarction (no infarction vs. infarction). An analysis of leptin levels before, during, and after DCI together with the ratio of leptin CSF to plasma concentration, was conducted.
Outcome definition
The primary endpoint was defined as the outcome assessed by the extended Glasgow outcome scale (GOSE) after 12 months. The GOSE was dichotomized as conventional, into unfavorable (GOSE1-4) and favorable (GOSE5-8) outcome. Additional outcome parameters were the association between BMI or leptin levels and DCI occurrence, the occurrence of refractory DCI (necessitating endovascular treatment), DCI-related infarction, DCI-related mortality, and overall mortality.
Statistical analysis
Descriptive statistics are represented as mean and standard deviation for continuous variables unless non-normally distributed in which case median and interquartile range is provided. Categorical data is depicted as frequencies and percentages. Normality testing was done via plotting and the Shapiro–Wilk test. Parametric continuous data were tested using T-tests and for non-parametric variables using the Mann–Whitney U test. Paired continuous non-parametric data were analyzed via the Wilcoxon signed-rank test, and categorical data were compared using Pearson’s χ2 test. To assess the predictive value of leptin levels on DCI and outcome, a receiver operating characteristics (ROC) analysis with calculation of the area under the curve (AUC) was performed. To ascertain the correlation between BMI and leptin levels in both compartments in this cohort, the Spearman correlation coefficient was calculated. The alpha significant level was set at a two-sided P value below .05, and all statistical analyses were performed using SPSS v. 25 (IBM, Chicago, IL). Illustrations were created with GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Participants
A total of 305 admitted SAH patients were analyzed for inclusion between 2010 and 2018. In 37 cases, long-term clinical outcome was missing due to a loss in follow-up. Five patients were excluded because of relevant prior comorbidity. The recruitment process is illustrated in Fig. 1. The mean age of the remaining 263 patients was 53.9 ± 12.7 years with a female to male ratio of 192 (73.0%) to 71 (27.0%). The aneurysm was secured with surgical clipping in 119 (45.2%) patients and via endovascular access in 141 (53.6%) cases. Three patients received a combination of both occlusion techniques. The median BMI was 24.8 ± 5.4 kg/m2. Five patients were underweight (1.9%), 133 presented with a normal weight (50.6%), 90 were overweight (34.2%), 25 were classified as obesity grade 1 (9.5%), and 10 as obesity grade 2 (3.8%). No significant differences in outcome-relevant baseline characteristics such as H&H and mFisher grading or aneurysm treatment modality were observed between BMI categories (Suppl. Table 1).

Flowchart of patient recruitment. BMI was categorized according the WHO criteria with the addition of an obesity subdivision resulting in five categories: underweight (< 18.5 kg/m2), normal weight (≥ 18.5 to < 25 kg/m2), overweight (≥ 25 to < 30 kg/m2), obesity grade 1 (≥ 30 to < 35 kg/m2), and obesity grade 2 (≥ 35 kg/m2). SAH, subarachnoid hemorrhage
A total of 136 (51.7%) patients developed DCI of which 72 (27.4%) developed DCI-related cerebral infarctions. DCI was the direct cause of death in 27 (10.3%) patients. A favorable outcome was achieved after 12 months in 162 (61.6%) cases. For the analysis of serum and CSF concentrations of leptin, 24 patients were selected from this cohort. Although small, this subgroup proved for the main outcome relevant baseline characteristics comparable to the entire cohort, apart from age. Patients in the subgroup with serum/CSF leptin analyses were on average 5.4 years older (P = 0.018). All relevant baseline characteristics are depicted in Table 1.
BMI, DCI, and outcome
When comparing BMI in subgroups based on DCI occurrence (no DCI = 25.2 kg/m2 ± 4.4 vs. DCI = 26.0 kg/m2 ± 5.1; P = .095), DCI-related infarction (no infarction: 25.7 kg/m2 ± 4.9 vs. infarction: 25.5 kg/m2 ± 4.6; P = .522), and overall mortality (survivors: 25.6 kg/m2 ± 4.8 vs. died: 25.7 kg/m2 ± 4.9; P = .652), no significant differences were identified. Similarly, non-significant results were obtained when comparing patients reaching unfavorable (GOSE1-4) or favorable outcome (GOSE5-8), and between outcome groups, the mean BMI for both was almost identical (GOSE1-4: 24.8 kg/m2 ± 5.6 vs. GOSE5-8: 24.9 kg/m2 ± 5.2; P = .995) (Table 2). The incidence of DCI varied between BMI categories between 40.0 and 80.0%. No significant differences were noted between obesity categories regarding incidence of DCI χ2 (4, 263) = 3.969, P = .410; DCI-related infarction χ2 (4, 263) = 3.473, P = .301 or DCI-related mortality χ2 (4, 263) = 2.983; P = .304. Even in the extreme BMI categories, favorable outcome was reached in three out of five underweight patients and in five out of ten severely obese patients (Table 3).
Early leptin levels
There was a statistically significant positive correlation between early serum leptin levels and BMI, (rs(22) = 0.503; P = .012) as well as early CSF leptin levels and BMI (rs(22) = 0.766; P < .001). In a ROC analysis, early leptin concentration in serum (AUC = 0.662; 95% CI: 0.378 to 0.947, P = .258) and CSF (AUC = 0.712; 95% CI: 0.454 to 0.971; P = .159) showed no predictive value in identifying patients at risk of unfavorable outcome. Similarly, early leptin levels in serum (AUC = 0.688; 95% CI: 0.419 to 0.956; P = .137) and CSF (AUC = 0.571; 95% CI: 0.263 to 0.880; P = .626) were not associated with later DCI occurrence.
Leptin levels around DCI
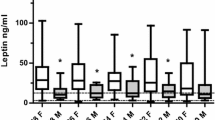
Of the 14 patients who developed DCI, leptin levels in serum were compared before and during DCI occurrence. There was a non-significant increase of systemic concentrations in patients developing DCI from a median level of 5.97 ng/ml IQR 11.4 to 7.22 IQR 16.3 ng/ml (P = .410). In CSF, a statistically significant increase of leptin levels from 326.0 pg/ml IQR 171.90 to 579.2 pg/ml IQR 211.9 (P = .049) was observed. The comparison of both time points is depicted in Fig. 2.

Comparison of leptin levels in serum (a) and CSF (b) before and after the occurrence of DCI (n = 14). As pre-DCI, leptin measurements available between 2 and 4 days prior to the initial DCI diagnosis were used. A statistically significant increase of leptin levels in CSF from 326.0 pg/ml IQR 171.90 to 579.2 pg/ml IQR 211.9 (P = .049) was observed. CSF, cerebrospinal fluid; DCI, delayed cerebral ischemia; pre-DCI, measurement 2–4 days prior to DCI diagnosis
Discussion
This observational study was conducted to assess whether obesity, presented by a higher BMI and higher systemic leptin levels, is protective against DCI and results in better clinical outcomes. Our results illustrate no such association neither between BMI—either as a continuous or categorized variable—nor leptin concentrations, with DCI or clinical outcome.
The obesity paradox concerning clinical outcome in SAH emerged on the basis of oftentimes inconclusive studies. The two largest studies were based on patient registries yielding considerable case numbers but came to diverging results with one trial documenting reduced in-hospital mortality and the other rejecting the obesity paradox [6, 10]. The methodological integrity of these trials was critically addressed in a recent systematic review [31]. The majority of trials so far have focused on early in-hospital mortality independent of the cause or relation to DCI. In this trial, a rigorous clinical and multimodal definition was applied surpassing mere angiographic vasospasm, as in previous studies. Our results did not identify a beneficial effect of higher body fat mass against DCI occurrence. It remains however conceivable that a protective effect of mild obesity exists—unspecific to SAH or stroke—potentially related to overall caloric reserves which may prove advantageous in the light of long ICU stays [21, 27]. For example, overweight defined by a body mass index between 25 and 30 is associated with lower mortality rates in patients with severe sepsis [41]. This effect was however not detected in our cohort.
A positive influence of obesity on the occurrence of DCI and hereby caused cerebral infarction was however observed in prior research. Rinaldo et al. reported a significant reduction of DCI in patients with higher BMI and postulated a possible neuroprotective effect of leptin [33]. These data served as an incentive to substantiate the correlation of BMI and DCI with systemic and CSF levels of leptin as the possible mediator of the proposed positive effect of obesity [32]. Although a control group with healthy subjects was not available, measured leptin concentration in SAH patients transcended, in any of the evaluated time frames, the proposed normal reference range [14]. To our knowledge, there have been four previous trials assessing human leptin levels after SAH, all focusing on samples taken during the first post-hemorrhage days [11, 16, 23, 25]. All trials consistently demonstrated higher baseline leptin serum levels compared to healthy individuals. Fan et al. were the first to assess serum leptin levels in SAH patients in comparison to healthy volunteers [11]. Leptin levels proved higher in patients who suffered SAH, and the height of leptin concentrations was positively correlated with the severity of disease as measured by the WFNS scale. In another observational study investigating 96 women, leptin levels remained independent of the initial clinical grade as measured by the H&H scale but gender inequalities in leptin levels may have led to bias in this investigation [16]. After SAH, serum leptin levels increase rapidly to peak after 24 h. Thereafter, the serum concentrations decrease gradually but remain substantially higher compared to healthy controls during the following 7 days [25]. This further illustrates the systemic nature of SAH as a disease where multiple organ systems become involved. However, leptin levels were not predictive of DCI or clinical outcome in our cohort.
Notably, DCI itself might affect the concentrations of leptin in CSF, which were higher in patients after onset of DCI. As there are no indications of leptin production within the central nervous system, CSF levels are expected to be the result of transport across the blood–brain or blood-CSF barrier. Increased levels during DCI may reflect a passive accumulation of leptin mediated by ischemic injury to the blood-CSF barrier. As leptin has been proposed to also possess an immunomodulatory function, some role of leptin in the progression or internal compensatory mechanisms within the inflammatory cascade initiate by ischemic injury cannot be excluded but would certainly warrant further studies once the complex DCI cascade is better understood [3, 4, 18].
Limitations
In common with most clinical SAH studies, the most important limitation of our study is the small number of patients that could be recruited. Thus, even though the sample size was comparable to or even exceeds that of many previous studies published in this context [17, 19, 20, 33], the deleterious effect of underweight or severe obesity might have been underestimated due to the limited number of patients in these categories and caution is warranted to extrapolate our results to more extreme BMI values. Even more importantly, the small overall number of patients means that, like in most previous studies, our analysis was notably underpowered and thus unable to detect small effects of obesity. Our findings should therefore only be regarded as evidence for the lack of a strong association between BMI or leptin levels and the development of DCI. As exemplified in a recent systematic review [31], a much larger number of patients in all obesity categories would be required to detect more subtle protective effects of body fat mass in SAH patients. Also, leptin is a hormone susceptible to diurnal variations related to feelings of hunger and satiety. These fluctuations have not been described in ICU patients and our results are based on a single daily measurement. A further limitation is the handpicked inclusion of patients in which leptin levels were assessed. As this was not performed at random, a selection bias might be present even if its character is unknown. The sample was selected to reflect the baseline characteristics of the entire cohort, but the number of patients remains fairly small increasing the chance of an error of the second kind. The difference identified between leptin levels before and after DCI was only marginally significant and, taking into account that we did not correct for the increased error probability due to multiple comparisons, is unlikely to represent a truly significant difference. Finally, as with other regulators of the inflammatory response, leptin function may be modulated by local leptin concentration, the ratio between free and bound leptin, the expression of different forms of the receptors, the ratio between signaling and non-signaling receptors, and the presence of specific inhibitors [12].
Conclusion
In this analysis, obesity or increased BMI was not associated with a reduced rate of DCI and better clinical outcomes. Based on these results, obesity seems not to play a relevant role in producing better or worse clinical outcomes. Similarly, early leptin levels do not differ between patients developing DCI or not. After developing DCI, leptin levels in CSF increase either by active transport or disruption of the blood-CSF barrier.
Data availability
The raw data of this analysis can be made available by the authors to any qualified researcher.
Code availability
Not applicable.
References
Albanna W, Weiss M, Müller M, Brockmann MA, Rieg A, Conzen C, Clusmann H, Höllig A, Schubert GA (2017) Endovascular rescue therapies for refractory vasospasm after subarachnoid hemorrhage: a prospective evaluation study using multimodal, continuous event neuromonitoring. Neurosurgery 80(6):942–949
Anzueto A, Frutos-Vivar F, Esteban A, Bensalami N, Marks D, Raymondos K, Apezteguia C, Arabi Y, Hurtado J, Gonzalez M, Tomicic V, Abroug F, Elizalde J, Cakar N, Pelosi P, Ferguson ND (2011) Influence of body mass index on outcome of the mechanically ventilated patients. Thorax 66(1):66–73
Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, Lai CF, Tartaglia LA (1996) The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci USA 93(16):8374–8378
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL et al (1996) Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 334(5):292–295
Dangayach NS, Grewal HS, De Marchis GM, Sefcik RK, Bruce R, Chhatlani A, Connolly ES, Falo MC, Agarwal S, Claassen J, Schmidt JM, Mayer SA (2018) Does the obesity paradox predict functional outcome in intracerebral hemorrhage? J Neurosurg 129(5):1125–1129
Dasenbrock HH, Nguyen MO, Frerichs KU, Guttieres D, Gormley WB, Ali Aziz-Sultan M, Du R (2017) The impact of body habitus on outcomes after aneurysmal subarachnoid hemorrhage: a Nationwide Inpatient Sample analysis. J Neurosurg 127(1):36–46
de Oliveira MAL, Jaja BN, Germans MR, Yan H, Qian W, Kouzmina E, Marotta TR, Turkel-Parrella D, Schweizer TA, Macdonald RL (2015) The VASOGRADE: a simple grading scale for prediction of delayed cerebral ischemia after subarachnoid hemorrhage. Stroke 46(7):1826–1831
de Rooij NK, Linn FH, van der Plas JA, Algra A, Rinkel GJ (2007) Incidence of subarachnoid haemorrhage: a systematic review with emphasis on region, age, gender and time trends. J Neurol Neurosurg Psychiatry 78(12):1365–1372
Dijkland S A, Jaja B N R, van der Jagt M, Roozenbeek B, Vergouwen M D I, Suarez J I, Torner J C, Todd M M, van den Bergh W M, Saposnik G, Zumofen D W, Cusimano M D, Mayer S A, Lo B W Y, Steyerberg E W, Dippel D W J, Schweizer T A, Macdonald R L, Lingsma H F. (2019). Between-center and between-country differences in outcome after aneurysmal subarachnoid hemorrhage in the Subarachnoid Hemorrhage International Trialists (SAHIT) repository. J Neurosurg 1–9
Elliott RS, Godoy DA, Michalek JE, Behrouz R, Elsehety MA, Hafeez S, Rios D, Seifi A (2017) The effect of morbid obesity on subarachnoid hemorrhage prognosis in the United States. World Neurosurg 105:732–736
Fan XF, Chen ZH, Huang Q, Dai WM, Jie YQ, Yu GF, Wu A, Yan XJ, Li YP (2013) Leptin as a marker for severity and prognosis of aneurysmal subarachnoid hemorrhage. Peptides 48:70–74
Fantuzzi G, Faggioni R (2000) Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol 68(4):437–446
Friedman JM, Halaas JL (1998) Leptin and the regulation of body weight in mammals. Nature 395(6704):763–770
Gijon-Conde T, Graciani A, Guallar-Castillon P, Aguilera MT, Rodriguez-Artalejo F, Banegas JR (2015) Leptin reference values and cutoffs for identifying cardiometabolic abnormalities in the spanish population. Revistaespanola de cardiologia (English ed) 68(8):672–679
Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM (1995) Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269(5223):543–6
Huang WJ, Chen WW, Zhang X (2014) Characteristic changes in estradiol and leptin levels in patients with subarachnoid hemorrhage induced cerebral-cardiac syndrome. Eur Rev Med Pharmacol Sci 18(24):3954–3958
Hughes JD, Samarage M, Burrows AM, Lanzino G, Rabinstein AA (2015) Body mass index and aneurysmal subarachnoid hemorrhage: decreasing mortality with increasing body mass index. World Neurosurg 84(6):1598–1604
Janik JE, Curti BD, Considine RV, Rager HC, Powers GC, Alvord WG, Smith JW 2nd, Gause BL, Kopp WC (1997) Interleukin 1 alpha increases serum leptin concentrations in humans. J Clin Endocrinol Metab 82(9):3084–3086
Juvela S, Siironen J, Kuhmonen J (2005) Hyperglycemia, excess weight, and history of hypertension as risk factors for poor outcome and cerebral infarction after aneurysmal subarachnoid hemorrhage. J Neurosurg 102(6):998–1003
Kagerbauer SM, Kemptner DM, Schepp CP, Bele S, Rothorl RD, Brawanski AT, Schebesch KM (2010) Elevated premorbid body mass index is not associated with poor neurological outcome in the subacute state after aneurysmal subarachnoid hemorrhage. Cent Eur Neurosurg 71(4):163–166
Kim BJ, Lee SH, Ryu WS, Kim CK, Lee J, Yoon BW (2011) Paradoxical longevity in obese patients with intracerebral hemorrhage. Neurology 76(6):567–573
Kim Y, Kim CK, Jung S, Yoon BW, Lee SH (2015) Obesity-stroke paradox and initial neurological severity. J Neurol Neurosurg Psychiatry 86(7):743–747
Kubo Y, Koji T, Kashimura H, Otawara Y, Ogawa A, Ogasawara K (2014) Appetite loss may be induced by lower serum ghrelin and higher serum leptin concentrations in subarachnoid hemorrhage patients. Nutr Neurosci 17(5):230–233
Lin C, Huang SJ, Wang N, Shen ZP (2012) Relationship between plasma leptin levels and clinical outcomes of pediatric traumatic brain injury. Peptides 35(2):166–171
Lindgren C, Naredi S, Söderberg S, Koskinen LO, Hultin M (2016) Leptin levels after subarachnoid haemorrhage are gender dependent. Springerplus 5(1):667
Okorodudu DO, Jumean MF, Montori VM, Romero-Corral A, Somers VK, Erwin PJ, Lopez-Jimenez F (2010) Diagnostic performance of body mass index to identify obesity as defined by body adiposity: a systematic review and meta-analysis. Int J Obes (Lond) 34(5):791–799
Olsen TS, Dehlendorff C, Petersen HG, Andersen KK (2008) Body mass index and poststroke mortality. Neuroepidemiology 30(2):93–100
Platz J, Guresir E, Schuss P, Konczalla J, Seifert V, Vatter H (2013) The impact of the body mass index on outcome after subarachnoid hemorrhage: is there an obesity paradox in SAH? A retrospective analysis. Neurosurgery 73(2):201–208
Pobereskin LH (2001) Incidence and outcome of subarachnoid haemorrhage: a retrospective population based study. J NeurolNeurosurg Psychiatry 70(3):340–343
Rass V, Helbok R (2019) Early brain injury after poor-grade subarachnoid hemorrhage. CurrNeurolNeurosci Rep 19(10):78
Rautalin I, Kaprio J, Korja M (2020) Obesity paradox in subarachnoid hemorrhage: a systematic review. Neurosurg Rev 43(6):1555–1563
Rinaldo L, Rabinstein A A, Lanzino G, In Reply: Increased body mass index associated with reduced risk of delayed cerebral ischemia and subsequent infarction after aneurysmal subarachnoid hemorrhage, Neurosurgery (2018).
Rinaldo L, Rabinstein AA, Lanzino G (2019) Increased body mass index associated with reduced risk of delayed cerebral ischemia and subsequent infarction after aneurysmal subarachnoid hemorrhage. Neurosurgery 84(5):1035–1042
Ryu WS, Lee SH, Kim CK, Kim BJ, Yoon BW (2011) Body mass index, initial neurological severity and long-term mortality in ischemic stroke. Cerebrovascular diseases (Basel, Switzerland) 32(2):170–176
Singer P, Blaser AR, Berger MM, Alhazzani W, Calder PC, Casaer MP, Hiesmayr M, Mayer K, Montejo JC, Pichard C, Preiser JC, van Zanten ARH, Oczkowski S, Szczeklik W, Bischoff SC (2019) ESPEN guideline on clinical nutrition in the intensive care unit. Clinical nutrition (Edinburgh, Scotland) 38(1):48–79
Tawk RG, Grewal SS, Heckman MG, Navarro R, Ferguson JL, Starke EL, Rawal B, Hanel R, Miller D, Wharen RE, Freeman WD (2015) Influence of body mass index and age on functional outcomes in patients with subarachnoid hemorrhage. Neurosurgery 76(2):136–141
van Donkelaar CE, Bakker NA, Birks J, Veeger N, Metzemaekers JDM, Molyneux AJ, Groen RJM, van Dijk JMC (2019) Prediction of outcome after aneurysmal subarachnoid hemorrhage. Stroke 50(4):837–844
Vergouwen MD, Etminan N, Ilodigwe D, Macdonald RL (2011) Lower incidence of cerebral infarction correlates with improved functional outcome after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab 31(7):1545–1553
Vergouwen MD, Ilodigwe D, Macdonald RL (2011) Cerebral infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke J Cereb Circ 42(4):924–929
Vergouwen MD, Vermeulen M, van Gijn J, Rinkel GJ, Wijdicks EF, Muizelaar JP, Mendelow AD, Juvela S, Yonas H, Terbrugge KG, Macdonald RL, Diringer MN, Broderick JP, Dreier JP, Roos YB (2010) Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke J Cereb Circ 41(10):2391–2395
Wang S, Liu X, Chen Q, Liu C, Huang C, Fang X (2017) The role of increased body mass index in outcomes of sepsis: a systematic review and meta-analysis. BMC Anesthesiol 17(1):118
Weir CB, Jan A (2020) BMI classification percentile and cut off points, StatPearls, StatPearls Publishing Copyright © 2020. StatPearls Publishing LLC., Treasure Island (FL)
Weiss M, Conzen C, Mueller M, Wiesmann M, Clusmann H, Albanna W, Schubert GA (2019) Endovascular rescue treatment for delayed cerebral ischemia after subarachnoid hemorrhage is safe and effective. Front Neurol 10:136
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the START-Program of the Faculty of Medicine, RWTH Aachen, Germany, grant number 691540.
Author information
Authors and Affiliations
Contributions
The design and conception of this trial were developed by MV, MW, and WA. MV, MW, and WA were involved in data acquisition. All authors were involved in the interpretation of data. The manuscript was drafted by MV, MW, and WA, and illustrations were created by MV. The final manuscript was critically revised and approved by all authors.
Corresponding author
Ethics declarations
Ethical statement
The trial was conducted in accordance with the recommendations of the ethics committee of the Medical Faculty of the RWTH Aachen University. (EK 062/14).
Consent to participate
All patients or their legal representatives provided written informed consent.
Consent for publication
All authors agree.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Table 1
Comparison of baseline characteristics between BMI categories. AC = anterior circulation; BMI = body mass index; DM2 = type 2 diabetes; mFisher = modified Fisher scale; PC = posterior circulation; SD = standard deviation. *Three patients were treated with a combination of endovascular occlusion and surgical clipping. (DOCX 15 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Veldeman, M., Weiss, M., Simon, T.P. et al. Body mass index and leptin levels in serum and cerebrospinal fluid in relation to delayed cerebral ischemia and outcome after aneurysmal subarachnoid hemorrhage. Neurosurg Rev 44, 3547–3556 (2021). https://doi.org/10.1007/s10143-021-01541-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10143-021-01541-1