
Abstract
Rice blast, caused by the fungus Magnaporthe oryzae, is a highly damaging disease. Introducing genes, which confer a broad spectrum resistance to the disease, such as Pib, makes an important contribution to protecting rice production. However, little is known regarding the mechanistic basis of the products of such genes. In this study, transcriptome of the cultivar Lijiangxintuanheigu (LTH) and its monogenic IRBLb-B which harbors Pib treated with M. oryzae were compared. Among the many genes responding transcriptionally to infection were some encoding products involved in the metabolism of ROS (reactive oxygen species), in jasmonate (JA) metabolism, and WRKY transcription factors, receptor kinases, and resistance response signal modulation. The down-regulation of genes encoding peroxiredoxin and glutathione S transferases implied that the redox homeostasis is essential for the expression of Pib-mediated resistance. The up-regulation of seven disease resistance-related genes, including three encoding a NBS-LRR protein, indicated that disease resistance-related genes are likely tend to support the expression of Pib resistance. These data revealed that potential candidate genes and transcriptional reprogramming were involved in Pib-mediated resistance mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice is a staple food for half of the world’s population. Like all crops, its production is constrained by a number of both biotic and abiotic stresses. Among the various stresses, rice blast, which is caused by fungal pathogen Magnaporthe oryzae, is the most devastating disease in rice production. It occurs almost in all rice planting areas, generally resulting in rice yield loss of 10 to 30% annually (Dean et al. 2012). An increase in understanding the molecular mechanisms of the rice–M. oryzae interaction may be useful to develop novel strategies for blast control.
To resist pathogen invasion, plants have developed a complex system of defense against microbial pathogens such as M. oryzae: the first layer involves pattern recognition receptors (PRRs), which trigger immunity after they recognize the presence of the pathogen-associated molecular patterns (PAMPs); the second layer is effectors trigger immunity (ETI), which relies on resistance (R) proteins recognize pathogen avirulence factors, after which a hypersensitive response (HR) is rapidly mounted (Sakulkoo et al. 2018). The latter mode of resistance has proven to be a highly effective means of endowing rice cultivars with immunity against blast (Fernandez and Orth 2018). To date, more than 100 rice R genes have been mapped and 35 have been isolated (Wang et al. 2017). Most of these R genes have been found to encode proteins belonging to the nucleotide-binding site leucine-rich repeat (NBS-LRR) family; one of these is Pib, carriers of which are resistant to a lot of the M. oryzae strains found in Japan and some of those found in China. As a result, Pib is considered to give broad spectrum resistance (Ramkumar et al. 2015; Wang et al. 1999; Zhang et al. 2018b), and so has been used by breeders (Fjellstrom et al. 2004; Koide et al. 2010; Singh et al. 2015; Tanweer et al. 2015). Until now, it is unclear how Pib-mediated resistance is affected.
An effective way of gauging the effect of a single gene is to base comparisons on pairs of so-called near-isogenic lines (NILs), which, as a result of the back-crossing procedure used for their construction, differ from one another with respect to the target gene and just a small number of other genes (Fukuoka et al. 2015; Yuan et al. 2017). Thus, for example, transcriptomic comparisons between the Chinese cultivar LTH with its Pi1 NIL IRBL18 and Pi9 NIL IRBL22 have shown that a particular class of transcription factor is important for the expression of resistance determined by both resistance genes (Wei et al. 2013). Similarly, Jain et al. (2017) have been able to describe the set of genes which act to ensure the blast resistance determined by Pi9 (Jain et al. 2017).
Here, similar strategy was directed at understanding the mechanistic basis of Pib-mediated resistance. In this study, the relevant NIL in a background of LTH was the line IRBLb-B; IRBLb-B (carrying Pib) is obtained from the Pib resistance donor parent BL1 crossed with recurrent parent LTH (Telebanco-Yanoria et al. 2010). The intention was to reveal the transcriptional differences between LTH and IRBLb-B when the plants were infected with M. oryzae. We expect that our results will improve the understanding of mechanisms of Pib-mediated broad-spectrum resistance against M. oryzae and facilitate breeding broad-spectrum resistance varieties.
Materials and methods
Plant materials and growing conditions
Grains of the japonica type rice cultivar LTH and IRBLb-B were obtained from the China National Rice Research Institute’s National Mid-term Genebank. The oslox3#KO was generated by CRISPR/cas9 system as described previously (Ma et al. 2015). The vector was transformed into XH11 (Japonica) callus using the Agrobacterium-mediated transformation method. The grains were surface-sterilized by immersion in 70% ethanol for 1 min, after which they were imbibed at 30 °C for 2 days and then allowed to germinate in moist towels at 37 °C for 24 h, transplanted the same development seedlings to sterile soil. The resulting seedlings were grown for 21 days in greenhouse soil providing a 14-h photoperiod and a constant temperature of 28 °C.
The presence of the effective allele at Pib in IRBLb-B and of the ineffective one in LTH was validated using a PCR assay based on, respectively, the primer pairs Pibdom F/R and Lys145 F/R, as described (Fjellstrom et al. 2004); the relevant primer sequences were given in Table S1.
Fungal inoculation and RNA isolation
M. oryzae strain P131 was cultured on prune agar medium as described (Kou et al. 2017). Conidia were collected from the 7-day-old-culture by rinsing with sterile Milli-Q water and filtering through two layers of sterile miracloth (Millipore, Burlington, MA, USA). For the purpose of investigating the histological reaction of LTH or IRBLb-B plants to M. oryzae infection, M. oryzae conidial suspension of 105 per mL was used to inoculate 5-cm lengths of leaf sheath cut from each of the two host genotypes, and the explants were maintained at 28 °C for either 26 h or 36 h. To generate samples for the purpose of RNA-seq analysis, 3-week-old seedlings were sprayed with a suspension of 106 M. oryzae conidia per mL in water containing 0.01% w/v gelatin; the seedlings were kept in a dark humid chamber overnight at 22 °C, then transferred into a growth chamber providing a 16-h photoperiod and a constant temperature of 22 °C with 90% relative humidity. The spray used for mock inoculation was an aqueous solution of 0.01% w/v gelatin.
RNA was extracted from secondary leaves (three replicates per genotype) exposed to M. oryzae for 36 h; the harvested leaves were snap-frozen in liquid nitrogen, and their RNA was extracted using the TRIzol reagent according to the manufacturer’s (Invitrogen, Carlsbad, CA, USA) protocol. Genomic DNA was digested from the extracts using DNase I (TaKaRa, Dalian, China); the integrity of the RNA in the extracts was checked using a 2100 Bioanalyzer device (Agilent, Santa Clara, CA, USA) and its concentration quantified using a ND-2000 device (NanoDrop Technologies, www.thermofisher.com). The thresholds applied for accepting the samples for the RNA-seq procedure were OD260/280 1.8–2.2, OD260/230 ≥ 2.0, RIN ≥ 6.5, and 28S:18S ≥ 1.0.
Library preparation and sequencing
Each transcriptome library was constructed from an ~ 1-μg aliquot of total RNA and was processed following the protocol provided with a TruSeqTM RNA sample preparation kit (Illumina, Inc., San Diego, CA). The libraries were size selected (200–300 bp) by agarose gel electrophoresis and were then PCR amplified over 15 cycles using Phusion DNA polymerase (NEB, Ipswich, MA, USA). After quantified by TBS380, paired-end sequencing was carried out using a HiSeq 4000 device (Illumina) operated by the Majorbio Genome Center (Shanghai, China).
Differential transcription and functional enrichment analysis
The SeqPrep (github.com/jstjohn/SeqPrep) and Sickle (github.com/najoshi/sickle) software packages were used to control the quality of the raw reads, after which the reads were mapped onto the reference genome sequence of cv. Nipponbare (MSU release 7) using TopHat software (http://ccb.jhu.edu/software/tophat/downloads/) imposing a mismatch threshold of two nucleotides. To identify significant differential transcription between a pair of samples, transcript abundances were calculated using the “fragments per kilobase of exon per million mapped reads” method. A false discovery rate (FDR) threshold of 0.05, in conjunction with an absolute log2 fold change of 2, was imposed to call a gene significantly differentially expressed (hereafter referred to as SDEGs).
A functional enrichment analysis was applied to the full set of SDEGs, based on the Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases taking advantage of the software tools Goatools (github.com/tanghaibao/Goatools) and KOBAS (kobas.cbi.pku.edu.cn/home.do). A heatmap was constructed using the cluster method, based on an in-house Perl script. The absolute log2 fold change data were used in Mapman software to get the Mapman view of biotic stress.
Validation of differential transcription using quantitative real time PCR
Quantitative real time PCR (qRT-PCR) was used to validate differential transcription concluded from the RNA-seq data of twenty four of the SDEGs identified in the contrast IRBLb-B non-infected vs IRBLb-B infected with M. oryzae. Relevant PCR primers were designed using the GenScript Real-time PCR Primer Design tool (www.genscript.com/tools/real-time-pcr-tagman-primer-design-tool) and are shown in Table S1. The template for each qRT-PCR was a preparation of cDNA synthesized from total RNA by M-MLV reverse transcriptase (TaKaRa) and diluted 1:5 with RNAase free water to give a concentration of ~ 20 ng/μL. Each 10 μL reaction contained 5 μL TB Green Master (TaKaRa), 0.2 μL of each primer (10 mM), 2 μL templates, and 2.6 μL RNAase-free water. The sequence of the rice Tubulin gene (LOC_Os03g13170) was used as the reference (Liao et al. 2019). Relative transcript abundances were calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001).
DAB and Trypan staining
The DAB (3, 3′-Diaminobenzidine) and Trypan were performed, as previously described (Zhu et al. 2013). The relative hydrogen peroxide level by ImageJ software (https://imagej.nih.gov/ij/), Briefly, the picture was reversed, and the coloring points turn to white. Then, the gray value of the area to be measured is determined. The relative hydrogen peroxide value was calculated according to the gray value. The LTH of 24 hpi was used as the reference.
Results
The response of LTH and IRBLb-B to M. oryzae infection
To verify the sequence identity of the Pib alleles present in IRBLb-B and LTH, the PCR assay along with DNA sequencing directed at Pib locus confirmed that IRBLb-B harbors the allele conferring blast resistance (Fig. 1a). When rice seedlings were inoculated with M. oryzae strain P131, LTH samples developed extensive lesions (Fig. 1b), while the IRBLb-B samples showed no reaction (Fig. 1b). We further investigated the penetrate pattern of M. oryzae in Pib-mediated incompatible interaction by rice sheath infection assay. M. oryzae appressoria were able to penetrate the sheath of both host genotypes by 26-hour post inoculation (hpi) (Fig. 1c). By 36 hpi, invasive hyphae had extended into the cells surrounding the penetration point in LTH, but in IRBLb-B, invasive hyphal growth was limited. In addition, the abundance of Pib transcript (LOC_Os02g57310) at 36 hpi was almost nine fold greater in the leaves of IRBLb-B inoculated with M. oryzae than mock (Fig. 1d). These results indicated that Pib expression was induced by rice blast infection and Pib resistance on fungal growth was detectible by 36 hpi.

The response of LTH and IRBLb-B to infection by M. oryzae strain P131. a IRBLb-B harbors the resistance allele at Pib. The PCR product of PibdomF/R indicated that IRBLb-B contains the resistant Pib allele; The PCR product of Lys145F/R showed that LTH harbors the susceptible Pib allele. b The development of blast disease symptoms on the leaves of LTH and IRBLb-B with inoculation of M. oryzae strain p131 and Mock. The images were taken at 7 dpi (days post inoculation). c Micrographs of LTH and IRBLb-B leaf sheaths infected with M. oryzae strain P131 taken at 26 hpi and 36 hpi. Bar = 10 μm. At 36 hpi, the invasive hyphae extended to neighboring cells in the LTH, while the invasive hyphae were limited in the first invaded cell in the IRBLb-B. d The expression of Pib (LOC_Os02g57310) was induced almost nine times by the infection of M. oryzae at 36 hpi. The error bar means the standard deviation for triplicate assays
RNA-seq profiling of M. oryzae-inoculated LTH and IRBLb-B seedlings
To further investigate the mechanism of Pib-mediated rice blast resistance, the strategy employed to acquire the transcriptomes of M. oryzae-inoculated seedlings is illustrated in Fig. 2a. Approximately 5 × 106 clean reads were obtained from each biological replicate (Table S2), over 95% of which were successfully aligned with the cv. Nipponbare genome sequence. A Pearson correlation analysis conducted between the biological replicates revealed strong positive coefficients for both two genotypes (Tables S3, S4). The number of genes which were not transcriptionally altered by M. oryzae infection and were represented in both IRBLb-B and LTH was 15,745 (Fig. 2b). The number of SDEGs identified in LTH was 4718, of which 2286 were up-regulated by M. oryzae infection and 2432 were down-regulated; the equivalent numbers in IRBLb-B were 2958 SDEGs (815 up- and 2143 down-regulated) (Figs. 2c and S1). Among the two sets of SDEGs, 2137 (588 up- and 1549 down-regulated) were shared between LTH and IRBLb-B (Fig. 2c). The numbers of two genotype-specific SDEGs were 2581 (LTH) and 821 (IRBLb-B) (Fig. 2c, d).

The RNA-seq workflow and global profiling. a The RNA-seq workflow of IRBLb-B and LTH with M. oryzae strain P131 infection. Redox homeostasis, phytohormone, receptor kinase, transcriptional factor, and disease resistance related genes were analyzed to reveal the Pib-mediated blast resistance. b The RNA-seq based identification of differential transcription in seedlings of IRBLb-B and LTH sampled 36 hpi with M. oryzae strain P131. Venn diagram illustrating the number of genes detected present in both genotypes and/or treatments, and those present in specific genotype and/or treatment combinations. c The venn diagram of SDEGs (FDR adjusted p ≤ 0.05 and absolute log2 fold ≥ 2) across LTH and IRBLb-B. d The heatmap of SDEGs across LTH and IRBLb-B
qRT-PCR validation of differential transcription deduced from the RNA-seq data
In order to verify the results obtained by RNA-seq analysis, a sample of 24 IRBLb-B specific M. oryzae-induced SDEGs, all experiencing a fold change in transcript abundance of at least four (Table S5), was used to validate the RNA-seq data using qRT-PCR. This set of SDEGs included five ROS-related genes, four JA-related genes, four WRKY transcription factors, four receptor kinase, and seven disease-resistant related genes. The outcome of the qRT-PCR assay was almost same pattern with the conclusion based on the RNA-seq data (Fig. 3; Table S5, Table S14).

Validation of RNA-seq based on identification of differential transcription using qRT-PCR. Twenty-four SDEGs, including ROS, JA, WRKY, Receptor kinase, and defense related genes, were selected for validation. The heatmap was carried out by TBtools software based on FPKM and 2−ΔΔCT data. Tubulin gene (LOC_Os03g13170) was chosen as the reference sequence. The LTH Mock as the reference sample
GO enrichment and KEGG pathway analysis of the SDEGs
To explore signaling pathways involved in the Pib-mediated blast resistance, a summary of the GO analysis of the 821 M. oryzae-infection induced IRBLb-B SDEGs (Table S6, S7): the enriched GO terms related to 28 molecular functions, seven cellular components, and 41 biological processes. The GO analysis showed that most enriched GO terms were related to redox balance, including oxidoreductase activity, monooxygenase activity, oxidation-reduction process, glutathione conjugation reaction, glutathione transferase activity, and response to oxygen-containing compound. Moreover, stress response GO terms, including response to stimulus, response to stress, and response to abiotic stimulus, were enriched. These results indicated that redox balance and stress response-related genes may be involved in Pib-mediated blast resistance (Fig. 4a). In order to analyze metabolic pathways involved in the Pib-mediated blast resistance, a KEGG pathway analysis identified that both primary (including starch and sucrose metabolism, amino acid metabolism, and fatty acid metabolism) and secondary (phenylpropanoid synthesis) metabolism, as well as plant–pathogen interactions, were enriched (Fig. 4b; Table S8). To further elucidate the different pathways and the genes in them that get induced in the resistant and susceptible lines, a KEGG analysis was carried out and found that the secondary metabolites, plant hormone signal transduction, MAPK signaling pathway, and plant-pathogen interaction were enriched in both resistant and susceptible lines. Peroxisomes and some primary metabolism including carbon metabolism and nitrogen metabolism were specifically enriched in resistant lines, suggesting that energy metabolism and peroxisome involved in resistance of IRBLb-B to rice blast (Table S13).

GO and KEGG enriched analysis of the 821 SDEGs detected uniquely in M. oryzae-inoculated IRBLb-B. a The various cellular components, biological processes, and molecular functions identified by GO enriched analysis. b KEGG enrichment analysis. The significance of enrichment with the P ≤ 0.05, 0.01, and 0.001 are marked by *, **, and ***, respectively
Redox homeostasis
ROS (such as superoxide, hydrogen peroxide, hydroxyl radical, and singlet oxygen) is the earliest and rapid defense response to limit M. oryzae spread (Kou et al. 2019; Lehmann et al. 2015). ROS were fine-tuned by several enzymes, including NADPH oxidase and scavenging enzymes (redoxin, peroxidase, catalase, and glutathioneredoxin) (Camejo et al. 2016). Our study showed that genes encoding three categories of respiratory burst proteins (redox state, peroxidases, and glutathione S-transferases) were represented among the set of IRBLb-B specific M. oryzae-induced SDEGs (Fig. 5; Table S9). Seven of these (five up-regulated by M. oryzae infection and two down-regulated) encoded peroxidase precursors. Five of the SDEGs encoded ROS scavenging compounds, namely, thioredoxin (LOC_Os07g48510), glutaredoxin (LOC_Os01g47760, LOC_Os02g51370, and LOC_Os05g48930), and peroxiredoxin (LOC_Os06g09610), all of which down-regulated by M. oryzae infection. Fourteen of the SDEGs (all down-regulated by M. oryzae infection) encoded a glutathione S-transferase. These result showed that redox homeostasis–related genes were modulated in IRBLb-B with rice blast infection.

MapMan overview of the SDEGs, unique to IRBLb-B upon M. oryzae infection, involved in biotic stress pathway. Values represent log2 fold changes. Red cells indicate up-regulated genes and green cells down-regulated genes. ABA abscisic acid, JA jasmonate, ERF ethylene response factor, bZIP basic region-leucine zipper, DOF DNA binding with one finger, MAPK mitogen-activated protein kinase. PR-protein pathogenesis-related protein
Next, we determined the ROS accumulation in IRBLb-B with rice blast infection. DAB was used to stain the infected leaves of LTH and IRBLb-B to detect the production of hydrogen peroxide. As shown in Fig. 6, no DAB staining was observed in LTH leaves with M. oryzae infection at 24 hpi, while obvious DAB staining was observed in that of IRBLb-B. By 36 hpi, both LTH and IRBLb-B leaves showed obvious DAB staining, while the leaves of IRBLb showed more and clear DAB staining (Fig. 6b). These results indicated that the resistance of IRBLb-B to rice blast was related to the accumulation of more hydrogen peroxide.

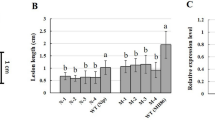
DAB staining analysis of LTH and IRBLb-B with M. oryzae infection at 24 hpi and 36 hpi and the phenotype of lesions on WT XH11 and lox3 knockout mutant inoculation by enshi2-2 strain. a DAB stain showed that IRBLb-B accumulated a relative high level of hydrogen peroxide compared with LTH at 24 hpi and 36 hpi. b The relative hydrogen peroxide level by ImageJ software, the LTH 24 hpi as the reference. The error bar represents the standard deviation for triplicate assays. c Leaf phenotype of XH11 and lox3#KO after inoculation by enshi2-2 strain. d The relative lesion area of XH11 and lox3#KO after inoculation by enshi2-2 strain. e The trypan blue stain of XH11 and lox3#KO after inoculation by enshi2-2 strain
Phytohormone-related genes
Plant hormones play important roles in response to biotic stress (Spoel and Dong, 2008). The set of IRBLb-B specific M. oryzae-induced SDEGs included four down-regulated genes associated with JA metabolism, namely, two encoding a 12-oxophytodienoate reductase (LOC_Os01g27230 and LOC_Os06g11280), one a lipoxygenase (LOC_Os12g37320) and one a CBS domain-containing membrane protein (LOC_Os09g02710), along with one up-regulated, encoding a lipoxygenase (LOC_Os03g49260) (Figs. 5 and 6; Table S10). Oslox3#KO was more susceptible to rice blast, suggesting that LOX3 played a positive regulatory role in rice blast resistance. A further twelve of the SDEGs were associated with ethylene (ET)-mediated processes: seven (three up- and four down-regulated) encoded AP2/ERF domain-containing proteins, one (down-regulated) ET synthesis-related gene (LOC_Os08g30080), and four (three up- and one down-regulated) genes (LOC_Os09g02710, LOC_Os10g31240, LOC_Os05g41760, and LOC_Os12g36630) regulated by ET. Three of the SDEGs encoded auxin metabolism products: these comprised two auxin synthesis-related proteins and one protein regulated by auxin. Four genes associated with abscisic acid (ABA) metabolism were identified as IRBLb-B specific M. oryzae-induced SDEGs (all were down-regulated): three encoded proteins related with ABA synthesis and one a protein regulated by ABA.
Kinase-mediated signaling-related genes
Pathogen signal perception and activation of downstream defense signaling molecules, including receptor kinases (RKs), MAPK, and calmodulin-dependent calcium sensor protein, are vital for plant defense (Jain et al. 2017). We determined whether RKs are involved in defense response against M. oryzae in IRBLb-B. In all, thirty-seven genes encoding a receptor kinase were classified as IRBLb-B specific M. oryzae-induced SDEGs (eighteen were up- and nineteen down-regulated by M. oryzae infection); this set included fourteen genes encoding members of the leucine-rich receptor subfamily (eleven up-regulated by M. oryzae infection), four members of the leucine-rich kinase 10L subfamily (one up-regulated by M. oryzae infection), nine members of the lectin-like receptor kinase subfamily (four up-regulated by M. oryzae infection), five members of the S-domain receptor-like protein kinase subfamily (one up-regulated by M. oryzae infection), and five members of the cell wall-associated kinase subfamily (one up-regulated by M. oryzae infection) (Fig. 5; Table S11). In addition, four genes encoding a calmodulin-dependent protein were down-regulated in response to M. oryzae infection, and two encoding a G protein were different-regulated in response to M. oryzae infection.
Transcription factor–related genes
It is well known that transcription factors, such as WRKY, MYB, and AP2/ERF, play important roles in response to pathogens in plant (Amorim et al. 2017). A total of twenty eight of the IRBLb-B specific M. oryzae-induced SDEGs encoded a transcription factor; these included WRKY26, WRKY32, and WRKY17 (which were all up-regulated by M. oryzae infection) and WRKY100 (down-regulated); MYB106, MYB103L, ARM1, and MYB7 (which were all up-regulated); and MYB61 (down-regulated). A further fifteen SDEGs (three up- and twelve down-regulated) encoded members of the AP2/ERF family, along with four (three up- and one down-regulated) encoding members of the bHLH family (Fig. 5; Table S12). Collectively, these results indicated that WRKY, MYB, AP2/ERF, and bHLH transcription factors may be involved in signal transduction of Pib to regulate the expression of defense genes in IRBLb-B.
Disease resistance–related genes
To investigate disease resistance–related genes related to M. oryzae infection in IRBLb-B, we searched for resistance-related genes in SDEGs unique to IRBLb-B. Seven putative disease resistance–related genes were among the set of IRBLb-B specific M. oryzae-induced SDEGs: these included Pib itself, three encoding a type of NBS-LRR protein (LOC_Os11g39160, LOC_Os11g45050, and LOC_Os12g13550), two PRM1 genes (LOC_Os11g11770 and LOC_Os11g12340), and one encoding a resistance-associated protein (LOC_Os08g42700) (Table 1). The abundance of Pib transcript was 4.3 fold higher in M. oryzae-infected than in mock-inoculated IRBLb-B plants (Table S6).
Discussion
Characterization of the rice-M. oryzae interaction has succeeded in detecting the activity of a number of R genes (Meng et al. 2019; Wang et al. 2017). Here, an attempt was made to reveal the mechanistic basis of Pib-mediated resistance by comparing the RNA-seq-derived transcriptomes of M. oryzae-inoculated LTH and its Pib NIL IRBLb-B. The number of genes in LTH which were differentially transcribed in the blast susceptible cultivar LTH as a result of M. oryzae infection was 4718, while a smaller number (2958) was induced in the blast resistant IRBLb-B plants. A further analysis of the latter set of genes suggested that a substantial number of them encoded redox homeostasis, WRKY transcription factors, and receptor kinases, along with proteins involved in either phytohormone metabolism or defense signaling. To our surprise, more than half of the genes were down-regulated upon M. oryzae infection in IRBLb-B. There may be some antagonism between growth and disease resistance, and plants that exhibit better resistance often sacrifice certain growth (Jiang et al. 2017).
The synthesis of ROS is an important component of the early phase of the defense mounted by rice against M. oryzae (Lehmann et al. 2015). These compounds act both to strengthen the plant’s cell walls and/or to trigger a number of downstream defense genes (Dangol et al. 2018; Gadjev et al. 2006). ROS typically accumulate to a higher level when the host and the pathogen are incompatible (Gupta et al. 2012; Jain et al. 2017). Here, the ROS accumulation in IRBLb-B plants infected with the avirulent M. oryzae strain P131 was higher than that in LTH (Fig. 6), and the ROS level was also accumulated in LTH at 36 hpi compared with 24 hpi, which indicated that ROS participated in the basic resistant of compatibility and incompatibility. Maintaining ROS content is likely basic requirement for Pib-mediated resistance.
Phytohormones, especially salicylic acid (SA), JA, and ET, are intimately involved in the host’s response to pathogen invasion (Pieterse et al. 2009; Spoel et al. 2003). JA and ethylene-related genes were induced during M. oryzae infection, while most of gene-related JA or ethylene were down-regulated in IRBLb-B, this may be due to the different sampling time. Some related genes were up-regulated in the early stage of infection and down-regulated in the late stage of infection. For example, OsLOX2, OsLOX5, OsCOL1b, and OsCOL2 show this expression pattern (Zhang et al. 2018a). After knock out the gene LOX3 which is significant induced by rice blast in IRBLb-B, we found that lox3#KO was more susceptible to rice blast, which indicated that the resistance of IRBLb-B required the participation of JA-related genes. More recently, other plant hormones, including ABA and auxin have also emerged as crucial regulator of plant–pathogen interactions (Anderson et al. 2004; Jiang et al. 2017; Jiang et al. 2013; Yazawa et al. 2012). In this study, three genes encoding auxin metabolism products and five IRBLb-B SDEGs encoding products associated with ABA metabolism were found differentially expressed in IRBLb-B and LTH. Overall, it is suggested that at least four kinds of phytohormones participate in Pib-mediated resistance to rice blast.
Pathogen signal perception, followed by the activation of downstream defense signaling molecules (receptor kinases, mitogen-activated protein kinases, and calmodulin-dependent calcium sensor proteins), is a fundamental part of the plant defense response (Jain et al. 2017). An analysis of the durably M. oryzae-resistant rice cultivar Digu has revealed many examples of the differential transcription of receptor kinase-encoding genes (Li et al. 2016), as is also the case for IRBL18, IRBL22 (Wei et al. 2013), and PB1+Pi9 (Jain et al. 2017). Here, thirty-seven receptor kinase encoding genes were present among the set of IRBLb-B SDEGs responding to M. oryzae infection. Some transcription factors have been reported to play important roles during perception of pathogen infection by receptor kinase. In particular, WRKY, MYB, and AP2/ERF genes can all be triggered when plants are exposed to a pathogen (Amorim et al. 2017; Dong et al. 2003; Fernandez-Calvo et al. 2011; Singh et al. 2002; Wiermer et al. 2005). In the present study, the set of IRBLb-B SDEGs responding to M. oryzae infection included four genes encoding WRKY factors, five MYB factors, fifteen AP2/ERF factors, and four bHLH factors, underlining the importance role of these transcription factors in signal transduction during the Pib-mediated defense response.
Plants dynamically reprogram their transcriptome upon pathogen attack (Pandey and Somssich 2009). Here, it has been shown that the plants harboring the broad spectrum blast resistance gene Pib activated a cascade of defense-related genes when challenged with the pathogen (Fig. 3). A model of the Pib-mediated defense response was given in Fig. 7. Upon exposure to M. oryzae, Pib is induced, resulting in the activation of a suite of downstream genes following signal transduction via both kinase-mediated and phytohormone-interacting pathways. Various transcription factors function to regulate the expression of key disease resistance–related and/or ROS-related genes.

A putative mechanistic model of the broad spectrum resistance mediated by Pib. The fungal avirulence factor is recognized by the Pib protein, which triggers kinase-mediated and hormone signaling to transduce the appropriate signal to a suite of downstream transcription factors. The activation of these transcription factors regulates genes encoding products involved in ROS metabolism and various defense-related proteins, leading eventually to the expression of the hypersensitive response. ET ethylene, JA jasmonate, TF transcription factor, ROS reactive oxygen species, HR hypersensitive response
Conclusions
Here, the transcriptomes of LTH and its NIL IRBLb-B have been compared, specifically with a view to reveal the transcriptional impact of the presence of the rice blast pathogen. The analysis identified that 821 genes were significant differentially transcribed in IRBLb-B when challenged by an incompatible race of M. oryzae. Many of these gene-encoded products are involved in ROS metabolism, phytohormone-mediated signaling, transcription factors-mediated signaling, and kinase-mediated signaling. The set of IRBLb-B SDEGs responding to M. oryzae infection included seven disease resistance–related genes. Thus, this study revealed that IRBLb-B plants were able to modulate some important candidate genes transcriptomic reprogramming upon M. oryzae attack.
References
Amorim LLB, dos Santos RD, Neto JPB, Guida-Santos M, Crovella S, Benko-Iseppon AM (2017) Transcription factors involved in plant resistance to pathogens. Curr Protein Pept Sc 18:335–351. https://doi.org/10.2174/1389203717666160621122405
Anderson JP et al (2004) Antagonistic interaction between abscisic acid and jasmonate-ethylene signaling pathways modulates defense gene expression and disease resistance in arabidopsis. Plant Cell 16:3460–3479. https://doi.org/10.1105/tpc.104.025833
Camejo D, Guzman-Cedeno A, Moreno A (2016) Reactive oxygen species, essential molecules, during plant-pathogen interactions. Plant Physiol Biochem 103:10–23. https://doi.org/10.1016/j.plaphy.2016.02.035
Dangol S, Chen Y, Hwang BK, Jwa NS (2018) Iron- and reactive oxygen species-dependent ferroptotic cell death in rice-Magnaporthe oryzae interactions. Plant Cell 31:189–209. https://doi.org/10.1105/tpc.18.00535
Dean R et al (2012) The Top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 13:414–430. https://doi.org/10.1111/j.1364-3703.2011.00783.x
Dong JX, Chen CH, Chen ZX (2003) Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol Biol 51:21–37. https://doi.org/10.1023/A:1020780022549
Fernandez J, Orth K (2018) Rise of a cereal killer: the biology of Magnaporthe oryzae biotrophic growth trends. Microbiol 26:582–597. https://doi.org/10.1016/j.tim.2017.12.007
Fernandez-Calvo P et al (2011) The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 23:701–715. https://doi.org/10.1105/tpc.110.080788
Fjellstrom R, Conaway-Bormans CA, McClung AM, Marchetti MA, Shank AR, Park WD (2004) Development of DNA markers suitable for marker assisted selection of three Pi genes conferring resistance to multiple Pyricularia grisea pathotypes. Crop Sci 44:1790–1798
Fukuoka S, Saka N, Mizukami Y, Koga H, Yamanouchi U, Yoshioka Y, Hayashi N, Ebana K, Mizobuchi R, Yano M (2015) Gene pyramiding enhances durable blast disease resistance in rice. Sci Rep 5:7773. https://doi.org/10.1038/srep07773
Gadjev I et al (2006) Transcriptomic footprints disclose specificity of reactive oxygen species signaling in Arabidopsis. Plant Physiol 141:436–445. https://doi.org/10.1104/pp.106.078717
Gupta SK, Rai AK, Kanwar SS, Chand D, Singh NK, Sharma TR (2012) The single functional blast resistance gene Pi54 activates a complex defence mechanism in rice. J Exp Bot 63:757–772. https://doi.org/10.1093/jxb/err297
Jain P et al (2017) Understanding host-pathogen interactions with expression profiling of NILs carrying rice-blast resistance Pi9 gene. Front Plant Sci 8:3389. https://doi.org/10.3389/fpls.2017.00093
Jiang CJ et al (2013) Cytokinins act synergistically with salicylic acid to activate defense gene expression in rice. Mol Plant-Microbe Interact 26:287–296. https://doi.org/10.1094/MPMI-06-12-0152-R
Jiang CJ, Liu XL, Liu XQ, Zhang H, Yu YJ, Liang ZW (2017) Stunted growth caused by blast disease in rice seedlings is associated with changes in phytohormone signaling pathways. Front Plant Sci 8:8. https://doi.org/10.3389/fpls.2017.01558
Koide Y et al (2010) Development of pyramided lines with two resistance genes, Pish and Pib, for blast disease (Magnaporthe oryzae B. Couch) in rice (Oryza sativa L.). Plant Breed 129:670–675. https://doi.org/10.1111/j.1439-0523.2010.01781.x
Kou Y, Tan YH, Ramanujam R, Naqvi NI (2017) Structure–function analyses of the Pth11 receptor reveal an important role for CFEM motif and redox regulation in rice blast. New Phytol 214:330–342. https://doi.org/10.1111/nph.14347
Kou Y, Qiu J, Tao Z (2019) Every coin has two sides: reactive oxygen species during Rice–Magnaporthe oryzae interaction. Int J Mol Sci 20:1191. https://doi.org/10.3390/ijms20051191
Lehmann S, Serrano M, L'Haridon F, Tjamos SE, Metraux JP (2015) Reactive oxygen species and plant resistance to fungal pathogens. Phytochemistry 112:54–62. https://doi.org/10.1016/j.phytochem.2014.08.027
Li W et al (2016) The durably resistant rice cultivar Digu activates defence gene expression before the full maturation of Magnaporthe oryzae appressorium. Mol Plant Pathol 17:354–368. https://doi.org/10.1111/mpp.12286
Liao Z et al (2019) SLR1 inhibits MOC1 degradation to coordinate tiller number and plant height in rice. Nat Commun 10:2738. https://doi.org/10.1038/s41467-019-10667-2
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Ma X et al (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284. https://doi.org/10.1016/j.molp.2015.04.007
Meng Q et al. (2019) Proteomics of rice—magnaporthe oryzae interaction: what have we learned so far? Frontiers in Plant Science 10 doi: https://doi.org/10.3389/fpls.2019.01383
Pandey SP, Somssich IE (2009) The role of WRKY transcription factors in plant immunity. Plant Physiol 150:1648–1655. https://doi.org/10.1104/pp.109.138990
Pieterse CMJ, Leon-Reyes A, Van der Ent S, Van Wees SCM (2009) Networking by small-molecule hormones in plant immunity. Nat Chem Biol 5:308–316. https://doi.org/10.1038/nchembio.164
Ramkumar G, Madhav MS, Devi SJSR, Prasad MS, Babu VR (2015) Nucleotide variation and identification of novel blast resistance alleles of Pib by allele mining strategy. Physiol Mol Biol Plants 21:301–304. https://doi.org/10.1007/s12298-015-0284-4
Sakulkoo W et al (2018) A single fungal MAP kinase controls plant cell-to-cell invasion by the rice blast fungus Science 359:1399-+. https://doi.org/10.1126/science.aaq0892
Singh KB, Foley RC, Onate-Sanchez L (2002) Transcription factors in plant defense and stress responses. Curr Opin Plant Biol 5:430–436. https://doi.org/10.1016/S1369-5266(02)00289-3
Singh AK, Singh PK, Arya M, Singh NK, Singh US (2015) Molecular screening of blast resistance genes in Rice using SSR markers. Plant Pathol J 31:12–24. https://doi.org/10.5423/PPJ.OA.06.2014.0054
Spoel SH et al (2003) NPR1 modulates cross-talk between salicylate- and jasmonate-dependent defense pathways through a novel function in the cytosol. Plant Cell 15:760–770. https://doi.org/10.1105/tpc.009159
Tanweer FA, Rafii MY, Sijam K, Rahim HA, Ahmed F, Ashkani S, Latif MA (2015) Introgression of blast resistance genes (Putative Pi-b and Pi-kh) into elite rice cultivar MR219 through marker-assisted selection. Front Plant Sci 6:1002. https://doi.org/10.3389/fpls.2015.01002
Telebanco-Yanoria MJ, Koide Y, Fukuta Y, Imbe T, Kato H, Tsunematsu H, Kobayashi N (2010) Development of near-isogenic lines of Japonica-type rice variety Lijiangxintuanheigu as differentials for blast resistance. Breed Sci 60:629–638. https://doi.org/10.1270/jsbbs.60.629
Wang ZX et al (1999) The Pib gene for rice blast resistance belongs to the nucleotide binding and leucine-rich repeat class of plant disease resistance genes. Plant J 19:55–64. https://doi.org/10.1046/j.1365-313X.1999.00498.x
Wang BH, Ebbole DJ, Wang ZH (2017) The arms race between Magnaporthe oryzae and rice: Diversity and interaction of Avr and R genes. J Integr Agric 16:2746–2760. https://doi.org/10.1016/S2095-3119(17)61746-5
Wei T, Ou B, Li J, Zhao Y, Guo D, Zhu Y, Chen Z, Gu H, Li C, Qin G, Qu LJ (2013) Transcriptional profiling of rice early response to Magnaporthe oryzae identified OsWRKYs as important regulators in rice blast resistance. PLoS One 8:e59720. https://doi.org/10.1371/journal.pone.0059720
Wiermer M, Feys BJ, Parker JE (2005) Plant immunity: the EDS1 regulatory node Current Opinion In Plant Biology 8:383-389. https://doi.org/10.1016/j.pbi.2005.05.010
Yazawa K, Jiang CJ, Kojima M, Sakakibara H, Takatsuji H (2012) Reduction of abscisic acid levels or inhibition of abscisic acid signaling in rice during the early phase of Magnaporthe oryzae infection decreases its susceptibility to the fungus. Physiol Mol Plant Pathol 78:1–7. https://doi.org/10.1016/j.pmpp.2011.12.003
Yuan G, Wan Y, Li X, He B, Zhang Y, Xu B, Wang S, Xie W, Zhou X, Wu Q (2017) Development of near-isogenic lines in a parthenogenetically reproduced thrips species, Frankliniella occidentalis. Front Physiol 8:8. https://doi.org/10.3389/fphys.2017.00130
Zhang X et al (2018a) Magnaporthe oryzae induces the expression of a microRNA to suppress the immune response in rice. Plant Physiol 177:352–368. https://doi.org/10.1104/pp.17.01665
Zhang X et al (2018b) A positive-charged patch and stabilized hydrophobic core are essential for avirulence function of AvrPib in the rice blast fungus. Plant J 96:133–146. https://doi.org/10.1111/tpj.14023
Zhu Y, Du B, Qian J, Zou B, Hua J (2013) Disease resistance gene-induced growth inhibition is enhanced by rcd1 independent of defense activation in Arabidopsis. Plant Physiol 161:2005–2013. https://doi.org/10.1104/pp.112.213363
Acknowledgements
The authors would like to thank Youliang Peng (China Agricultural University, Beijing) for the gift of M. oryzae strain P131. We also thank Dr. Yazhou Su and Jane Sadhna Jagernath for proof reading of this manuscript.
Funding
This project was supported by the Chinese Academy of Agricultural Sciences under the “Elite Youth” program; the Agricultural Sciences and Technologies Innovation Program, Zhejiang Provincial Natural Science Foundation of China, grant numbers “LQ19C130007 and LQ19C140004”; and key R&D project of Zhejiang Province, grant number “2019C02018.”
Author information
Authors and Affiliations
Contributions
JQ and YK designed the experiments. JQ, FL, MX, and SM performed the experiments. JQ analyzed the data. JQ wrote the manuscript. YK proofed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 13 kb)
ESM 2
(DOCX 17 kb)
ESM 3
(DOCX 16 kb)
ESM 4
(DOCX 16 kb)
ESM 5
(XLSX 10 kb)
ESM 6
(XLSX 214 kb)
ESM 7
(XLSX 30 kb)
ESM 8
(XLSX 14 kb)
ESM 9
(XLSX 11 kb)
ESM 10
(XLSX 11 kb)
ESM 11
(XLS 37 kb)
ESM 12
(XLSX 12 kb)
ESM 13
(XLSX 11 kb)
ESM 14
(XLSX 11 kb)
Fig. S1
Volcano plot illustrating genes which were differentially transcribed in both LTH and IRBLb-B as a result of M. oryzae infection (JPG 1175 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Qiu, J., Lu, F., Xiong, M. et al. Comparative transcriptomic analysis reveals the mechanistic basis of Pib-mediated broad spectrum resistance against Magnaporthe oryzae. Funct Integr Genomics 20, 787–799 (2020). https://doi.org/10.1007/s10142-020-00752-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-020-00752-x