
Abstract
The increasing amount of available expressed gene sequence data makes whole-transcriptome analysis of certain crop species possible. Potato currently has the second largest number of publicly available expressed sequence tag (EST) sequences among the Solanaceae. Most of these ESTs, plus other proprietary sequences, were combined and used to generate a unigene assembly. The set of 246,182 sequences produced 46,345 unigenes, which were used to design a 44K 60-mer oligo array (Potato Oligo Chip Initiative: POCI). In this study, we attempt to identify genes controlling and driving the process of tuber initiation and growth by implementing large-scale transcriptional changes using the newly developed POCI array. Major gene expression profiles could be identified exhibiting differential expression at key developmental stages. These profiles were associated with functional roles in cell division and growth. A subset of genes involved in the regulation of the cell cycle, based on their Gene Ontology classification, exhibit a clear transient upregulation at tuber onset indicating increased cell division during these stages. The POCI array allows the study of potato gene expression on a much broader level than previously possible and will greatly enhance analysis of transcriptional control mechanisms in a wide range of potato research areas. POCI sequence and annotation data are publicly available through the POCI database (http://pgrc.ipk-gatersleben.de/poci).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Potato (Solanum tuberosum) is an important food crop throughout the world. Breeding strategies in the last few decades have primarily focused on the improvement of yield, pest and disease resistance, and tuber quality traits. As consumer demands for food quality increase, knowledge pertaining to the natural variation and genetic regulation of quality traits will become an ever more important focus of potato research. In recent years, major efforts have been made in the construction of extensive potato expressed sequence tag (EST) libraries, sequencing of which has facilitated the identification of genes relevant to a wide variety of aspects of potato physiology and metabolism (Crookshanks et al. 2001; Flinn et al. 2005; Nielsen et al. 2005; Rensink et al. 2005a; Ronning et al. 2003). As a tool for potato functional genomics research, different noncommercial microarrays have been constructed using the available EST data, including a spotted complementary deoxyribonucleic acid (cDNA) array from The Institute for Genomics Research (TIGR) containing around 12,000 cDNA clones (Rensink et al. 2005b) and a dedicated cDNA array containing around 2,000 cDNA clones specific for the study of the potato tuber lifecycle (Kloosterman et al. 2005). Neither of these arrays has fully utilized the available sequence information within potato EST databases, and both have some other major limitations.
The TIGR cDNA microarray has been used to study gene expression in cold-, heat-, and salt-stressed potato plants (Rensink et al. 2005c). Restrepo et al. (2005) used the same TIGR potato cDNA array to conduct gene profiling following late blight (Phytophthora infestans) infection. In addition, expression data from 52 microarray experiments conducted at TIGR using ribonucleic acid (RNA) from various Solanaceous species are available on the web (http://www.tigr.org/tigr-scripts/tdb/sol/study/sol_study.pl).
In this paper, we present the construction of a novel potato microarray platform based on the Agilent (www.chem.agilent.com) 60-mer oligo synthesis technology, comprising a total of 44,290 features of which 42,034 are derived from potato sequences. This microarray is the most complete representation of the current status of the sequenced potato transcriptome. All publicly available EST data and additional ESTs contributed from a consortium of participating laboratories named the Potato Oligo Chip Initiative (POCI) has provided the largest potato EST collection available thus far. The functionality of the POCI array has been tested by analysis of RNA targets from a tuber developmental series. The formation and growth of a potato tuber is a complex process that can be divided into discrete physiological events. A major challenge still is to establish a coherent sequence of molecular events leading up to the formation and subsequent growth of the tuber and to identify the set of genes involved. The initial formation of a tuber requires the induction of cell division in the subapical stolon region, and therefore, transcriptional regulation of cell cycle genes is expected during these stages. During further tuber growth, starch, storage proteins, and a wide range of metabolites accumulate within the tuber. Final tuber size, morphology, and content represent different aspects of tuber quality. These traits are largely dependent on the potato genotype and environmental conditions and involve the highly coordinated expression of many genes. Large-scale transcriptional studies are essential for a full understanding of the processes involved in tuber development. The expression data obtained in this study can serve as a valuable resource for identifying potential candidate genes involved in the initiation of tuber growth, tuber development, and a variety of tuber quality traits.
Materials and methods
Description of EST libraries
A collection of 246,182 S. tuberosum EST sequences were kindly provided from several research institutes (Table 1). The ESTs were derived from approximately 37 libraries derived from various potato tissues, developmental stages, and physiological conditions including exposure to common scab, Phytophthora infestans, as well as salt, cold, heat, and drought stress (Supplementary Table S1). The majority of sequences in these libraries have been previously described (Flinn et al. 2005; Rensink et al. 2005c; Ronning et al. 2003). The EST sequences were produced from the cultivars Bintje, Kennebec, Kuras, Shepody, Agria, and Solara (Table 1).
EST processing and assembling
Base calling, cleaning, masking, clustering, and assembly of the EST sequences was conducted using bioinformatics methods described previously (Flinn et al. 2005). In addition, Phytophthora nucleotide sequences from the Phytophthora Genome Consortium EST database at the Virginia Bioinformatics Institute (phytophthora.vbi.vt.edu) were used for the removal of contaminant late blight pathogen sequences.
Functional annotation of unigene sequences
Homology searches of unigenes were conducted using Basic Local Alignment Search Tool (BLAST) procedures (Altschul et al. 1990). The unigene set was searched using BLASTX and BLASTN against a local copy of the GenBank nonredundant (nr) protein and nucleotide databases (www.ncbi.nlm.nih.gov). A putative function was assigned to a sequence using an E value cutoff of less than 1 × 10−10.
Orientation of unigene sequences
Unigenes were orientated in the 5′-to-3′ direction using three separate criteria. First, unigenes were orientated according to a BLAST hit against the National Center for Biotechnology Information’s (NCBI) nr protein database (E value cutoff of less than 1 × 10−5). Unigenes with no significant BLAST hit were orientated according to the sequencing direction of the underlying ESTs, provided the unigenes were composed of more than one EST, (i.e., they were not singletons). The remaining sequences were screened for polyadenylation tails with a minimum tail length of ten bases, and these were defined as the 3′ ends of the transcripts. Any remaining singletons were orientated according to the sequencing direction of the EST.
Gene Ontology annotation
A Gene Ontology (GO; www.geneontology.org) term was assigned to individual unigenes based on sequence similarity, using BLASTX, against the Uniref100 protein database (http://www.ebi.ac.uk/uniprot). The GO annotation is inferred from the unigenes top BLAST hit (E value cutoff of less than 1.0 × 10−10) to UniRef100 GO-annotated sequences (Camon et al. 2004).
POCI database and annotation tool
For establishment of the POCI database, the Oracle database management system was used (www.oracle.com). A web front end for data management and data visualization has been implemented with Oracle Application Express, which also provides a user management system with various user groups. This way, registered users can edit data where guests are only allowed to view and download information. The online blast against POCI unigenes was implemented using Oracle blast implementation (Stephens et al. 2005).
Sample preparation and hybridization
The potato tuber developmental range used for the transcriptional analysis was derived from a previously described study (Kloosterman et al. 2005) comprising eight developmental stages of which six were used in the present study. The two additional stages were left out of this study to allow for an optimal loop design and to reduce the number of required hybridizations. Total RNA was purified using RNeasy MinElute Spin Columns (Qiagen) and DNAseI treated before first- and second-strand synthesis following the MessageAmp™ II aRNA amplification kit protocol (Ambion) using the following minor modifications. For first-strand synthesis, 2 μg of total RNA was used in the reaction followed by second-strand synthesis with half of the recommended enzyme concentrations. Purified cDNA was used for in vitro transcription using an indirect amino-allyl-labeling method with a 1:1 incorporation ratio of uridine triphosphate (UTP)/5-(3-aminoallyl)-UTP. In vitro transcription was performed at 37°C with a lid temperature of 42°C for a period of 12 h, after which samples were purified according to the manufacturer’s protocol. Purified complementary RNA (cRNA) samples were prepared in 2-μg aliquots and dried using a speed vac and stored at −80°C. For sample labeling, 2 μg of cRNA was resuspended in 5 μl freshly made carbonate buffer (0.1 M Na2CO3) adjusted to pH 9. Equal amounts of either Cy3 or Cy5 dye (GE Healthcare) were added and incubated for 1.5 h at room temperature. Samples were purified using RNeasy MinElute Spin Columns (Qiagen). Dye incorporation was measured using a ND-100 Spectrophotometer v3.3 (Nanodrop Technologies) and 1 μg of both Cy3- and Cy5-labeled cRNA samples was used for each hybridization. Hybridization and slide processing were performed using the 1 × 44 k POCI array according to Agilent specifications using the Agilent hybridization chamber and oven, including the stabilization and drying wash step. Slides were scanned on the Agilent DNA Microarray Scanner, and data were extracted using the feature extraction software package (v9.1.3.1) using a standard protocol. The POCI array design includes 2,256 positive and negative control features that are set up for use with the Agilent’s Two-color spike-in kit (www.chem.agilent.com). In future experiments, the POCI array will only become available in the 4 × 44 k format (www.chem.agilent.com).
Array data analysis
The data were imported in Microsoft Excel, and 2log Cy5/Cy3 ratios were calculated after passing quality check and minimal expression levels. 2log ratios for all 14 hybridizations were exported into Genstat® v9.2 for statistical analysis and calculation of expression estimates and standard errors for each feature during the six developmental stages. Features with a residual variance greater than 2.0 were rejected from the data set. Calculated estimates and standard errors of 31,293 features for the six developmental stages were finally imported in the GenemathsXT 1.6 (Applied Maths) software package for additional analysis.
Results and discussion
Potato unigene assembly
A total of 219,507 ESTs passed through the sequence-cleaning pipeline and were used in the unigene assembly. The ESTs were grouped into 18,343 clusters, and the assembly yielded 26,474 contigs with an average length of 991 bp. The contigs were comprised of 199,636 ESTs. Of the remaining 19,871 singleton ESTs, 9,800 sequences clustered together with other sequences. The singleton ESTs had an average length of 561 bp. The combined contigs and singletons produced a total of 46,345 unique sequences with an average length of 806 bp. From these unigenes, 60-mer oligos were designed for 42,034 of the sequences using Agilent’s proprietary methods. The sequences on the POCI array represent a total of 184,620 ESTs. A comparison of the sequences on the POCI array to those on the TIGR cDNA array indicated that 19,986 (47.5%) of the POCI sequences were not present on the TIGR array. In contrast, only 80 (0.7%) of the TIGR chip sequences were unique at the same BLAST E value (<1 × 10−10). To give POCI users access to all relevant data concerning sequences on the POCI chip, we set up a searchable POCI database (http://pgrc.ipk-gatersleben.de/poci). Sequences of synthesized oligo nucleotides as well as unigenes can be viewed, downloaded, and searched by their unigene identifier. The ESTs identifiers that were used to generate the unigene set are available, together with their membership relation to the unigene identifiers. ESTs that did not cluster with any other are marked as singletons.
A self-BLASTN was also conducted to allow microarray users to identify similar genes on the POCI array. Designed oligos were also compared with the unigenes to identify similar genes. This information is useful for the study of candidate genes and pathways, as well as for explaining similar gene expression profiles. A summary of the self-BLAST and oligo-BLAST data is presented in Table 2, and the complete data set can be searched on the POCI website. The majority (84.3%) of the oligos did not have significant similarity to nontarget unigenes (Table 2). Only 120 oligo sequences perfectly matched nontarget unigenes, and most (6,346 out of 6,588) of those oligos with significant nontarget hits had less than 90% similarity (data not shown).
POCI gene annotation
The POCI unigenes were blasted against the NCBI nr database and the Uniref100 protein database for functional annotation and GO. Figure 1 summarizes the biological functional annotation of the POCI sequences. Of the total number of unigenes on the array, 13,900 (33.1%) did not have a significant BLAST hit against the protein nr database. The computed annotation of the POCI unigenes with NCBI nr and GO is also available and searchable by keywords in the previously mentioned POCI database (http://pgrc.ipk-gatersleben.de/poci). Moreover, complete results of alignments against several relevant databases are available. In detail, these are:
-
NCBI nr polypeptides
-
Arabidopsis polypeptides
-
The potato gene index of TIGR (Quackenbush et al. 2001)
-
Tomato unigenes from the Solanaceous genomic network (Mueller et al. 2005)

Biological process functional annotation of the POCI assembly-unique sequences for those whose putative function could be determined
To find possible homologous sequences, the algorithms blastn and blastx were used for Solanaceae databases and less related organisms, respectively. For further investigation, whole-sequence alignments can be downloaded. Because all these results are precomputed and stored, a reverse search by database identifiers is also possible. This is especially helpful to find representatives/homologues of candidate genes derived from other platforms on the POCI chip. If the respective identifier is not known, direct BLAST searches (blastn and tblastx) using single sequences can be performed against the POCI unigene set.
The correct functional annotation of all features on the POCI array is important if one wants to infer biological meaning from obtained expression data. Therefore, the POCI database also contains a POCI annotation tool that works as a platform for manual curation of the POCI annotation. With this tool, the precomputed annotation for a unigene can be changed by every registered user with an editing account. Every annotation that has not been curated manually is marked with “NOT VALIDATED.” This tool will facilitate ongoing improvement of the annotations of sequences on the chip.
Transcriptional changes during potato tuber development
The POCI array design is based on a large EST collection containing genes from a wide variety of tissues, developmental stages, and treatments allowing for a diverse range of gene transcription studies covering all aspects of potato biology. In this paper, we describe a first use of the POCI array in a study to follow transcriptional changes during the process of potato tuber formation to identify genes driving potato tuber initiation and growth. Since the tuber development range has been studied previously using a small dedicated spotted cDNA microarray (Kloosterman et al. 2005), the newly obtained data can be assessed through comparison with the previously published data set. The tuber development range includes the various physiological events leading up to the induction, formation, and early growth stages of a potato tuber (Fig. 2a). The original developmental range described in Kloosterman et al. (2005) contained two additional developmental stages (stages 6 and 8) that were excluded from the present study to allow for an optimal loop design (Fig. 2b) and to reduce the number of hybridizations. Stages that were included are: stage 1, stolon tip grown under long day conditions; stage 2, stolon tip under short day (SD) conditions; stage 3, subapical stolon swelling 6–7 days after the switch to SD conditions; stage 4, tuber initiation (7–8 days); stages 5–7, tuber growth stages (9.5 and 11 days). The six developmental stages were labeled and hybridized to the array, and the data were analyzed as described in the “Materials and methods.”

Experimental setup for the expression profiling of the potato tuber developmental range (a–b). a Representation of the potato tuber development stages (1–7) used for expression profiling represented in days after switch to SD conditions. Stage 1, stolon tip under long-day conditions; stage 2, stolon tip under SD conditions; stage 3, subapical stolon swelling; stage 4, tuber initiation; stages 5–7, tuber growth stages, 9–10 and 15 days after switch to SDs, respectively. b Hybridization scheme comprising a loop design. Arrows indicate individual hybridizations with the arrowhead representing the incorporation of the Cy5 dye and the tail incorporation of the Cy3 dye
Estimated log ratios of 31,293 features passed quality check and significant expression levels and were selected for further analysis (Supplementary Table S2). Principal component analysis (PCA) showed a clear influence of the developmental time range with the first component (PC1), explaining 67.8% of the variance (PC1 = 67.8%, PC1 + 2 = 89.9%, PC1 + 2 + 3 = 95.2%). A similar distribution of the developmental stages was found in the previous study in which the time component (X-axis) explains a large percentage of the observed variance (Fig. 3). The second component (Y-axis) explains 22.1% of the observed variation. This component can probably be attributed to transcriptional changes during stages 3 and 4 during which the transition from stolon to a tuber takes place.

PCA analysis of the six tuber developmental stages (a) and expressed genes (b). b Genes are colored based on their assigned expression profile during tuber development. Light green, early downregulated; dark green, late downregulated; blue, transient upregulated; yellow, transient downregulated; red, early upregulated; dark red, late upregulated; purple, other differentially expressed genes (p < 0.005); gray, nondifferentially expressed genes (p > 0.005); white, constitutive expressed genes during tuber development
Genes were tested for differential expression within each of the six developmental stages (p < 0.05) based on the estimated log values and standard errors. A total of 15,959 features showed differential hybridization in at least one of the six developmental stages, while 15,334 features were not significantly differentially expressed (Table 3). The latter group is, however, likely to include genes that are differentially expressed from a biological standpoint and therefore still interesting but could not be classified as such due to large standard deviations. The inclusion of more biological replicates could potentially impart more significance to such data.
Genes exhibiting differential expression during the process of potato tuber formation exhibit both unique and common expression profiles. To identify the most common expression profiles, self-organizing maps were calculated. The most common expression profiles found were categorized in six major groups with one other group containing all remaining profiles (Table 3). Within the nondifferentially expressed class, we were able to identify 1,258 features that showed very low variation in transcript levels during the developmental time range. Such genes were classified as constitutively expressed during tuber development, and the average profile of this group was used for comparison to other expression profiles (Fig. 4).

Average expression of the major identified profiles during potato tuber development plotted in days after switch to SD conditions (see Fig. 2). Differential expression profiles are presented relative to the average expression of constitutively expressed genes (gray; a–f). a Early downregulated; strong downregulation at first visible stolon swelling (stage 3; 6–7 days). b Late downregulated; downregulation after initial tuber has been formed (stage 4; 7–8 days). c Early upregulated; upregulation prior to visible swelling. d Late upregulated; upregulation after initial tuber has been formed. e Transient upregulated; temporary upregulation during the stages of tuber formation (Stage 3 and 4). f Transient downregulated; temporary downregulation during the stages of tuber formation (Stage 3 and 4)
It is interesting to note that the most common expression profile found among the genes analyzed follows a downregulated expression pattern, which is particularly strong at tuber onset (Fig. 4a) and represents 3,778 features (Table 3). A much smaller set of genes (862 features) also follow a downregulated expression pattern with a delay compared to the first group, dropping in expression from stages 4, 5, and onward (Fig. 4b). In general, these genes showed a less pronounced decrease in comparison to the early downregulated subset (Fig. 4a,b).
The second largest group (Table 3; 3,544 features) exhibits a temporarily upregulated expression pattern at tuber onset (Fig. 4e). As stolon growth is inhibited and cells in the subapical stolon region follow a transition to the tuber state, the expression of new sets of genes and proteins are required to facilitate this transition and initiation of cell division and expansion. Based on the GO classification of genes represented in the POCI array, the majority of genes in the transient upregulated group could be unequivocally assigned to a specific GO classification associated with translation (52%; GO; 0006412) and cell cycle (24%; GO: 0007049; data not shown). Similarly, a large proportion of genes within this specific profile group are, based on sequence homology, associated with the ribosome (33%; GO: 0005840), with respect to their cellular process (data not shown).
In sharp constrast, few genes exhibiting a transient downregulated expression profile (Fig. 4f) are associated with translational activity or cell cycle (data not shown). During the transition of a stolon to a tuber, the plane of cell division and expansion is reorientated to facilitate radial swelling (Xu et al. 1998b). The cytoskeleton has to be reorientated in a similar fashion during these stages. In fact, the largest proportion of genes associated with the cytoskeleton (32%) follow a transient downregulated trend (data not shown). The transient downregulated profile group, however, is much smaller (1,492 features) in comparison to the transient upregulated profile (3,544 features; Table 3).
Analogous to the early and late downregulated profiles, we could make a distinction between early and late upregulated transcript profiles. The early upregulated genes (Fig. 4c) exhibit an increase in transcription levels during stage 2, often prior to visible swelling, indicating early transcriptional control and a changing metabolic status, as many sucrose-related genes are strongly induced. The import of sucrose, initially apoplastic and subsequently symplastic (Viola et al. 2001), is undoubtedly a major factor in potato tuber formation. In vitro tuberization requires a certain threshold of sucrose concentration, and a clear link with gibberellic acid (GA) metabolism has been made previously (Xu et al. 1998a). Overall, genes in the early upregulated category are strongly induced during stages 3, 4, and 5 after which transcript levels remain relatively stable during further tuber growth. On average, the upregulated genes exhibiting a somewhat delayed increase in transcript levels (stages 4–7), clearly visible from stage 4 onward (Fig. 4d), continue to show a strong increase until the last included developmental stage at which tuber filling is thought to be at its peak.
The time point and developmental stages at which transcriptional changes occur provide clues as to their biological function. They also indicate a shift in the metabolic status during tuber transition and subsequent tuber growth that can be analyzed in more detail. The expression level of a gene that has been shown to be important for tuber formation (StGA2ox1) serves as a good example of the set of genes that are early induced at tuber onset. StGA2ox1 has recently been shown to be strongly upregulated at tuber onset in the subapical stolon region and is thought to be involved in the orientation of initial cell divisions and expansion at tuber organogenesis (Kloosterman et al. 2007). StGA2ox1 was first identified using the small dedicated cDNA microarray, and its strong upregulated expression pattern was later confirmed by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR; Kloosterman et al. 2005). StGA2ox1 expression data obtained with the POCI array provides a similar expression pattern but achieves a much higher level of sensitivity when compared with the cDNA array results (Fig. 5). Similarly, expression profiles of other candidate genes that exhibit strong transcriptional control at tuber onset have been shown to correlate well with qRT-PCR data. Expression data of differentially expressed genes and other gene expression profiles represented on the array can be found in Supplementary Table S2.

Comparison of the relative expression levels of the StGA2ox1 gene during the potato tuber development range using different expression profiling platforms; filled triangles, qRT-PCR; empty diamonds, POCI array; filled squares, tuber lifecycle cDNA array
Biological function of expression profiles
Based on the observed common expression profiles, gene function of individual members or individual metabolic routes that are over-represented within a particular profile can be assessed. As mentioned above, genes associated with the cell cycle (GO: 0007049) are over-represented (24%) within the transient upregulated profile group (data not shown). This is not surprising, as during the stages of tuber initiation, an increase in cell division has been reported (Xu et al. 1998b), followed by a growth period of active cell division and expansion until the final tuber size is reached. The bulk of the mature tuber is formed by the perimedullary region, which originates from a small band of cells around the vascular bundle during tuber growth (Xu et al. 1998b). The level of cell mitotic activity together with cell expansion occurs particularly during the stages of stolon swelling and initial tuber growth, allowing for a rapid growth period of the newly formed tuber. The transient increase in transcript levels of cell cycle-related genes serve as a good marker for the increase in overall cell cycle activity during stolon swelling (Fig. 6).

Heat map of the expression data from a subset of genes exhibiting a common transient upregulated expression profile that is based on their sequence homology are associated with cell cycle (GO: 0007049) or shows homology to StEBP1 (EBP1-like)
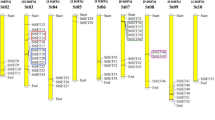
The processes and genes involved in the initiation of cell division in the subapical region of the stolon, as well as the time point of inhibition of further cell divisions, is still largely unknown. We have recently shown that an auxin-dependent regulator of cell growth (EBP1) is transiently upregulated during tuber development. StEBP1 was shown to be required for dose-dependent expression of cell cycle genes in potato and Arabidopsis (Horvath et al. 2006). Using the self-BLAST information, we identified eight unigenes present on the POCI array that show high homology to the StEBP1 nucleotide sequence of which six gave a significant signal throughout the tuber development range (Fig. 6). The unigene MICRO.127.C2 shares the highest homology to the cloned StEBP1 (99%) and confirms the transient upregulated profile published previously (Horvath et al. 2006). The remaining unigenes could either represent closely related family members, different allelic variations, or nonoverlapping 5′- and 3′-derived ESTs from the same transcribed gene and are referred to here as EBP1-like genes. MICRO.127.C1, MICRO.127.C3, PPCAS07TH, BPLI4F10TH, and MICRO.127.C4 have regions of high sequence homology with the coding sequence of StEBP1/MICRO.127.C2, but variation in the nucleotide sequence is significant and is particularily evident in the 3′-untranslated region allowing differentiating probe designs (Fig. 7). MICRO.127.C4 has a unique 73-bp insertion that is absent in the homologous unigenes and StEBP1. Based on the oligo design for this probe, cross-hybridization is unlikely, and therefore, it is interesting to note that MICRO.127.C4 exhibits a similar expression pattern as MICRO.127.C2 (StEBP1; Fig. 6) and thus may reveal allelic variation or the existence of homologous genes that could fulfil redundant or indeed novel functional roles in controlling cell cycle-related processes. Similarily, MICRO.127.C1 codes for a predicted protein of 377 amino acids, sharing 92% sequence similarity to StEBP1, but it exhibits a more upregulated profile during development very similar to the expression profile of singleton PPCAS07TH. Singleton BPLI4F10TH and MICRO.127.C3 show a similar expression pattern with elevated transcript levels at stage 1 and stages 4 and 5 (Fig. 6). In addition, the BPLI4F10TH sequence reveals a 16-bp deletion in a relatively conserved region on which the probe design was targeted (Fig. 7). Hence, by designing the unique potato 60-mer oligo in regions of high sequence variation, closely related family members or large allelic variations can be distinguished. This provides additional levels of gene expression information less likely to be obtained through conventional spotted cDNA microarrays. However, verification of observed expression profiles of highly homologous genes should always be confirmed by qRT-PCR using gene- or allele-specific primers.

Sequence alignment of StEBP1 and StEBP-like unigenes sharing regions of high sequence similarity (dark-shaded areas), good similarity (gray), and low similarity (no shading). Black boxes indicated unique 60-mer oligo design sequence representing the respective unigene on the POCI array
Metabolic pathway analysis
In a previous study (Kloosterman et al. 2005), gene expression profiles of enzymes active in the starch biosynthesis route were assessed during potato tuber development and provided an overview of the key metabolic steps. The POCI array comprises roughly 20 times the number of unigenes compared to the dedicated cDNA array that were implemented in the previous developmental range study. This should allow the analysis of most relevant metabolic routes to potato researchers. As an example, the biosynthesis route of carotenoids during potato tuber development is analyzed in more detail (Fig. 8). Individual enzyme reactions are often represented by multiple features on the POCI array and are likely to represent either the same gene based on the on nonoverlapping 5′ and 3′ ESTs oligo design (gene redundancy) or allelic variants or closely related gene family members. Within the schematic representation (Fig. 8), only genes with a significant homology to known proteins at the amino acid level (>83%) are represented. Various carotenoids accumulate in different concentrations depending on genotype (Morris et al. 2004). It is interesting to note that the most common expression profile of genes in the carotenoid pathway is transient upregulation at tuber organogenesis (Fig. 8, stages 3–4: phytoene synthase, phytoene desaturase, lycopene beta-cyclase, lycopene epsilon-cyclase, carotenoid hydroxylase 1, 9-cis-epoxidase). This indicates a shift in the metabolic flux within this pathway at these stages, and this is in line with high total carotenoid content early in tuber development, as described by Morris et al (2004).

Overview of major enzymatic steps involved in the carotenoid biosynthesis pathway and the expression profiles of genes catalyzing these reactions during early tuber development. Based on sequence homology, multiple probes can be shown for a single enzymatic reaction that can either represent the same gene, allelic variation, or closely related gene family members. GGPP Geranylgeranyl pyrophosphate, PSY phytoene synthase, PDS phytoene desaturase, CrtISO carotenoid isomerase, ZDS zeta-carotene desaturase, LCY-b lycopene beta-cyclase, LCY-e lycopene epsilon cyclase, CHY1, CHY2 carotenoid hydroxylase 1 and 2, Zdep zeaxanthin de-epoxidase, ZEP zeaxanthin epoxidase, NXS neoxanthin synthase, 9-cisEpox 9-cis-epoxidase, CCD carotenoid cleavage deoxygenase. The asterisk indicates the ambiguity with respect to the gene coding for the biosynthesis of neoxanthin as described in the text
A second expression pattern that can be observed for a number of genes within the pathway is characterized by an increase in transcription levels starting at tuber onset (Fig. 8, stage 3: zeta-carotene desaturase, carotenoid isomerase, LCY-b, carotenoid hydroxylase 2 [CHY2], neoxanthin synthase [NXS], and carotenoid cleavage dioxygenase [CCD]). In particular, CHY2 shows a very strong increase in transcript levels during the later stages of tuber growth. It is interesting to note that the CHY2 gene has been mapped to chromosome 3, directly under a quantitative trait locus for flesh color within a diploid potato population and is considered to be a likely candidate for the observed variation in carotenoid content (data not shown). A recent study by Diretto et al. (2007) shows that silencing of both beta-carotene hydroxylase 1 and 2 results in a dramatic shift in carotenoid concentrations. Potato genes with high homology to both a tomato LCY-b (88%) and a potato NXS (89%) show a strong upregulation similar to CHY2. Based on the sequence homology, it currently remains unclear which of the two reactions is catalyzed by the predicted protein and is, therefore, represented in both reactions (Fig. 8).
The oxidative cleavage of several major carotenoids leads to the production of apocarotenoids. These reactions are catalyzed by a family of CCDs (Auldridge et al. 2006). Control of abscisic acid hormone levels is thought to be at least partially controlled by carotene cleavage dioxygenases, and several studies have shown that the loss of these cleavage enzymes has an effect on plant development (reviewed in Auldridge et al. 2006). It is, however, important to note that the current analysis is performed in S. tuberosum cv. Bintje, which is generally considered to contain low amounts of carotenoids, and therefore, expression profiles may deviate significantly in comparison with high-carotenoid content tubers as shown by Morris et al. (2004). We have presented the biosynthesis route of the major carotenoids as an example of transcriptional control analysis using the POCI array; however, other metabolic routes could be equally well analyzed in a similar fashion using gene annotation and GO classification (http://pgrc.ipk-gatersleben.de/poci).
Summary
Within the POCI consortium, we have constructed a new platform for transcriptional analysis of potato using in situ synthesized 60-mer oligo arrays. The oligo design is based on the extended potato unigene set comprising more than 246,000 EST reads from a wide variety of sources. The oligo array contains 42,034 potato features, and its application was first tested through the hybridization of a potato tuber developmental range and provided novel insights in the genes driving this complex process. The design, annotation, and expression data will be made available and will serve as a database resource for potato researchers worldwide (http://pgrc.ipk-gatersleben.de/poci). With the potato genome-sequencing project well underway (www.potatogenome.net), the availability of such a high-throughput transcriptional analysis system will prove important in functional gene annotation and unraveling of complex metabolic or developmental pathways such as tuber development.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Auldridge ME, McCarty DR, Klee HJ (2006) Plant carotenoid cleavage oxygenases and their apocarotenoid products. Curr Opin Plant Biol 9:315–321
Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R (2004) The Gene Ontology Annotation (GOA) Database: sharing knowledge in uniprot with gene ontology. Nucleic Acids Res 32:D262–D266
Crookshanks M, Emmersen J, Welinder KG, Nielsen KL (2001) The potato tuber transcriptome: analysis of 6077 expressed sequence tags. FEBS Lett 506:123–126
Diretto G, Welsch R, Tavazza R, Mourgues F, Pizzichini D, Beyer P, Giuliano G (2007) Silencing of beta-carotene hydroxylase increases total carotenoid and beta-carotene levels in potato tubers. BMC Plant Biol 7:11
Flinn B, Rothwell C, Griffiths R, Lague M, DeKoeyer D, Sardana R, Audy P, Goyer C, Li XQ, Wang-Pruski G, Regan S (2005) Potato expressed sequence tag generation and analysis using standard and unique cDNA libraries. Plant Mol Biol 59:407–433
Horvath BM, Magyar Z, Zhang Y, Hamburger AW, Bako L, Visser RG, Bachem CW, Bogre L (2006) EBP1 regulates organ size through cell growth and proliferation in plants. EMBO J 25:4909–4920
Kloosterman B, Vorst O, Hall RD, Visser RGF, Bachem CW (2005) Tuber on a chip: differential gene expression during potato tuber development. Plant Biotechnol J 3:505–519
Kloosterman B, Navarro C, Bijsterbosch G, Lange T, Prat S, Visser RG, Bachem CW (2007) StGA2ox1 is induced prior to stolon swelling and controls GA levels during potato tuber development. Plant J 52:362–373
Li X, Griffiths R, Lague M, DeKoeyer D, Rothwell C, Haroon M, Stevens B, Xu C, Gustafson V, Bonierbale M, Regan S, Flinn B (2007) EST sequencing and analysis from cold-stored and reconditioned potato tubers. Acta Hortic 745:491–493
Morris WL, Ducreux L, Griffiths DW, Stewart D, Davies HV, Taylor MA (2004) Carotenogenesis during tuber development and storage in potato. J Exp Bot 55:975–982
Mueller LA, Solow TH, Taylor N, Skwarecki B, Buels R, Binns J, Lin C, Wright MH, Ahrens R, Wang Y, Herbst EV, Keyder ER, Menda N, Zamir D, Tanksley SD (2005) The SOL Genomics Network: a comparative resource for Solanaceae biology and beyond. Plant Physiol 138:1310–1317
Nielsen KL, Gronkjaer K, Welinder KG, Emmersen J (2005) Global transcript profiling of potato tuber using LongSAGE. Plant Biotechnol J 3:175–185
Quackenbush J, Cho J, Lee D, Liang F, Holt I, Karamycheva S, Parvizi B, Pertea G, Sultana R, White J (2001) The TIGR Gene Indices: analysis of gene transcript sequences in highly sampled eukaryotic species. Nucleic Acids Res 29:159–164
Rensink W, Hart A, Liu J, Ouyang S, Zismann V, Buell CR (2005a) Analyzing the potato abiotic stress transcriptome using expressed sequence tags. Genome 48:598–605
Rensink WA, Iobst S, Hart A, Stegalkina S, Liu J, Buell CR (2005b) Gene expression profiling of potato responses to cold, heat, and salt stress. Funct Integr Genomics 5:201–207
Rensink WA, Lee Y, Liu J, Iobst S, Ouyang S, Buell CR (2005c) Comparative analyses of six solanaceous transcriptomes reveal a high degree of sequence conservation and species-specific transcripts. BMC Genomics 6:124
Restrepo S, Myers KL, del Pozo O, Martin GB, Hart AL, Buell CR, Fry WE, Smart CD (2005) Gene profiling of a compatible interaction between Phytophthora infestans and Solanum tuberosum suggests a role for carbonic anhydrase. Mol Plant Microbe Interact 18:913–922
Ronning CM, Stegalkina SS, Ascenzi RA, Bougri O, Hart AL, Utterbach TR, Vanaken SE, Riedmuller SB, White JA, Cho J, Pertea GM, Lee Y, Karamycheva S, Sultana R, Tsai J, Quackenbush J, Griffiths HM, Restrepo S, Smart CD, Fry WE, Van Der Hoeven R, Tanksley S, Zhang P, Jin H, Yamamoto ML, Baker BJ, Buell CR (2003) Comparative analyses of potato expressed sequence tag libraries. Plant Physiol 131:419–429
Stephens SM, Chen JY, Davidson MG, Thomas S, Trute BM (2005) Oracle database 10 g: a platform for BLAST search and regular expression pattern matching in life sciences. Nucleic Acids Res 33:D675–D679
Viola R, Roberts AG, Haupt S, Gazzani S, Hancock RD, Marmiroli N, Machray GC, Oparka KJ (2001) Tuberization in potato involves a switch from apoplastic to symplastic phloem unloading. Plant Cell 13:385–398
Xu X, van Lammeren AA, Vermeer E, Vreugdenhil D (1998a) The role of gibberellin, abscisic acid, and sucrose in the regulation of potato tuber formation in vitro. Plant Physiol 117:575–584
Xu X, Vreugdenhil D, van Lammeren AAM (1998b) Cell division and cell enlargement during potato tuber formation. J Exp Bot 49:573–582
Acknowledgments
We wish to thank Robin Buell (TIGR), Xiu-Qing Li (AAFC), and Monique Wordragen (A&F) for their contribution of EST sequences for the POCI assembly and Marc Ghislain (International Potato Center CIP) and Mark Taylor (SCRI) for their participation in the POCI consortium. In addition, we greatly appreciate the bioinformatics support provided by Martin Lagüe (AAFC) and the helpful discussions with the people from Agilent Technologies. This work was partially supported by Genome Canada, the Atlantic Innovation Fund of the Atlantic Canada Opportunities Agency (1004889-1), the New Brunswick Department of Agriculture, Fisheries and Aquaculture (AFA03/04-096), The Scottish Government Rural and Environment Research and Analysis Directorate, and the EU-SOL project (PL 016214-2 EU-SOL). Parts of this project were carried out within the research program of the Centre of BioSystems Genomics (CBSG), which is part of The Netherlands Genomics Initiative/Netherlands Organization for Scientific Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Bjorn Kloosterman and David De Koeyer contributed equally
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table S1
Libraries used to generate ESTs and number of raw ESTs used for the POCI EST assembly (DOC 96.5 KB)
Supplementary Table S2
Relative gene expression data of the six tuber developmental stages (XLS 20.5 KB)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kloosterman, B., De Koeyer, D., Griffiths, R. et al. Genes driving potato tuber initiation and growth: identification based on transcriptional changes using the POCI array. Funct Integr Genomics 8, 329–340 (2008). https://doi.org/10.1007/s10142-008-0083-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-008-0083-x